
- Home
- Protocols

-

Intact in situ Preparation of Drosophila melanogaster Lymph Gland for Comprehensive Analysis of Larval Hematopoiesis
Published: May 20, 2021 DOI: 10.21769/BioProtoc.4052 Views: 4103
Original research article
The authors used this protocol in:

Feb 2021
Abstract
Blood cells have a limited lifespan and are replenished by a small number of hematopoietic stem and progenitor cells (HSPCs). Adult vertebrate hematopoiesis occurs in the bone marrow, liver and spleen making a comprehensive analysis of the entire HSPC pool nearly impossible. The Drosophila blood system is well studied and has developmental, molecular and functional parallels with the vertebrate system. Unlike vertebrates, post-embryonic hematopoiesis in Drosophila is essentially restricted to the larval lymph gland (LG), a multi-lobed organ that flanks the dorsal vessel. The anterior-most or primary lobes of the LG are easy to dissect out and hence their cellular and molecular characteristics have been studied in considerable detail. The 2-3 pairs of posterior lobes are more delicate and fragile and hence have largely been ignored. However, posterior lobes harbor a significant blood progenitor pool and several hematopoietic mutants show differences in phenotype between the anterior and posterior lobes. Hence a comprehensive analysis of the LG is important for a thorough understanding of Drosophila hematopoiesis. Our studies showed that a larval fillet method of dissection allows preparation of the whole LG with intact posterior lobes, which is important for meaningful analysis. Here we provide a detailed protocol for larval fillet preparation to access and analyze the complete LG lobes along with the dorsal vessel and pericardial cells. We demonstrate that tissue architecture and integrity is maintained and provides methods of quantitative analysis. This protocol can be used to quickly and effectively isolate complete LGs from first instar larval to pupal stages and can be implemented with ease.
Keywords: Drosophila hematopoiesisBackground
Vertebrate hematopoietic stem and progenitor cells (HSPCs) give rise to various kinds of mature blood cell types. HSPCs can be identified by surface marker phenotype, staining properties of vital dyes, proliferative ability and in vivo differentiation potential (Crisan and Dzierzak, 2016; Ema et al., 2014; Granick et al., 2012). Murine and Zebrafish in vivo models have proved extremely useful in understanding various aspects of vertebrate HSPC biology. Adult mouse HSPCs primarily reside in the bone marrow although recent studies show that HSPCs can circulate in the peripheral blood (Granick et al., 2012; Wright et al., 2001). Bone marrow is primarily located in the flat bones of the pelvis, vertebrae, ribs, and cranium; and in the long bones of the tibia, femur and humerus. However, for post-embryonic analysis HSPCs are obtained mainly from the long bones of the tibia and femur that represent a subset of the entire progenitor population. Distribution of HSPCs across various anatomical sites makes it difficult to study the entire progenitor pool especially in post-embryonic stages and in larger animals such as mouse and human. The Drosophila hematopoietic system has proved helpful in addressing various aspects of hematopoiesis owing to conserved signaling mechanisms and transcriptional factors that regulate hematopoiesis (Banerjee et al., 2019).
Drosophila hematopoiesis occurs in two successive waves. First, blood cell progenitors emerge from the procephalic/ head mesoderm in the early embryo and give rise to larval circulating and sessile hemocytes, which persist until adulthood (Ghosh et al., 2015; Holz et al., 2003; Honti et al., 2010; Sanchez et al., 2019; Tepass et al., 1994). The second wave of hematopoiesis takes place in a specialized larval hematopoietic organ the lymph gland (LG), located dorsally, flanking the anterior cardiac tube/ dorsal vessel (Grigorian et al., 2013; Lanot et al., 2001; Mandal et al., 2004; Rugendorff et al., 1994). Blood cell progenitors that form the LG are derived from the embryonic dorsal mesoderm and clonal analysis suggests the presence of hemangioblast precursor cells that can give rise to LG blood cells and cells of the dorsal vessel (Mandal et al., 2004). By stage 11, Odd-skipped (Odd) is expressed in the thoracic and the abdominal segments, T1-A6 (Ward and Skeath, 2000). The thoracic clusters form the LG and the abdominal clusters give rise to the pericardial cells (Mandal et al., 2004). At stage 11-12, expression of the homeotic gene, Antennapedia is restricted to segment T3 (Mandal et al., 2007). By stage 13-16, Odd+ cells in the thoracic segment (T1-T3) coalesce to form the LG, whereas, Antennapedia is expressed in 5-6 cells at the posterior boundary of the LG primordium (Mandal et al., 2007). Two Collier expressing clusters appear in the thoracic segment T2 and T3 that coalesce following germ-band retraction (Crozatier et al., 2004). Collier expression is maintained at high levels at the posterior tip of the developing LG in 3-5 cells whereas, the remaining LG cells express Collier at low levels (Crozatier et al., 2004). In the late embryo, the LG consists of a single pair of lobes called the primary/ anterior lobes containing approximately 20 cells in each lobe that express Serpent and Odd (Jung et al., 2005). At the first instar larval stage, primary lobe cells proximal to the dorsal vessel express Serpent, Notch, Dorothy, STAT92e and lack expression of domeless (dome) and are termed as pre-progenitors (Banerjee et al., 2019; Dey et al., 2016; Jung et al., 2005). By the second instar primary lobes have increased in size, consisting of approximately 200 cells in each lobe. Additionally, 2-3 pairs of smaller lobes are formed posterior to the primary lobes referred to as the secondary, tertiary and quaternary lobes (Banerjee et al., 2019; Jung et al., 2005; Rodrigues et al., 2021).
Based on morphology and molecular marker analysis, third instar larval LG primary lobes are compartmentalized into three zones. The posterior signaling center (PSC) acts as the signaling niche. The medullary zone (MZ) towards the cardiac tube consists of multi-potent progenitors. A peripheral cortical zone (CZ) mainly harbors phagocytic plasmatocytes and a few crystal cells. Intermediate zone (IZ) progenitors reside in the region between the MZ and the CZ and are identified by the expression of progenitor and early differentiation markers and lack the expression of late markers (Banerjee et al., 2019; Jung et al., 2005). The multiple posterior lobes harbor progenitors that resist differentiation upon immune challenge (Rodrigues et al., 2021). Under steady state conditions, blood cells produced in the LG are released in circulation only at the pupal stage and contribute to the pupal and adult blood cell populations (Ghosh et al., 2015; Grigorian et al., 2011; Holz et al., 2003; Sanchez et al., 2019).
While primary lobes are well characterized, the identity of the posterior lobes was ill-characterized till recently (Rodrigues et al., 2021). Based on the expression of a limited set of markers and mutant analysis, a few studies proposed that the secondary lobes are essentially composed of blood cell progenitors that differentiate at the larval/pupal transition (Benmimoun et al., 2015; Grigorian et al., 2011; Jung et al., 2005; Kulkarni et al., 2011). Studies on secondary/posterior lobes used preparation of lymph gland samples detached from their brain/ring gland anterior attachment site with thin tungsten needles and placed on glass slides (Lanot et al., 2001). This method of sample preparation causes damage to the delicate organ and might be the reason for the partial analysis of LG lobes. To obtain the entire intact LG, we use the larval fillet method of dissection described in this protocol that helps maintain intact primary and posterior lobes. This protocol has been invaluable for a comprehensive analysis in our previous studies (Khadilkar et al., 2014; Kulkarni et al., 2011; Rodrigues et al., 2021; Sinha et al., 2013; Sinha et al., 2019). For instance, we could show that depletion of asrij, arf1 or garz and overexpression of arf1GAP1 leads to severe phenotypes of hyperproliferation and premature differentiation in the posterior lobes as compared to the primary lobes (Khadilkar et al., 2014; Kulkarni et al., 2011). We also employed this method of dissection for whole LG proteomic analysis, which provided a resource to identify novel regulators of hematopoiesis (Sinha et al., 2019). Further, differential RNA sequencing analysis for the primary and the posterior lobes helped identify novel progenitor markers and regulators of hematopoiesis and unveiled the molecular heterogeneity as well as functional compartmentalization of the LG progenitor pool present in the different lobes (Rodrigues et al., 2021). Our studies thus far suggest that analysis of the whole LG is crucial for exploring the complete application of Drosophila LG hematopoiesis. Here we describe detailed protocols for whole LG sample preparation that can be used for GFP expression screens, immunostaining, RNA in situ and high-throughput analyses.
Materials and Reagents
Fly stocks
Canton-S was used as the wild type reference strain
dome-Gal4,UAS2xEGFP (provided by Utpal Banerjee, University of California Los Angeles)
srpHemo-Gal4-UAS-GFP (National Centre for Biological Sciences (NCBS), Fly Facility).
Materials
Fine paint brush (number- 2)
Glass cavity dish 40 x 40 mm (Atom Scientific, catalog number: SDCE4040-1)
Sylgard (Sigma-Aldrich, catalog number: 761036) or equivalent
Micro test plate- 96 well (Tarsons, catalog number: 941196) or equivalent
35 mm Petri dish (Tarsons, catalog number: 460035) or equivalent
Reagents
Tissue dissection and fixation
10x Phosphate Buffered Saline (PBS, pH 7) stock (see Recipe)
Paraformaldehyde (Fisher Scientific, catalog number: 23995)
Immunostaining and mounting
Triton X-100 (Sigma-Aldrich, catalog number: T8787)
Normal Goat serum (GeNei, catalog number: NS1)
Primary antibodies: mouse anti-P1/NimC1 and mouse anti-Hemese (provided by Istvan Ando, Biological Research Center of the Hungarian Academy of Sciences), rabbit anti-Asrij (Kulkarni et al., 2011).
Secondary antibodies: Alexa fluor 568 goat anti-mouse (Invitrogen, catalog number: A11004)
Alexa fluor 488 goat anti-rabbit (Invitrogen, catalog number: A11008)
Phalloidin conjugated to Alexa fluor 633 (Invitrogen, catalog number: A22284)
DAPI (4',6-Diamidino-2-Phenylindole, Dihydrochloride, ThermoFisher Scientific, catalog number: D1306)
Glycerol (Merck, catalog number:BP229-1)
Neutral red (Merck, catalog number: N4638)
RNA in situ hybridization
In situ hybridization probe: tep4 (Rodrigues et al., 2021)
20x Saline-Sodium Citrate buffer (20x SSC, pH-7) (see recipe)
Methanol (Merck, catalog number: 34860)
Tween-20 (Sigma-Aldrich, catalog number: P9416)
Molecular grade water-UltraPure DNase/RNase-free distilled water (Invitrogen, catalog number: 10977015)
Formamide (Sigma-Aldrich, catalog number: 11814320001)
tRNA (Sigma-Aldrich, catalog number: 10109517001)
Heparin (Sigma-Aldrich, catalog number: H3149)
Roche blocking agent (Sigma-Aldrich, catalog number: 11096176001)
CHAPS (Sigma-Aldrich, catalog number: C9426)
EDTA (Fisher Scientific, catalog number: S311-100)
NaCl (Fisher Scientific, catalog number: S25542)
MgCl2 (Fisher Scientific, catalog number: BP214-500)
Anti-DIG conjugated to alkaline phosphatase (Sigma-Aldrich, catalog number: 11093274910)
NBT/BCIP (Promega, catalog number: S3771)
FastRed substrate kit (Abcam, catalog number: ab64254)
Equipment
Stereomicroscope (Olympus SZ51, magnification range 0.8x-4x)
Fine forceps (Fine Science Tools, Dumont #5, catalog number: 11252-20)
Spring scissors 2.5 mm cutting edge (Fine Science Tools, catalog number: 15000-08)
Insect pins (Fine Science Tools, Minutien, 0.1 mm, stainless steel, catalog number: 26002-10)
Confocal microscope (Zeiss, LSM 880)
Software
ImageJ
Adobe Photoshop CS5 (Adobe Systems)
Procedure
Larval fillet preparation for obtaining intact lymph gland (LG):
Whole LG preparations can be obtained for the first, second, third instar larvae and pupae using this method of dissection. Fly breeding and crosses were performed using standard protocols and larvae were reared to the appropriate stage on standard cornmeal agar medium under non saturating density. Figure 1A shows the relative size of the first, second and third instar larvae.

Figure 1. Schematic representation of larval LG dissection. (A) Representative image of first, second and third instar larvae. (B-F) Step-wise schematic representation of LG dissection from wandering third instar larvae. BL: brain lobes, VNC: ventral nerve cord, LG: lymph gland and PC: pericardial cells. Number of pericardial cells may vary between LG lobes.Larvae and LG images are not depicted to scale but enlarged for clarity.Using a fine paint brush transfer the larvae to a cavity dish/ Petri plate containing water, then rinse the larvae to get rid of any food particles.
Transfer the larvae to a clean cavity dish and place on ice for 20-30 min to immobilize the larvae. Immobilization (optional) helps to pin the larvae (see step 4).
Place cooled larva with the dorsal side up on the Sylgard dish and view it through a stereomicroscope at magnification 4x (zoom range 0.8x-4x), focusing on the dorsal cuticle. All subsequent steps are to be performed while viewing larval tissue under the microscope.
Restrain larva by inserting insect pins firmly through it near the anterior and the posterior spiracles and through the Sylgard dish (Figure 1B). Add a drop of PBS (about 200 µl) to prevent the larva from desiccating.
Using fine dissection scissors make a small incision in the cuticle on the right side near the posterior end. Insert the scissors into the incision end and slit the cuticle laterally (Figure 1C, D).
Lift the loose end of the cuticle with the help of fine forceps, extend it to the left side and cut along the left lateral edge of the cuticle (Figure 1E).
Carefully remove the viscera. Locate the LG that is attached to the brain lobes in the anterior, flanking the dorsal vessel, followed by rows of pericardial cells at the posterior end (Figure 1F). Side illumination with dual goose-neck light source can help distinguish the refringent LG.
Add a fresh drop of PBS and gently remove it to get rid of any tissue debris. Repeat if required.
Replace PBS with 200 µl fixative (4% paraformaldehyde, see recipe) and incubate for 20 min at room temperature. Remove the fixative, wash three times with 1x PBS then carefully remove the pins, slowly lift the LG preparation (with the underlying ventral cuticle) from the posterior end and transfer to a 96-well plate or a 1.5 ml microcentrifuge tube for immunostaining or in situ procedures.
Pupal fillet preparation for obtaining intact lymph gland (LG):
Whole LG can be prepared from 0 to 20h after pupa formation (APF). In our experience, most lobes histolyze by 15h APF (Rodrigues et al., 2021).
Place pupa on the Sylgard dish with the dorsal side facing up. All subsequent steps are to be performed while viewing the pupal tissue under the stereomicroscope.
Insert a fine insect pin firmly near the anterior spiracle through the Sylgard dish (Figure 2A). Add a drop of PBS (about 200 µl) to prevent the pupa from desiccating.
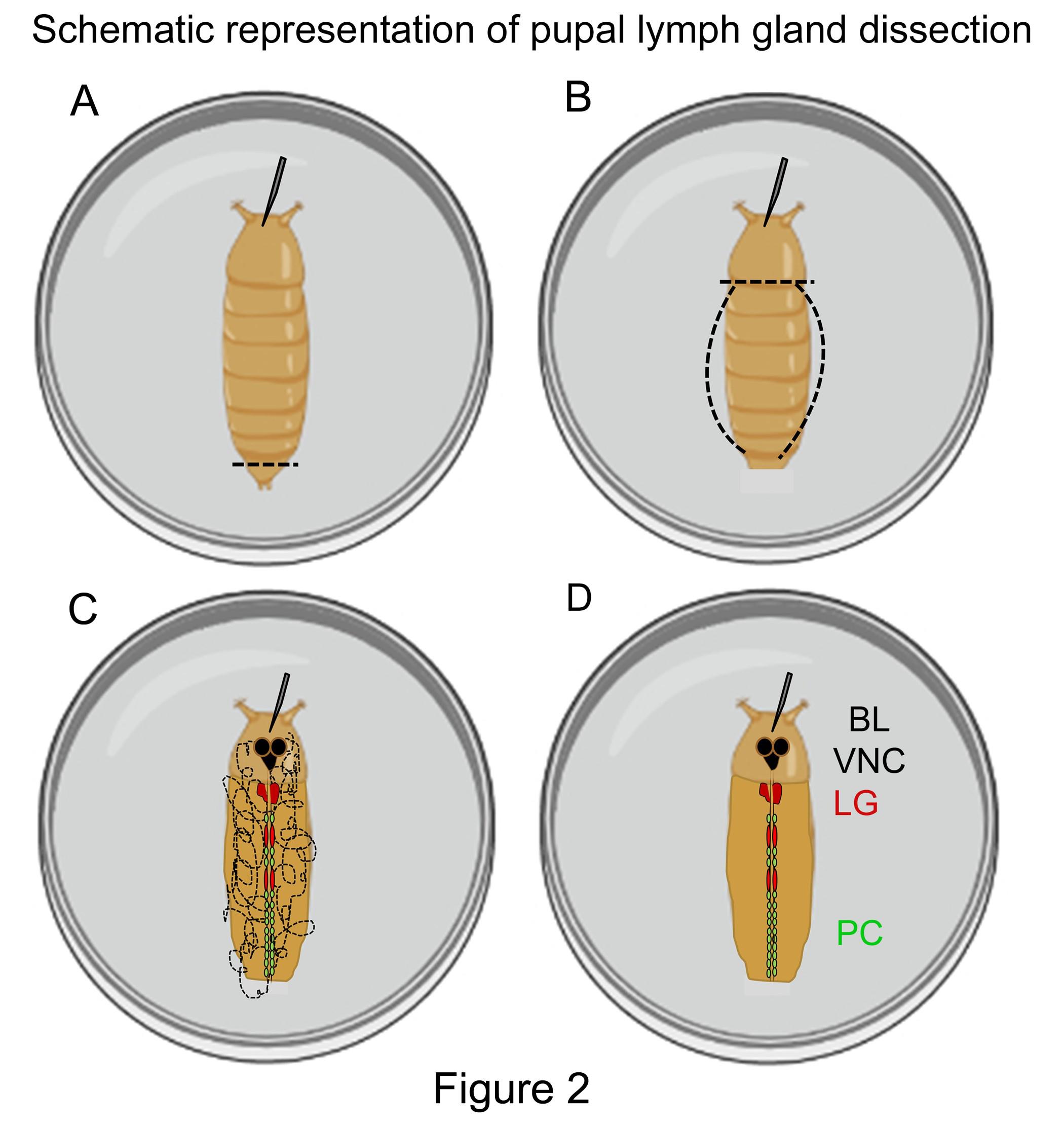
Figure 2. Schematic representation of pupal LG dissection. (A-D) Step-wise schematic representation of pupal LG dissection. BL: brain lobes, VNC: ventral nerve cord, LG: lymph gland and PC: pericardial cells. Number of pericardial cells may vary between LG lobes. Pupa and LG images are not depicted to scale but enlarged for clarity.Using fine dissection scissors cut horizontally along the posterior spiracles (Figure 2A).
Insert the scissor into the incised end and make a slit laterally along the right side of the cuticle, followed by incision along the anterior part as shown by the dotted lines (Figure 2B).
Lift the loose end of the cuticle and carefully extend it to the left side and cut along the left lateral edge of the cuticle along the dotted lines (Figure 2B).
Carefully remove the visceral organs without damaging the LG (Figure 2C). The cardiac tube and pericardial cells are a good landmark to locate the remaining LG lobes (Figure 2D). Add a fresh drop of PBS to remove any tissue debris, repeat if required.
Follow step 9 onwards of larval LG dissection and proceed for immunostaining or in situ procedures.
Neutral red staining of lymph gland (LG) for easier visualization:
Dissect and fix larval or pupal LG as described above. Wash thrice in 1x PBS for 8-10 min each.
Add 100 µl of 0.2% neutral red solution (0.2% neutral red in 1x PBS) and incubate at room temperature for 5-7 min or till LG is visibly stained.
Wash off neutral red with 1x PBS. Brain lobes and LG lobes should be easily visible. First, second, third instar and pupal LG can be easily identified by neutral red staining (Figure 3A-D).

Figure 3. LG preparations stained with neutral red. (A-D) Hemi-dissected fillet preparation with intact primary and posterior lobes stained with neutral red. LG lobes are intensely stained (red) as shown for first instar (A) second instar (B) third instar (C) larvae and pupa (D). BL: brain lobes, VNC: ventral nerve cord, LG: lymph gland, Pri: primary lobes, Sec: secondary lobes, Tert: tertiary lobes and asterisks indicate pericardial cells. Scale bar: 100μm
Immunostaining of lymph gland (LG):
All steps are performed by placing the 96-well dish on a flat bed rocker.
Wash 10-12 fixed LG preparations in a 96-well dish in 200 µl 1x PBS, three times for 10-15 min each.
Add 200 µl of 0.1% Triton X-100 in 1x PBS (0.1% PTX, see recipe) as a permeabilizing agent to the LG preparations incubate for 15-20 min.
Discard PTX, add 100 µl of 20% normal goat serum in 1x PBS as a blocking agent and, incubate for 30 min.
Remove the blocking agent and add 40 µl of primary antibody at the desired concentration diluted in 1x PBS (or blocking agent). Incubate at 4°C overnight.
Remove the primary antibody, wash the LG preparations with 1x PBS, three times for 10-15 min each.
Discard PBS, add 100 µl of 20% normal goat serum and incubate for 30 min.
Replace with 40 µl of secondary antibody (usually 1:400) in 1x PBS and incubate for 2 h at room temperature.
Remove secondary antibody, wash three times in 200 µl of 1x PBS for 10-15 min each. Proceed to the mounting step (see below, procedure section F).
In situ hybridization of lymph gland (LG):
Wash 15-20 fixed LG preparations in a microcentrifuge tube in 500 µl 1x PBS three times for 10-15 min each.
For long term storage, wash LG preparations 4 times in methanol and store in methanol at -20°C. Rinse the samples once with 50% methanol – 50% PBS and three times in 1x PBS to resume the experiment.
Wash LG preparations in 500 µl of 0.1% Tween-20 in 1x PBS (PBT) for three times, 10-15 min each (See recipe for 0.1% PBT).
Equilibrate LG preparations in 500 µl of equal volumes of 0.1% PBT and hybridization buffer (HB). (See recipe for hybridization buffer)
Discard the PBT-HB solution and, pre-incubate in 500 µl HB for 1 h at 65°C.
Remove HB and replace with 200 µl HB with 1 µl of Digoxigenin (DIG)-labelled RNA probe (Dilution of the probe needs to be adjusted empirically depending on the probe concentration and level of expression of the gene of interest). Hybridise overnight at 65°C.
Remove HB carefully and wash the LG preparations in 500-800 µl of HB for 1 h at 65°C.
Wash LG preparations in 500-800 µl equal volumes of 0.1% PBT and HB for 30 min at 65°C, followed by three quick washes in 0.1% PBT at room temperature.
Follow up with three washes in 0.1% PBT for 10 min each at room temp.
Block non-specific binding with 1% bovine serum albumin (BSA) or normal goat serum (NGS) in 0.1% PBT for 30 min.
Incubate with anti-DIG antibody coupled to Alkaline Phosphatase diluted (1:1000) in the blocking solution for 2 h at room temp.
Remove the anti-DIG blocking solution mixture, followed by three quick rinses in 0.1% PBT and then three 10 min washes in 0.1% PBT.
Equilibrate for 10 min in 500 μl freshly prepared staining buffer (SB) (see recipe for staining buffer).
Visualize with NBT/BCiP in 1 ml SB (6.5 μl NBT + 3.5 μl BCiP) at room temperature or 37°C until staining is visible.
Wash 3 times in 1x PBS and mount the LG preparations as indicated below.
Mounting intact lymph gland (LG):
Place the fixed and stained larval fillet preparation in a glass bottom 35 mm Petri dish. These can also be made by punching a hole of 1 cm diameter in a 35 mm Petri dish and gluing a coverslip to the bottom of the dish (Figure 4). Add a small drop (10-20 µl) of 70% glycerol with DAPI (1:500) or mounting medium to prevent the samples from desiccating.
Using a pair of forceps hold the filleted cuticle at the anterior end. With another fine forceps carefully detach the brain lobes from rest of the cuticle. The LG is attached via the ring gland to the brain lobes and is flanking the dorsal vessel (Figure 1F). This procedure will partially dislodge it from the cuticle.
Next, using a pair of forceps, hold the cuticle at the posterior end and with another forceps carefully detach and slide the posterior part of the dorsal vessel along with the pericardial cells on to the coverslip. The tissue is very fragile and the whole procedure has to be done gently while viewing through a stereomicroscope.
Once the LG is loosened from the anterior and the posterior ends carefully move the LG away from the ventral cuticle. Do not directly lift or hold the LG. Grasp the brain lobes at the anterior and the pericardial cells at the posterior end when moving the LG.
Multiple lymph glands can be mounted in this way in a single dish (Figure 4) and can be subjected to high resolution imaging analysis.
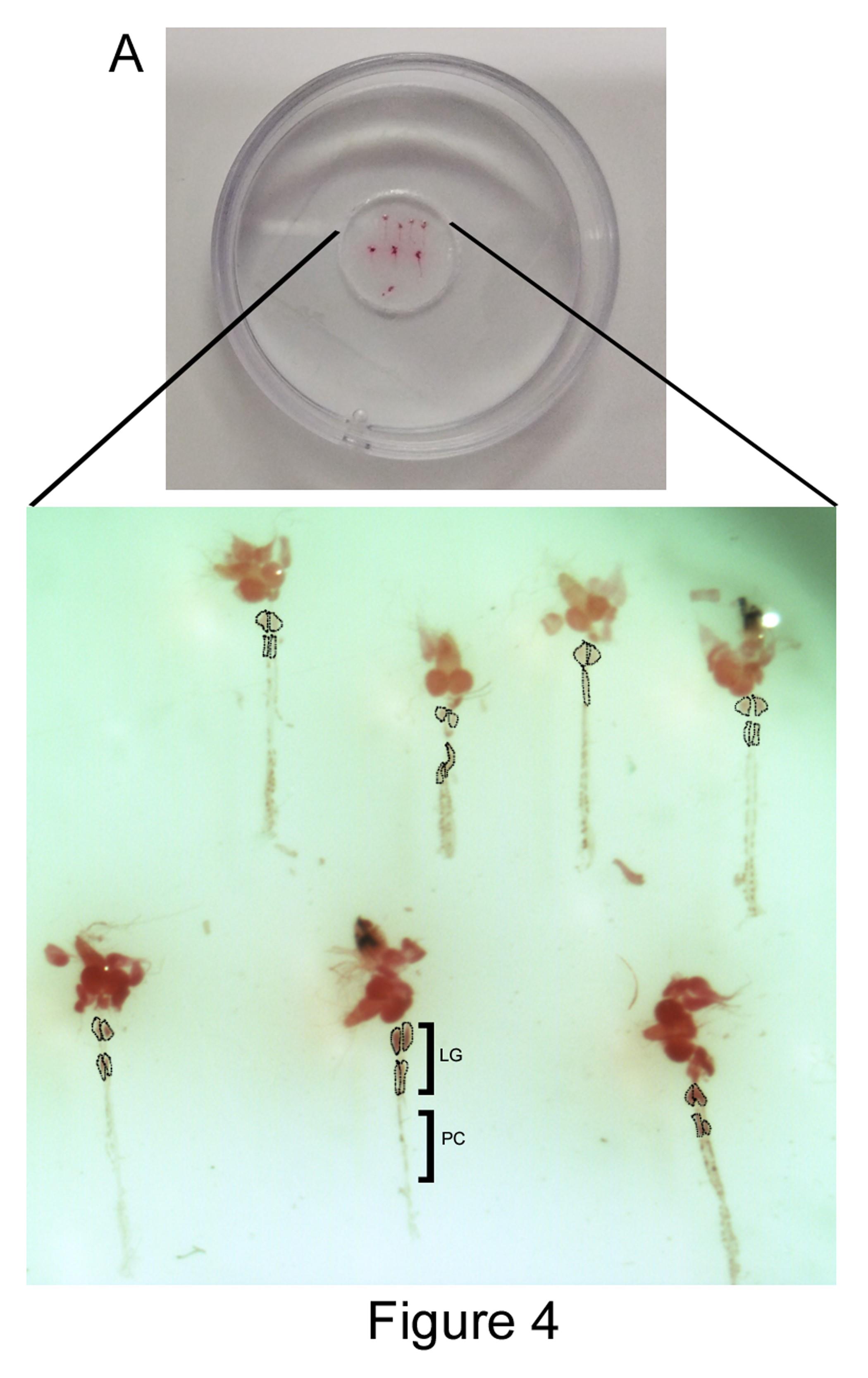
Figure 4. LG preparations mounted in a 35 mm coverslip bottom dish. 35 mm coverslip bottom dish shows seven third instar larval LG preparations stained with neutral red. Black dotted lines indicate LG lobes. LG: lymph gland and PC: pericardial cells.
Data analysis
Representative results
The Drosophila third instar larval LG is a multi-lobed organ that flanks the dorsal vessel. The first pair of lobes called as the primary lobes are followed by 2-3 pairs of posterior lobes- the secondary, tertiary and (rarely) quaternary lobes separated by pericardial cells that function as nephrocytes. For simplicity we refer to the secondary, tertiary and quaternary lobes as the posterior lobes. Rows of pericardial cells line the dorsal vessel at the posterior end.
To demonstrate that this method of dissection maintains tissue integrity, we analyzed markers for the whole LG, cardiac tube and the pericardial cells. Phalloidin marks actin and is useful for visualizing the integrity of LG lobes and the dorsal vessel (Figure 5A). Hemese, a generic blood cell marker (Kurucz et al., 2003), is expressed in all cells of the primary and most cells of the posterior lobes (Figure 5B). Asrij (Kulkarni et al., 2011) another pan-hemocyte marker marks LG hemocytes of the primary and the posterior lobes (Figure 5C). srpHemo-Gal4 containing a regulatory region of the serpent gene is active in the embryonic hemocytes (Bruckner et al., 2004) and additionally, is strongly expressed in the pericardial cells (Figure 5D). However, very few cells in the primary lobes express srpHemo-Gal4 (Figure 5D). With the help of the above-mentioned markers we confirm this method of dissection is useful for isolating intact LG primary and posterior lobes.
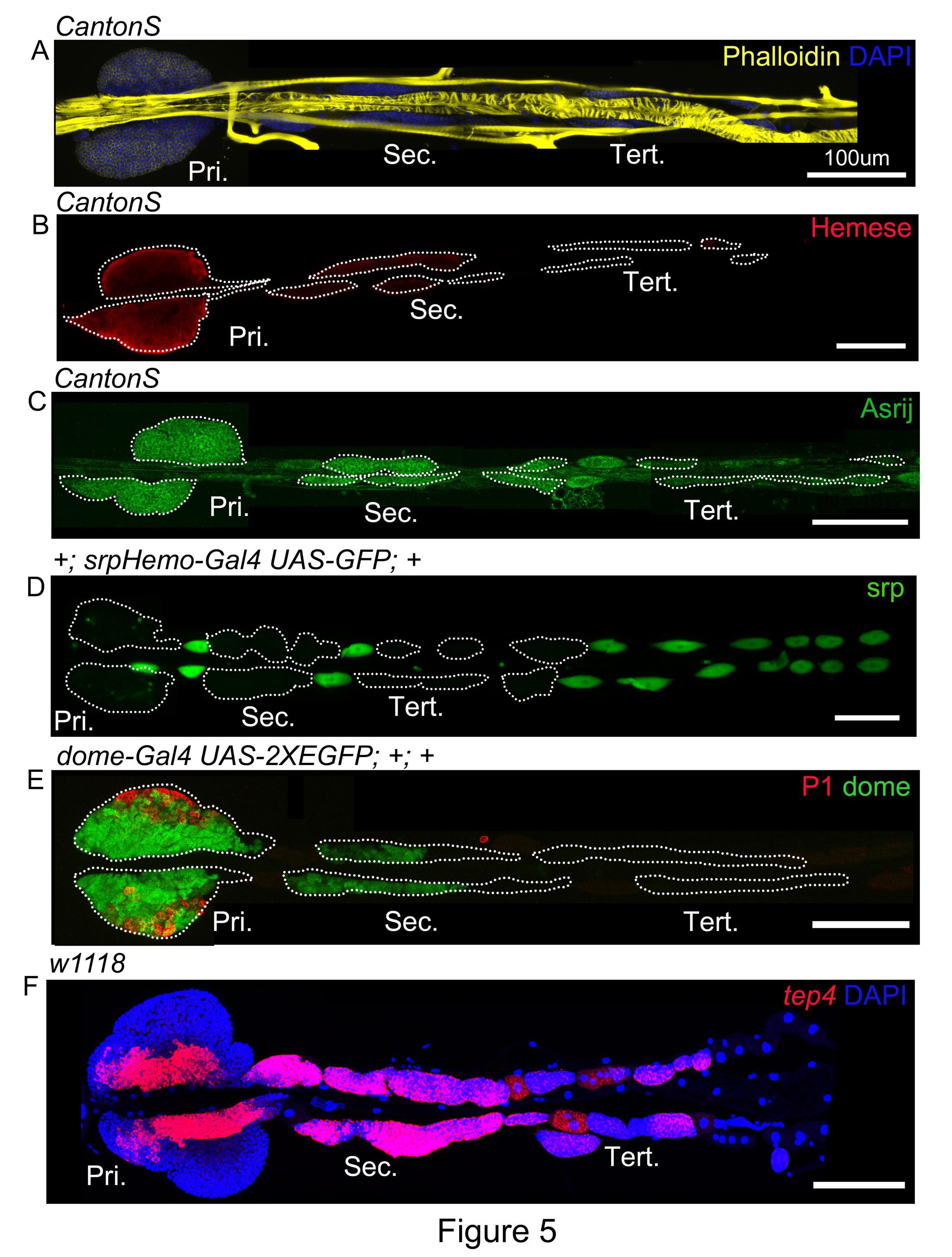
Figure 5. Validation of intact whole LG preparations using pan hemocyte, progenitor and differentiation markers. Whole LG preparations from wandering third larval instar include primary and posterior lobes. (A) Phalloidin (yellow) marks actin and is used for identifying LG blood cells and the cardiac tube. Pan hemocyte markers (B) Hemese (red) and (C) Asrij (green) are expressed in the LG lobes and help to identify intact lobes. (D) srpHemo-Gal4 (green) is strongly expressed in the pericardial cells. (E) dome (green) marks progenitors in the primary and the posterior lobes, P1/ Nimrod C1 (red) marks plasmatocytes. (F) in situ hybridization for tep4 (red) shows high expression in the MZ, secondary lobes and some cells of the tertiary lobes. Pri: primary lobes, Sec: secondary lobes, Tert: tertiary lobes. (A-F) Scale bar: 100μmFor in-depth analysis of tissue integrity, we further examined known progenitor (MZ) and differentiation (CZ) markers as defined in the anterior lobes. We performed reporter, immunostaining or RNA in situ analysis to analyze marker expression. As described previously, progenitor marker domeless (Jung et al., 2005) is expressed in the MZ, secondary lobes and in some cells of the tertiary lobes (Figure 5E). Differentiation marker, P1/ Nimrod C1 (Kurucz et al., 2007) expression is restricted to the CZ of the primary lobes (Figure 5E). RNA in situ for another progenitor marker, tep4 (Rodrigues et al., 2021) reveals high expression in the MZ, the secondary lobes and in some cells of the tertiary lobes (Figure 5F). Using the fillet method of LG dissection, we obtained intact primary and posterior lobes as observed with MZ and CZ markers.
Using this method of dissection, we have successfully separated primary and posterior lobes and performed RNA sequencing analysis (Figure 6). This has led to the identification of new markers of LG blood cells (Rodrigues et al., 2021). Maintaining tissue integrity and quality of the sample depends on critical steps in the dissection process. Though the dissections are demanding, identifying the LG and performing quick dissections (2-3 min) has been achieved routinely with 2-3 days of practice even by inexperienced summer interns. This detailed protocol aims to establish a standard method for LG dissection and comprehensive analysis that can also be adapted for isolating LG from early larval instars and pupal stages.
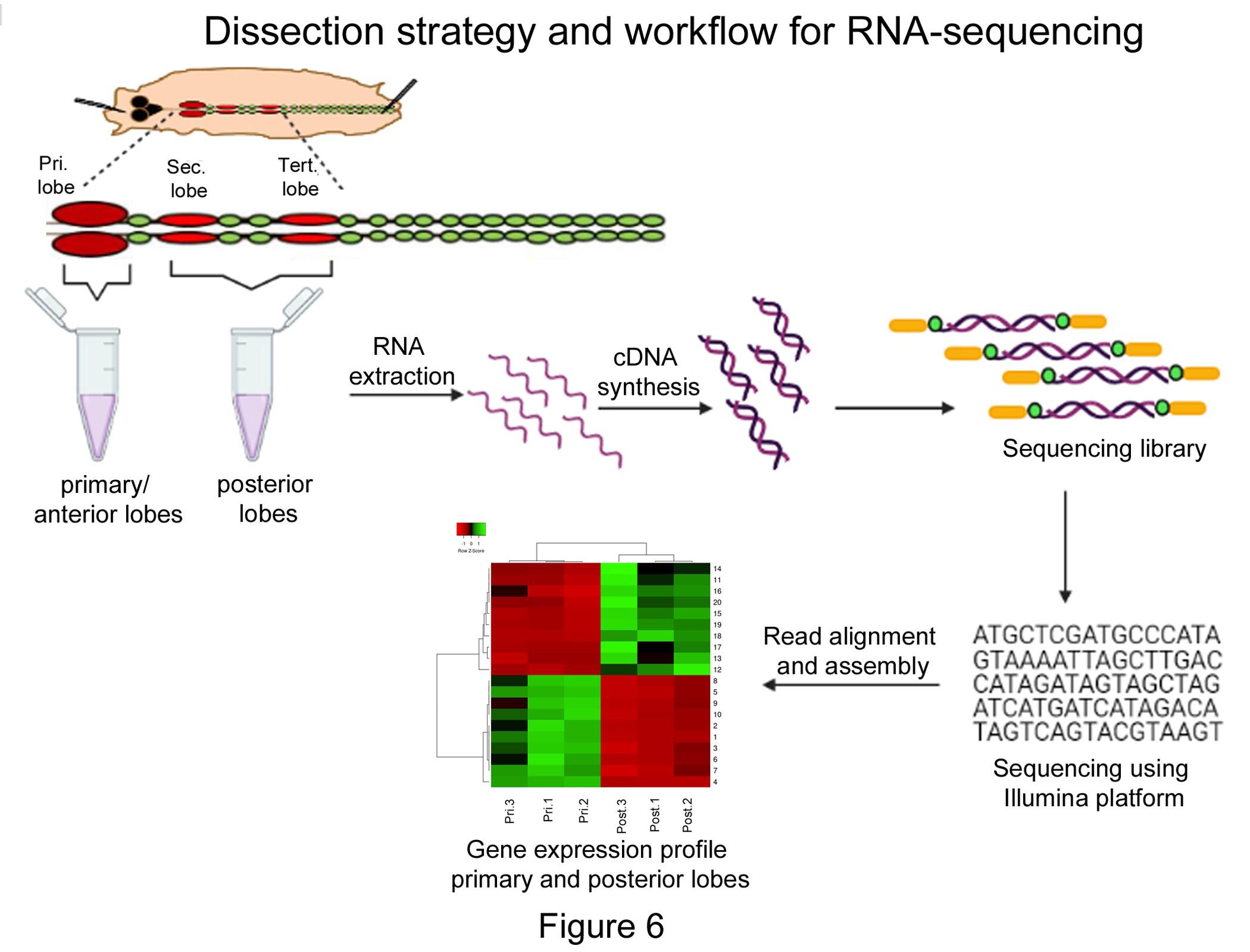
Figure 6. Schematic representation of dissection strategy and workflow for RNA-sequencing analysis. Step-wise representation of dissection strategy and RNA-sequencing for obtaining transcriptomic profiles for the primary and the posterior lobes separately. Pri: primary lobes, Sec: secondary lobes, Tert: tertiary lobes and Post: posterior lobes.Discussion
Unlike vertebrate HSPCs, most Drosophila blood progenitors reside in a single hematopoietic organ, the lymph gland, which also harbors their differentiated progeny. The small size of the LG (~1.5–2 mm in length) makes a complete and comprehensive in situ analysis of its resident progenitor and differentiated pools possible. Yet most of our understanding of larval LG hematopoiesis is derived from in-depth analysis restricted to the anterior-most lobes, called the primary lobes. Till recently, cellular and molecular mechanisms that operate in the primary lobe were thought to be applicable to the posterior lobes. However, anterior and posterior lobes have significant differences in gene expression and mutant phenotypes (Khadilkar et al., 2014; Kulkarni et al., 2011; Rodrigues et al., 2021). Also, the posterior lobes are physically separated from the anterior lobe PSC and differentiated hemocytes that are involved in regulating progenitor fate in the anterior lobes (Figure 1F). Further, a larger pool of blood progenitors resides in the posterior lobes (Rodrigues et al., 2021). Hence incomplete analysis of the LG overlooks important developmental and functional information regarding hematopoiesis that could be relevant to vertebrate hematopoiesis. To aid dissection and analysis of the complete intact LG, we describe a detailed protocol that will also be useful in analyzing the entire progenitor population. LG primary lobes are specified at the embryonic stage whereas, the posterior lobes form during the second instar larval stage and continue to develop till the wandering larval stage. Hence, this method of dissection allows simultaneous analysis of developmental hematopoiesis in a single animal (Rodrigues et al., 2021).
We demonstrated the utility of our method by analyzing pan-hemocyte, progenitor and differentiation markers at the RNA and protein level. Obtaining whole intact LG lobes is essential for understanding progenitor heterogeneity in physiological and immune conditions (Rodrigues et al., 2021). This protocol can be used for studying the ontogeny, development and immune aspects of LG blood cells. It can also be used for isolating distinct lobes for follow up biochemical and omics assays (Rodrigues et al., 2021; Sinha et al., 2019). Therefore, this protocol will be particularly valuable to gain new and more complete insights into the regulation of blood cell progenitor fate.
Notes
We use neutral red stained preparations only for the ease of visualization during training and practicing LG dissection and not for immunostaining or in situ hybridization experiments.
Avoid placing the coverslip on the LG preparation as this can damage the tissue. We prefer to use coverslip glass- bottom dishes for mounting. With experience, 8-10 LG preparations can be mounted in a single coverslip- bottom dish (Figure 4).
Fillet method of LG dissection retains the ventral larval cuticle containing sessile hemocyte clusters that can be retrieved at the sample mounting step (see procedure, section F, step 4), allowing simultaneous analysis of blood cells emerging from distinct anlage (Khadilkar et al., 2014).
This protocol helps to isolate 3 organ systems: dorsal vessel, pericardial cells and LG from the same animal in a single sample preparation.
As anterior/primary lobe progenitors are specified by the first instar and secondary and tertiary lobes at subsequent stages, this method allows analysis of developmental stages in a single animal.
Recipes
10x PBS
Dissolve sodium chloride (NaCl) –18.9 g, sodium hydrogen phosphate (Na2HPO4) – 2.48 g, Monosodium phosphate (NaH2PO4·2H2O) – 1.17 g in autoclaved distilled water, adjust volume to 250 ml. Adjust pH to 7. Store at room temperature.
4% Paraformaldehyde (PFA)
Dissolve 4 g PFA powder in 80ml of 1x PBS and adjust the volume to 100 ml with 1x PBS solution. Place the mixture at 60 °C in a water bath to dissolve the PFA. Store at 4°C.
0.1% Triton X-100 in PBS (PTX)
Add 0.1 ml Triton X-100 in 100 ml 1x PBS. Store at room temperature.
0.1% Tween-20 in PBS (PBS-T)
Add 0.1 ml Tween-20 in 100 ml 1x PBS. Store at room temperature.
20x Saline-sodium citrate buffer (SSC)
Dissolve sodium chloride (NaCl) - 43.82 g, sodium citrate (Na3C6H5O7) - 22.05 g, adjust volume to 200 ml with molecular grade (DNase/ RNase free) water. Adjust pH to 7. Autoclave and store at -20°C.
Hybridization buffer
Dissolve, Formamide- 50 ml, 20x SSC- 10 ml, yeast RNA (50 mg/ml)- 2 ml, heparin (0.05 g/ml)- 100μl, Roche blocking reagent (10%)- 20 ml (prepared according to manufacturer instruction), CHAPS (10%)- 1 ml, EDTA (0.5M)- 1 ml, Tween-20 (10%)- 1 ml, adjust the volume to 100 ml using molecular grade water (DNase/ RNase free). Store at -20°C.
Staining buffer
Dissolve, 5M sodium chloride (NaCl)- 200µl, 1M magnesium chloride (MgCl2)- 500μl, Tris-HCl pH 9.5- 1ml, Tween-20 (10%)- 1μl, adjust volume to 10ml with autoclaved distilled water.
Acknowledgments
We thank the Drosophila community for fly stocks and antibodies; National Centre for Biological Sciences, Fly Facility for stocks; JNCASR Imaging facility and our laboratory members for valuable inputs and suggestions. Schematics created with BioRender.com. This work was funded by the Indo-French Centre for the Promotion of Advanced Research (IFCPAR/CEFIPRA) grant to MSI and LW; MSI’s work was also supported by SERB grant, J C Bose award project and Jawaharlal Nehru Centre for Advanced Scientific Research.
Author contributions: DR, LW and MSI performed experiments; DR, KVR, LW and MSI wrote the manuscript; MSI and LW obtained funding and facilities for the work
Competing interests
The authors declare no conflict of interest
References
- Banerjee, U., Girard, J. R., Goins, L. M., Spratford, C. M. (2019). Drosophila as a Genetic Model for Hematopoiesis. Genetics 211(2): 367-417.
- Benmimoun, B., Polesello, C., Haenlin, M., Waltzer, L. (2015). The EBF transcription factor Collier directly promotes Drosophila blood cell progenitor maintenance independently of the niche. Proc Natl Acad Sci U S A 112(29): 9052-9057.
- Bruckner, K., Kockel, L., Duchek, P., Luque, C. M., Rorth, P., Perrimon, N. (2004). The PDGF/VEGF receptor controls blood cell survival in Drosophila. Dev Cell,7(1): 73-84.
- Crisan, M., Dzierzak, E. (2016). The many faces of hematopoietic stem cell heterogeneity. Development 143(24): 4571-4581.
- Crozatier, M., Ubeda, J. M., Vincent, A., Meister, M. (2004). Cellular immune response to parasitization in Drosophila requires the EBF orthologue collier. PLoS Biol 2(8): E196.
- Dey, N. S., Ramesh, P., Chugh, M., Mandal, S., Mandal, L. (2016). Dpp dependent Hematopoietic stem cells give rise to Hh dependent blood progenitors in larval lymph gland of Drosophila. Elife 5.
- Ema, H., Morita, Y., Suda, T. (2014). Heterogeneity and hierarchy of hematopoietic stem cells. Exp Hematol42(2): 74-82 e72.
- Ghosh, S., Singh, A., Mandal, S., Mandal, L. (2015). Active hematopoietic hubs in Drosophila adults generate hemocytes and contribute to immune response. Dev Cell 33(4): 478-488.
- Granick, J. L., Simon, S. I., Borjesson, D. L. (2012). Hematopoietic stem and progenitor cells as effectors in innate immunity. Bone Marrow Res 2012: 165107.
- Grigorian, M., Liu, T., Banerjee, U., Hartenstein, V. (2013). The proteoglycan Trol controls the architecture of the extracellular matrix and balances proliferation and differentiation of blood progenitors in the Drosophila lymph gland. Dev Biol 384(2): 301-312.
- Grigorian, M., Mandal, L., Hartenstein, V. (2011). Hematopoiesis at the onset of metamorphosis: terminal differentiation and dissociation of the Drosophila lymph gland. Dev Genes Evol221(3): 121-131.
- Holz, A., Bossinger, B., Strasser, T., Janning, W., Klapper, R. (2003). The two origins of hemocytes in Drosophila. Development 130(20): 4955-4962.
- Honti, V., Csordas, G., Markus, R., Kurucz, E., Jankovics, F., Ando, I. (2010). Cell lineage tracing reveals the plasticity of the hemocyte lineages and of the hematopoietic compartments in Drosophila melanogaster. Mol Immunol 47(11-12): 1997-2004.
- Jung, S. H., Evans, C. J., Uemura, C., Banerjee, U. (2005). The Drosophila lymph gland as a developmental model of hematopoiesis. Development 132(11): 2521-2533.
- Khadilkar, R. J., Rodrigues, D., Mote, R. D., Sinha, A. R., Kulkarni, V., Magadi, S. S.,Inamdar, M. S. (2014). ARF1-GTP regulates Asrij to provide endocytic control of Drosophila blood cell homeostasis. Proc Natl Acad Sci U S A 111(13): 4898-4903.
- Kulkarni, V., Khadilkar, R. J., Magadi, S. S., Inamdar, M. S. (2011). Asrij maintains the stem cell niche and controls differentiation during Drosophila lymph gland hematopoiesis. PLoS One 6(11): e27667.
- Kurucz, E., Markus, R., Zsamboki, J., Folkl-Medzihradszky, K., Darula, Z., Vilmos, P., Udvardy, A., Krausz, I., Lukacsovich, T., Gateff, E., Zettervall, C. J., Hultmark, D., Ando, I. (2007). Nimrod, a putative phagocytosis receptor with EGF repeats in Drosophila plasmatocytes. Curr Biol 17(7): 649-654.
- Kurucz, E., Zettervall, C. J., Sinka, R., Vilmos, P., Pivarcsi, A., Ekengren, S., Hegedus, Z., Ando, I., Hultmark, D. (2003). Hemese, a hemocyte-specific transmembrane protein, affects the cellular immune response in Drosophila. Proc Natl Acad Sci U S A 100(5): 2622-2627.
- Lanot, R., Zachary, D., Holder, F., Meister, M. (2001). Postembryonic hematopoiesis in Drosophila. Dev Biol 230(2): 243-257.
- Mandal, L., Banerjee, U., Hartenstein, V. (2004). Evidence for a fruit fly hemangioblast and similarities between lymph-gland hematopoiesis in fruit fly and mammal aorta-gonadal-mesonephros mesoderm. Nat Genet 36(9): 1019-1023.
- Mandal, L., Martinez-Agosto, J. A., Evans, C. J., Hartenstein, V., Banerjee, U. (2007). A Hedgehog- and Antennapedia-dependent niche maintains Drosophila haematopoietic precursors. Nature 446(7133): 320-324.
- Rodrigues, D., Renaud, Y., VijayRaghavan, K., Waltzer, L., Inamdar, M. S. (2021). Differential activation of JAK-STAT signaling reveals functional compartmentalization in Drosophila blood progenitors. Elife 10.
- Rugendorff, A., Younossi-Hartenstein, A., Hartenstein, V. (1994). Embryonic origin and differentiation of the Drosophila heart. Rouxs Arch Dev Biol 203(5): 266-280.
- Sanchez Bosch, P., Makhijani, K., Herboso, L., Gold, K. S., Baginsky, R., Woodcock, K. J., Alexander, B., Kukar, K., Corcoran, S., Jacobs, T., Ouyang, D., Wong, C., Ramond, E. J. V., Rhiner, C., Moreno, E., Lemaitre, B., Geissmann, F., Bruckner, K. (2019). Adult Drosophila Lack Hematopoiesis but Rely on a Blood Cell Reservoir at the Respiratory Epithelia to Relay Infection Signals to Surrounding Tissues. Dev Cell 51(6): 787-803 e785.
- Sinha, A., Khadilkar, R. J., S, V. K., Roychowdhury Sinha, A., Inamdar, M. S. (2013). Conserved regulation of the Jak/STAT pathway by the endosomal protein asrij maintains stem cell potency. Cell Rep 4(4): 649-658.
- Sinha, S., Ray, A., Abhilash, L., Kumar, M., Sreenivasamurthy, S. K., Keshava Prasad, T. S., Inamdar, M. S. (2019). Proteomics of Asrij Perturbation in Drosophila Lymph Glands for Identification of New Regulators of Hematopoiesis. Mol Cell Proteomics 18(6): 1171-1182.
- Tepass, U., Fessler, L. I., Aziz, A., Hartenstein, V. (1994). Embryonic origin of hemocytes and their relationship to cell death in Drosophila. Development 120(7): 1829-1837.
- Ward, E. J., Skeath, J. B. (2000). Characterization of a novel subset of cardiac cells and their progenitors in the Drosophila embryo. Development 127(22): 4959-4969.
- Wright, D. E., Wagers, A. J., Gulati, A. P., Johnson, F. L., Weissman, I. L. (2001). Physiological migration of hematopoietic stem and progenitor cells. Science 294(5548): 1933-1936.
![]() Rodrigues et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Rodrigues et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Category
Developmental Biology > Cell growth and fate > Differentiation
Immunology > Animal model > Other
Biological Sciences > Biological techniques
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.