

往期刊物2025
卷册: 15, 期号: 21
生物化学

Lipid-Mediated Sequential Recruitment of Proteins Via Dual SLIPT and Dual SLIPTNVOC in Live Cells
基于双SLIPT与双SLIPT-NVOC的脂质介导蛋白依序定位活细胞研究方法
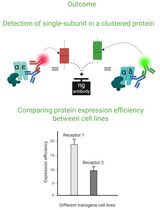
Cluster FLISA—A Method to Compare Protein Expression Efficiency Between Cell Lines and Subunit Clustering of Proteins
Cluster FLISA——用于比较不同细胞系蛋白表达效率及蛋白亚基聚集状态的方法
生物信息学与计算生物学
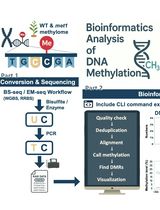
Computational Workflow for Genome-Wide DNA Methylation Profiling and Differential Methylation Analysis
全基因组DNA甲基化分析与差异甲基化分析的计算流程


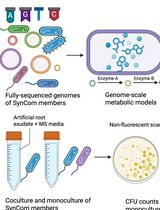
In Silico Prediction and In Vitro Validation of Bacterial Interactions in the Plant Rhizosphere Using a Synthetic Bacterial Community
基于合成细菌群落的植物根际细菌相互作用的计算预测与体外验证
生物工程

Production of Genetically Engineered Extracellular Vesicles for Targeted Protein Delivery
制备基因工程化细胞外囊泡用于靶向蛋白递送
细胞生物学
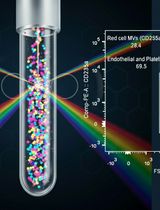
Protocol for the Isolation and Analysis of Extracellular Vesicles From Peripheral Blood: Red Cell, Endothelial, and Platelet-Derived Extracellular Vesicles
外周血中细胞外囊泡的分离与分析方法:红细胞、内皮细胞及血小板来源的细胞外囊泡
医学

A Quantitative Spectrophotometric Assay Matched With Environmental Scanning Electron Microscopy to Measure Calcium Crystals in Human Osteoarthritic Synovial Fluid
结合环境扫描电子显微镜的定量分光光度法检测人骨关节炎滑液中的钙晶体
微生物学
Optimized Protocol for the Collection, Cryopreservation, and In Vitro Cultivation of Human Gut Microbiota for Toxicomicrobiomics Applications
用于毒理微生物组学研究的人体肠道微生物采集、冷冻保存与体外培养优化方法

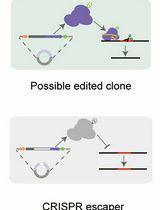
A Practical CRISPR-Based Method for Rapid Genome Editing in Caulobacter crescentus
基于CRISPR的快速基因组编辑方法在新月弧菌中的应用
分子生物学
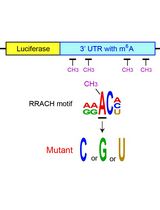
Examining the Roles of m6A Sites in mRNA Using the Luciferase Gene Fused With Mutated RRACH Motifs
利用融合突变RRACH基序的荧光素酶基因研究mRNA中m6A位点的功能
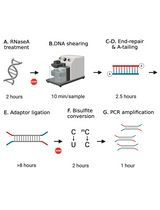
Library Preparation for Genome-Wide DNA Methylation Profiling
全基因组DNA甲基化分析的文库构建方法
神经科学
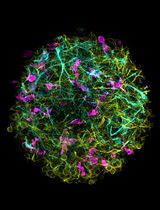
Generation of 3D Human iPSC-Derived Multi-Cell Type Neurospheres for Studying Neuron, Astrocyte, and Microglia Crosstalk
构建三维人源iPSC多细胞类型神经球以研究神经元、星形胶质细胞和小胶质细胞的相互作用
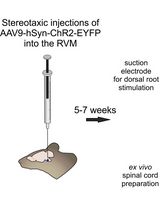
Optogenetic Approach for Investigating Descending Control of Nociception in Ex Vivo Spinal Cord Preparation
利用光遗传学研究离体脊髓中疼痛下行调控机制
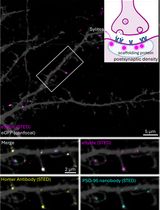
Labeling Postsynaptic Densities for Super-Resolution Microscopy With Minimal Signal-Loss and Offset
低信号损失与低偏移的突触后致密区超分辨显微标记方法
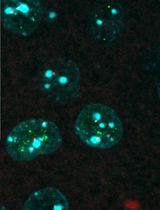
Colocalizing Telomeres With PML or γH2AX Foci by IF-FISH in Mouse Brain Neurons
利用 IF-FISH 技术在小鼠脑神经元中检测端粒与 PML 或 γH2AX 焦点的共定位
植物科学

Live-Cell Monitoring of Piecemeal Chloroplast Autophagy
活细胞监测叶绿体片段自噬过程

A Reliable In Planta Inoculation and Antifungal Screening Protocol for Rhizoctonia solani-Induced Sheath Blight in Rice
水稻纹枯病的体内接种与抗真菌筛选可靠方法
