

Bioinformatic Analysis for Profiling Drug-induced Chromatin Modification Landscapes in Mouse Brain Using ChlP-seq Data
利用ChIP-seq数据分析药物诱导小鼠大脑中染色质修饰发生改变的生物信息学方法
发布: 2017年02月05日第7卷第3期 DOI: 10.21769/BioProtoc.2123 浏览次数: 12818
评审: Oneil G. BhalalaSalma HasanSabine Le Saux
参见作者原研究论文
The authors used this protocol in:
Dec 2012
Advertisement









Abstract
Chromatin immunoprecipitation followed by massively parallel sequencing (ChIP-seq) is a powerful technology to profile genome-wide chromatin modification patterns and is increasingly being used to study the molecular mechanisms of brain diseases such as drug addiction. This protocol discusses the typical procedures involved in ChIP-seq data generation, bioinformatic analysis, and interpretation of results, using a chronic cocaine treatment study as a template. We describe an experimental design that induces significant chromatin modifications in mouse brain, and the use of ChIP-seq to derive novel information about the chromatin regulatory mechanisms involved. We describe the bioinformatic methods used to preprocess the sequencing data, generate global enrichment profiles for specific histone modifications, identify enriched genomic loci, find differential modification sites, and perform functional analyses. These ChIP-seq analyses provide many details into the chromatin changes that are induced in brain by chronic exposure to cocaine, and generates an invaluable source of information to understand the molecular mechanisms underlying drug addiction. Our protocol provides a standardized procedure for data analysis and can serve as a starting point for any other ChIP-seq projects.
Keywords: Chromatin immunoprecipitation (ChIP) (染色质免疫沉淀(ChIP))Background
Chromatin modification has been implicated in the molecular mechanisms of drug addiction and may hold the key to understanding multiple aspects of addictive behaviors (Robison and Nestler, 2011). Chromatin Immuno-Precipitation (ChIP) followed by massively parallel sequencing (ChIP-seq) is the current state of the art technology to profile the chromatin landscape. The typical procedure of ChIP-seq involves: 1) using the antibody against a protein of interest to pull down the binding DNA, which has been fixed to the protein and broken into smaller fragments, 2) the immunoprecipitated DNA is then purified and constructed into a library for high throughput sequencing of short reads (usually 50-100 bp) from the ends of insert DNA fragments, 3) the short reads are aligned to the genome and put through data analysis. Compared with its predecessor – ChIP-chip, ChIP-seq has unparalleled advantages such as unbiased coverage of the entire genome, single base resolution, and significantly improved signal-to-noise ratio (Park, 2009). It has proven to be an invaluable tool to understand numerous types of chromatin modifications.
Brief overview of ChIP-seq experiment: Please see original research articles for greater experimental details (Renthal et al., 2009; Lee et al., 2006). Briefly, adult mice received a standard regimen of repeated cocaine (7 daily IP injections of cocaine [20 mg/kg] or saline) and were used 24 h after the last injection (Robison and Nestler, 2011). The nucleus accumbens, a major brain reward region, was obtained by punch dissection and used for ChIP-seq. Chromatin IP was performed as described previously (Renthal et al., 2009; Lee et al. 2006; http://jura.wi.mit.edu/young_public/hES_PRC/ChIP.html) with minor modifications, using two antibodies, anti-H3K4me3 (tri-methylation of Lys4 in histone H4) (Abcam #ab8580) and anti-H3K9me3 (Abcam #ab8898). Sequencing libraries for each experimental condition were generated in triplicate, and were then sequenced on an Illumina sequencer.
Bioinformatic analysis: The sequencing data generated from libraries under treatment and corresponding control conditions will be used to identify the specific genomic regions that have undergone chromatin changes. We can then associate these regions with biological functions and select the regions of interest for further study. The basic procedure for this kind of bioinformatic analysis can be laid out as a pipeline of eight steps (Figure 1):
1. Perform quality control analyses on the sequencing data files;
2. Align the sequences to the genome;
3. Remove PCR duplicates;
4. Perform cross-correlation quality analysis;
5. Generate coverage files to be loaded into a genome browser;
6. Generate global coverage plots;
7. Perform peak detection and annotation;
8. Perform differential analysis and functional association.
This protocol shall explain each of these steps in more detail, including some of the principles and rationale underlying the bioinformatics analyses, and provide the necessary commands for execution in a Unix-like command-line environment. 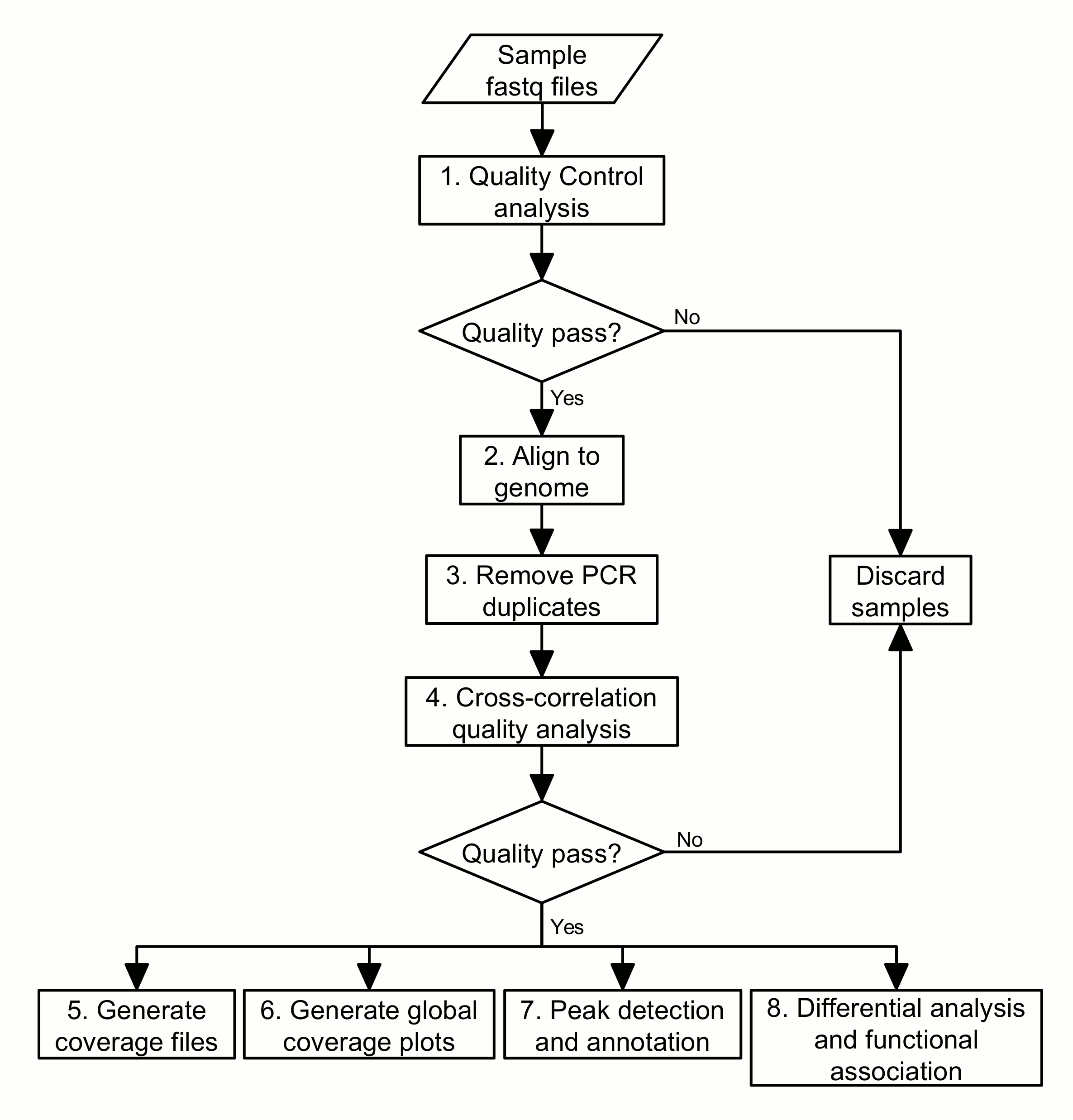
Figure 1. Flowchart of the ChIP-seq analysis pipeline
Equipment
- Personal computer or high performance computing cluster. All software mentioned here can be run under a Unix-like workstation, such as Linux and Mac. For Windows users, a terminal emulator, such as Cygwin (http://cygwin.com/), can be used.
Software
- FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
- Bowtie2 (Langmead and Salzberg, 2012; http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)
- Samtools (Li et al., 2009; http://samtools.sourceforge.net/)
- phantompeakqualtools (http://code.google.com/archive/p/phantompeakqualtools/)
- IGV and IGVtools (Robinson et al., 2011; http://software.broadinstitute.org/software/igv/)
- ngs.plot (Shen et al., 2014; http://github.com/shenlab-sinai/ngsplot)
- MACS (Zhang et al., 2008; http://github.com/taoliu/MACS)
- diffReps (Shen et al., 2013; http://github.com/shenlab-sinai/diffreps)
- bedtools (Quinlan and Hall, 2010; http://bedtools.readthedocs.io/en/latest/)
- region-analysis (Shao et al., 2016; http://github.com/shenlab-sinai/region_analysis)
- ChIPseqRUs (Loh et al., 2016; https://github.com/shenlab-sinai/chip-seq_preprocess)
- David (Dennis et al., 2003; http://david.ncifcrf.gov/)
- IPA (http://www.ingenuity.com/)
- GREAT (McLean et al., 2010; http://bejerano.stanford.edu/great/public/html/)
Procedure
文章信息
版权信息
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Loh, Y. E., Feng, J., Nestler, E. and Shen, L. (2017). Bioinformatic Analysis for Profiling Drug-induced Chromatin Modification Landscapes in Mouse Brain Using ChlP-seq Data. Bio-protocol 7(3): e2123. DOI: 10.21769/BioProtoc.2123.
分类
系统生物学 > 表观基因组学 > 测序 > ChIP-seq
系统生物学 > 表观基因组学 > 组蛋白修饰
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。