- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Selection of Genetically Modified Bacteriophages Using the CRISPR-Cas System

Published: Vol 7, Iss 15, Aug 5, 2017 DOI: 10.21769/BioProtoc.2431 Views: 11662
Reviewed by: Modesto Redrejo-RodriguezAnonymous reviewer(s)
Original research article
The authors used this protocol in:

0 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
We present a CRISPR-Cas based technique for deleting genes from the T7 bacteriophage genome. A DNA fragment encoding homologous arms to the target gene to be deleted is first cloned into a plasmid. The T7 phage is then propagated in Escherichia coli harboring this plasmid. During this propagation, some phage genomes undergo homologous recombination with the plasmid, thus deleting the targeted gene. To select for these genomes, the CRISPR-Cas system is used to cleave non-edited genomes, enabling isolation of the desired recombinant phages. This protocol allows seamless deletion of desired genes in a T7 phage, and can be expanded to other phages and other types of genetic manipulations as well.
Keywords: BacteriophageBackground
Bacteriophages (phages) are the most prevalent and widely distributed biological entity in the biosphere, highlighting their ecological importance (Suttle, 2007). Many studies also propose using phages for medical purposes (Weber-Dabrowska et al., 2001; Merril et al., 2003; Harper and Enright, 2011; Edgar et al., 2012; Bikard et al., 2014; Citorik et al., 2014; Yosef et al., 2014 and 2015). Unfortunately, only a few published methods detail genetic engineering of phage genomes (Selick et al., 1988; Marinelli et al., 2008; Pires et al., 2016) and in addition, some of these methods are tedious, and some cannot achieve desired results such as seamless deletions. A simple and efficient technique for seamless genetic engineering of phages is thus desired. In this protocol, we present a technique described in 2014 (Kiro et al., 2014) for deleting genes of the E. coli phage, T7. We first designed genetic constructs that facilitate desired homologous recombination events. We then used the CRISPR-Cas type I-E system to select desired engineered phages. Several other studies have also reported the use of CRISPR-Cas systems to engineer phages and the reader is referred to them for selecting the most appropriate and fitting protocols for the specific requirements (Martel and Moineau, 2014; Box et al., 2015; Lemay et al., 2017).
Materials and Reagents
- Materials
- 1.7 ml microfuge tube (Corning, Axygen®, catalog number: MCT-175-C )
- 15 ml tube (Corning, catalog number: 430052 )
- PCR tubes (Corning, Axygen®, catalog number: PCR-0208-C )
- Pipette tips
- Bacterial strains
- Electro-competent BL21-AI (Invitrogen, Genotype: F− ompT hsdSB(rB−, mB−) gal dcm araB::T7RNAP-tetA, tetr)
- Electro-competent NEB5α (New England Biolabs)
- Phage
- WT T7 phage (laboratory collection. Available at ATCC, catalog number: BAA-1025-B2 )
- Plasmids
- pUC19 (Yanisch-Perron et al., 1985)
- pWUR397 (Brouns et al., 2008. cas3 under T7 promoter, KanR)
- pWUR400 (Brouns et al., 2008. cascade genes under T7 promoter, StrR)
- pWUR477 (Brouns et al., 2008. pACYCDuet-1 (Novagen) cloned with control spacers under T7 promoter, camR)
- Enzymes
- DpnI restriction enzyme (New England Biolabs, catalog number: R0176S )
- T4 polynucleotide kinase (New England Biolabs, catalog number: M0201S ) used with T4 DNA ligase buffer (New England Biolabs, catalog number: M0202S )
- Kits
- Gel and PCR Clean-up Kit (MACHEREY-NAGEL, catalog number: 740609.50 )
- KAPA HiFi PCR Kit (Roche Diagnostics, catalog number: 07958935001 )
- Lamda Taq PCR Master mix (Lamda Biotech, catalog number: D123P-200 )
- Quick Ligation Kit (New England Biolabs, catalog number: M2200L )
- Reagents
- Agar (BD, DifcoTM, catalog number: 214010 )
- Ampicillin (Merck, catalog number: 171254 ; Stock 100 mg/ml in double-distilled water, filtered, -20 °C)
- Chloramphenicol (Merck, catalog number: 220551 ; Stock 35 mg/ml in ethanol, filtered, -20 °C)
- Isopropyl-β-D-thiogalactopyranoside (IPTG) (Bio-Lab, catalog number: 16242352 ; Stock 1 M in double-distilled water, filtered, -20 °C)
- Kanamycin (Merck, catalog number: 420311 ; Stock 50 mg/ml in 50% glycerol and double-distilled water, filtered, -20 °C)
- L-arabinose (Gold Bio, catalog number: A-300-1 ; Stock 20% in double-distilled water, filtered, RT)
- LB (Luria-Bertani) medium (10 g/L tryptone, 5 g/L yeast extract and 5 g/L NaCl) (Acumedia)
- Molecular biology water (Bio-Lab, catalog number: 232123 )
- PEG 8000 (polyethylene glycol) (Promega, catalog number: V3011 ; Stock 50% in double-distilled water, filtered, RT)
- Streptomycin (EMD Millipore, catalog number: 5711 ; Stock 50 mg/ml in double-distilled water, filtered, -20 °C)
- TAE buffer (Bio-Lab, catalog number: 20502323 )
- LB medium (see Recipes)
- LB agar plates (see Recipes)
- Soft agar (see Recipes)
- TAE buffer (see Recipes)
Equipment
- Cell density meter (OD600 reader) (Amersham Biosciences, model: Ultrospec 10 )
- Electroporation device (Bio-Rad Laboratories, model: MicroPulserTM )
- Gel electrophoresis apparatus (Cleaver Scientific, model: MultiSUB Choice )
- Micro-centrifuge (Eppendorf, model: MiniSpin® )
- NanoDrop spectrophotometer (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 2000c )
- Pipettes
- Shaker (for 50 ml tubes, at 37 °C) (Thermo Fisher Scientific, Thermo ScientificTM, model: MaxQTM 2000 )
- Thermal Cycler (Bio-Rad Laboratories, model: C1000 TouchTM )
- Thermoblock (Labnet International, model: AccuBlockTM Digital Dry Baths )
Procedure
- Plasmid construction
- Construction of pGeneX
This plasmid encodes DNA homologous to the upstream and downstream sequences of the target genetic locus. These DNA will recombine with the desired locus thus generating the desired recombinant phages (Figure 1). - Perform a PCR amplification reaction with pUC19 plasmid as a template, using primers with 60 bp 5’ overhangs homologous to sequences flanking the target gene to be deleted (hereafter, geneX). The forward primer should carry a 60 bp overhang sequence identical to the 60 bp downstream to geneX. The reverse primer should carry a 60 bp overhang matching the upstream sequence. See design in Figure 1.



Figure 1. Schematics of the pGeneX plasmid. A. An example of forward and reverse primers encoding sequences annealing to the pUC19 backbone (gray) with 60 bp 5’ overhang sequence identical to the upstream (red) and downstream (blue) flanking sequences of geneX. B. PCR amplification with these primers on the plasmid and self-ligation yield the pGeneX plasmid. - Add 1 µl of restriction enzyme DpnI directly to the PCR mixture to eliminate residues of the pUC19.
- Incubate for 1 h at 37 °C.
- Isolate DNA product according to size using gel electrophoresis.
- Purify the product using Gel and PCR Clean-up kit according to the manufacturer’s instructions. Elute in 20 µl.
- Phosphorylate the purified DNA:
- Incubate the purified DNA at 70 °C for 5 min.
- Cool on ice for 5 min.
- Add to the 20 µl purified DNA:
3 µl T4 DNA ligase buffer
1 µl T4 polynucleotide kinase
3 µl PEG-800 (50%)
3 µl molecular biology water - Incubate for 30 min at 37 °C.
Note: DNA oligos could be also synthesized with a 5’-phosphate. In such cases, step A1f should be skipped.
- Incubate the purified DNA at 70 °C for 5 min.
- Purify the product using Gel and PCR Clean-up kit according to the manufacturer’s instructions. Elute in 20 µl.
- Self-ligate the phosphorylated DNA:
- Add to 10 µl phosphorylated DNA:
10 µl Quick ligation buffer
1 µl Quick ligase enzyme - Incubate for 15 min at 25 °C.
- Add to 10 µl phosphorylated DNA:
- Purify the product using Gel and PCR Clean-up kit according to the manufacturer’s instructions. Elute in 20 µl.
- Construction of pAnti-GeneX
This plasmid encodes specific spacers that will serve to target the unmodified phage, thus enabling selection of the modified phage.- Perform a PCR amplification reaction with pWUR477 plasmid as a template using primers with 5’ overhangs that form a new spacer upon ligation. See Figure 2 below.


Note: Minimum reaction volume of 30 µl.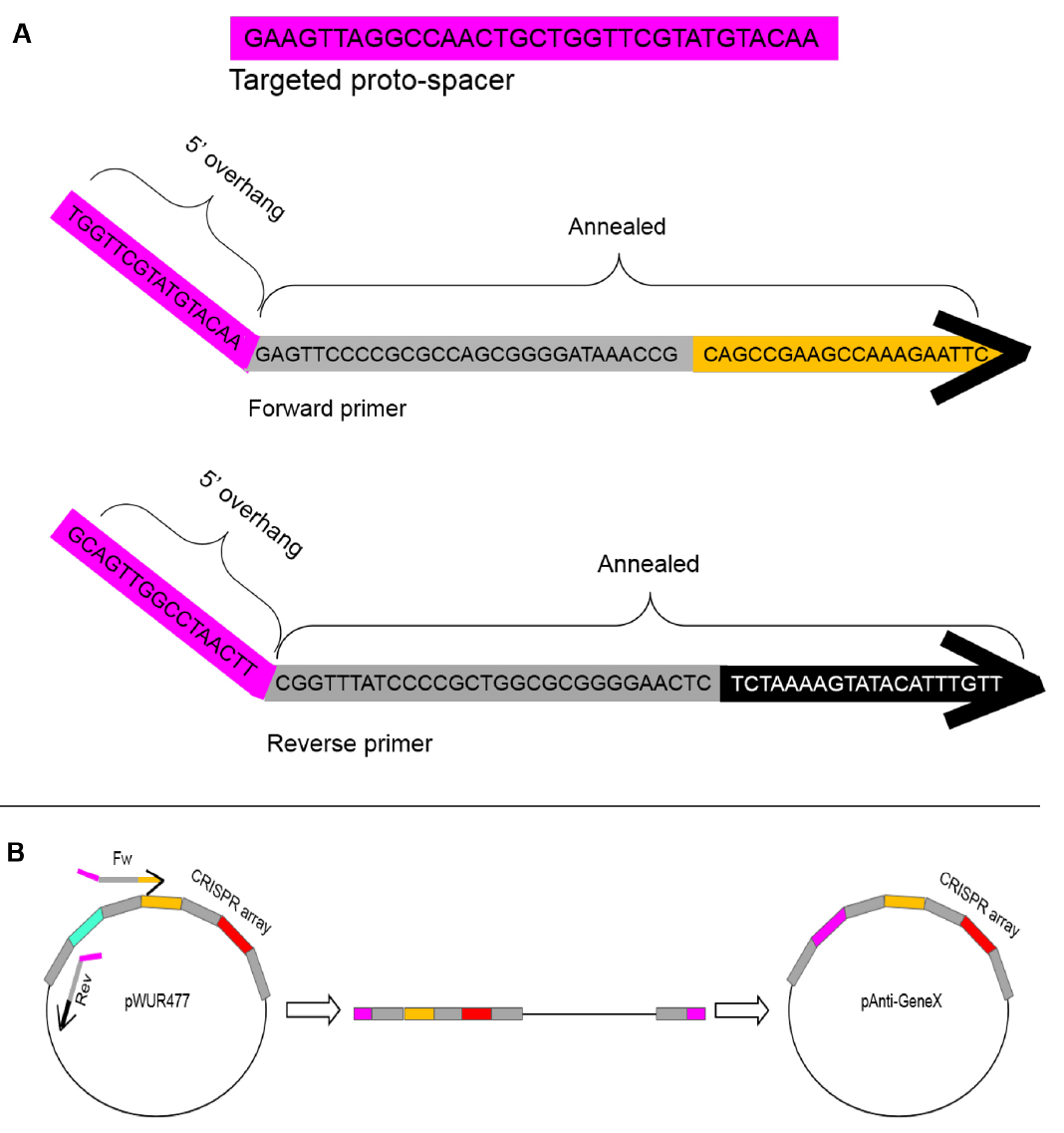
Figure 2. Schematics of the pAnti-GeneX construct. A. An example of a primer design yielding a spacer to target gene X. The targeted DNA is in pink. This protospacer is a 33 bp sequence in the gene of interest and is preceded by an AA, yielding a functional protospacer-adjacent motif (PAM). The forward and reverse primers encode annealing sequences to the pWUR477 template, and 5’ overhangs which together form the desired spacer sequence. B. PCR amplification with the forward and reverse primers on the pWUR477 plasmid and self-ligation to construct the pAnti-GeneX plasmid. Grey rectangles represent repeat sequences, pink represents the new spacer, orange represents spacer 2 and red represents spacer 3, and black represents the plasmid backbone. - Add 1 µl of restriction enzyme DpnI directly to the PCR mixture to eliminate residues of the pWUR477.
- Incubate for 1 h at 37 °C.
- Isolate DNA product according to size using gel electrophoresis.
- Purify the product using Gel and PCR Clean-up kit according to the manufacturer’s instructions. Elute in 20 µl.
- Phosphorylate the purified DNA:
- Incubate the purified DNA at 70 °C for 5 min.
- Cool on ice for 5 min.
- Add to the 20 µl purified DNA:
3 µl T4 DNA ligase buffer.
1 µl T4 polynucleotide kinase.
3 µl PEG-800 (50%).
3 µl molecular biology water. - Incubate for 30 min at 37 °C.
Note: DNA oligos could also be synthesized with a 5’-phosphate. In such cases, step A2f should be skipped.
- Incubate the purified DNA at 70 °C for 5 min.
- Purify the product using Gel and PCR Clean-up kit the according to the manufacturer’s instructions. Elute in 20 µl.
- Self-ligate the phosphorylated DNA:
- Add to 10 µl phosphorylated DNA:
10 µl Quick ligation buffer
1 µl Quick ligase enzyme - Incubate for 15 min at 25 °C.
- Add to 10 µl phosphorylated DNA:
- Purify the product using Gel and PCR Clean-up kit according to the manufacturer’s instructions. Elute in 20 µl.
- Perform a PCR amplification reaction with pWUR477 plasmid as a template using primers with 5’ overhangs that form a new spacer upon ligation. See Figure 2 below.
- Construction of pGeneX
- Preparation of recombinant T7 lysate
- Transform the pGeneX and the pUC19 plasmid (lacking the homologous sequences) as a control into NEB5α cells.
- Inoculate one colony of each transformant into 3 ml LB supplemented with 100 µg/ml ampicillin and shake at 37 °C, overnight.
- Dilute overnight cultures 1:100 into 6 ml fresh LB, supplemented with 100 µg/ml ampicillin.
- Shake at 37 °C to an OD600 ~1.
- Infect 5 ml of each culture with WT T7 phage at an MOI of ~0.1 (107-108 PFU). Leave the remaining 1 ml culture as an uninfected control.
- Shake the infected bacteria at 37 °C until lysis occurs (usually 2 h).
Note: Lysis can be recognized by ‘clear’ culture, strings of cell debris or a translucent appearance.
- Transform the pGeneX and the pUC19 plasmid (lacking the homologous sequences) as a control into NEB5α cells.
- Select T7 engineered phages
- Transform pWUR397, pWUR400 and targeting plasmid (pAnti-GeneX) or a control plasmid (pWUR477), into BL21-AI cells.
- Inoculate one colony of each transformant into 3 ml LB supplemented with 50 µg/ml streptomycin, 50 µg/ml kanamycin and 35 µg/ml chloramphenicol and shake at 37 °C, overnight.
- Prepare at least 4 LB agar plates (see Recipes) supplemented with 50 µg/ml streptomycin, 50 µg/ml kanamycin and 35 µg/ml chloramphenicol.
- Dilute overnight cultures 1:100 into 5 ml fresh LB supplemented with the appropriate antibiotics.
- Shake at 37 °C (without inducers) until OD600 ~1.
- Add 0.2% L-arabinose and 0.1 mM IPTG (final concentrations) to the cultures.
- Shake at 37 °C for an additional hour.
- Pre-warm soft agar (0.7% agar, see Recipes) to a liquid phase. Add 0.2% L-arabinose and 0.1 mM IPTG to the agar and keep at 50 °C.
- Pre-warm LB agar plates with the antibiotics prepared in step C3 for at least 1 h at 37 °C.
- For every combination of phage and bacteria (see Table 1), mix in a 15 ml tube:
100 µl bacteria (from step C7).
10 µl phage lysates prepared in ‘Preparation of recombinant T7 lysate’.
3.5 ml of the pre-warmed soft agar (0.7%) with inducers.
Table 1. Combinations of phage with bacteria. All four different combinations: targeting/non-targeting bacteria with recombinant/non-recombinant lysates.
- Vortex briefly and IMMEDIATELY pour onto the pre-warmed plates. Gently tilt and rotate plate to spread top agar evenly.
- Allow the plates to cool for 5 min, invert, and incubate overnight at RT or for 4-6 h at 37 °C (until plaques are visible).
- Transform pWUR397, pWUR400 and targeting plasmid (pAnti-GeneX) or a control plasmid (pWUR477), into BL21-AI cells.
- Isolation and validation
- Inoculate one colony of targeting bacteria (BL21-AI/pWUR397, pWUR400, pAnti-GeneX) into 3 ml LB supplemented with 50 µg/ml streptomycin, 50 µg/ml kanamycin and 35 µg/ml chloramphenicol.
- Shake at 37 °C overnight.
- Dilute plaques larger than 0.4 cm into 100 µl LB.
Note: Small plaques are expected even on the control plates. Desired plaques (i.e., ones that have the desired deletion mutation) should appear 3-5 times larger than the background plaques. - Perform a PCR amplification reaction on the diluted plaque for validation: Use primers upstream and downstream to geneX. Determine gene deletion according to PCR product size. Use wild-type T7 as a template for positive control. See Figure 3.
Note: Deletion of a fragment encoding the corresponding protospacer can result in isolated phages that do not encode the desired deletion (by escaping the CRISPR-Cas targeting).


Figure 3. Illustration for PCR validation. Forward and reverse primers anneal upstream and downstream to geneX (green). PCR amplification on the edited phage results in a smaller DNA product. PCR amplification of the WT phage or phage with deletion of a fragment encoding the corresponding protospacer results in a full length DNA product. Black square represents gel electrophoresis, white bands represent DNA products. - Pre-warm soft agar (0.7% agar) to a liquid phase. Add 0.2% L-arabinose and 0.1 mM IPTG (final concentrations) to the agar and keep at 50 °C.
- Pre-warm LB agar plates for at least 1 h at 37 °C. One plate for every modified phage.
- Streak positive deleted phages on the pre-warmed LB agar plate.
- For each modified phage, mix in a 15 ml tube:
200 µl of the targeting bacteria
3 ml of the pre-warmed soft agar with the inducers - Gently pour the soft-agar with the bacteria on the streaked plates (from step D6).
Note: Pour the soft-agar with the bacteria on the lower concentration area and let the fluid spread to the high concentration end. - Allow the plates to cool for 5 min, invert, and incubate overnight at RT or for 4-6 h at 37 °C (until plaques are visible).
- Pick one plaque from each plate and repeat steps D3-D9, 3 times.
- Perform a PCR amplification on an isolated phage and validate desired sequence with DNA sequencing.
- Dilute 1:50 targeting bacteria into 5 ml LB supplemented with 50 µg/ml streptomycin, 50 µg/ml kanamycin and 35 µg/ml chloramphenicol, 0.2% L-arabinose and 0.1 mM IPTG.
- Shake at 37 °C to an OD600 ~0.5.
- Infect the culture with 10 µl of the validated engineered phage.
- Shake the infected bacteria at 37 °C until lysis occurs (usually 2 h).
- Inoculate one colony of targeting bacteria (BL21-AI/pWUR397, pWUR400, pAnti-GeneX) into 3 ml LB supplemented with 50 µg/ml streptomycin, 50 µg/ml kanamycin and 35 µg/ml chloramphenicol.
Data analysis
A flow chart of the protocol is shown in Figure 4. An overview of the procedures for isolating desired recombinant bacteriophage and percentages of engineered cells can be found in the original paper (Kiro et al., 2014).
Figure 4. Flow chart of the protocol
Recipes
- LB medium
- Measure 20 g LB (Luria-Bertani) medium powder and fill up to 1 L with deionized water
- Autoclave solution and cool down
- Store at RT
- LB agar plates
- Measure 7.5 g agar and add sterile LB medium up to 500 ml
- Autoclave the solution, cool down and add antibiotics
- Pour into Petri dishes
- Wait until the agar solidifies
- Store at 4 °C
- Soft agar
- Measure 1.75 g agar and add sterile LB medium up to 250 ml
- Autoclave the solution and cool down
- Store at RT
- TAE buffer
- Pour 200 ml of TAE buffer (stock) and add deionized water up to 10 L
- Mix thoroughly
- Store at RT
Acknowledgments
The research leading to these results was funded by the European Research Council under the European Community’s Seventh Framework Programme (FP7/207-2013)/ERC grant agreement No. 336079 (EcCRISPR). The protocol described here is adapted from (Kiro et al., 2014).
References
- Bikard, D., Euler, C. W., Jiang, W., Nussenzweig, P. M., Goldberg, G. W., Duportet, X., Fischetti, V. A. and Marraffini, L. A. (2014). Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat Biotechnol 32(11): 1146-1150.
- Box, A. M., McGuffie, M. J., O'Hara, B. J. and Seed, K. D. (2015). Functional analysis of bacteriophage immunity through a type I-E CRISPR-Cas system in Vibrio cholerae and its application in bacteriophage genome engineering. J Bacteriol 198(3): 578-590.
- Brouns, S. J., Jore, M. M., Lundgren, M., Westra, E. R., Slijkhuis, R. J., Snijders, A. P., Dickman, M. J., Makarova, K. S., Koonin, E. V. and van der Oost, J. (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321(5891): 960-964.
- Citorik, R. J., Mimee, M. and Lu, T. K. (2014). Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat Biotechnol 32(11): 1141-1145.
- Edgar, R., Friedman, N., Molshanski-Mor, S. and Qimron, U. (2012). Reversing bacterial resistance to antibiotics by phage-mediated delivery of dominant sensitive genes. Appl Environ Microbiol 78(3): 744-751.
- Harper, D. R. and Enright, M. C. (2011). Bacteriophages for the treatment of Pseudomonas aeruginosa infections. J Appl Microbiol 111(1): 1-7.
- Kiro, R., Shitrit, D. and Qimron, U. (2014). Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol 11(1): 42-44.
- Lemay, M. L., Tremblay, D. M. and Moineau, S. (2017). Genome engineering of virulent lactococcal phages using CRISPR-Cas9. ACS Synth Biol.
- Marinelli, L. J., Piuri, M., Swigonova, Z., Balachandran, A., Oldfield, L. M., van Kessel, J. C. and Hatfull, G. F. (2008). BRED: a simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS One 3(12): e3957.
- Martel, B. and Moineau, S. (2014). CRISPR-Cas: an efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res 42(14): 9504-9513.
- Merril, C. R., Scholl, D. and Adhya, S. L. (2003). The prospect for bacteriophage therapy in Western medicine. Nat Rev Drug Discov 2(6): 489-497.
- Pires, D. P., Cleto, S., Sillankorva, S., Azeredo, J. and Lu, T. K. (2016). Genetically engineered phages: a review of advances over the last decade. Microbiol Mol Biol Rev 80(3): 523-543.
- Selick, H. E., Kreuzer, K. N. and Alberts, B. M. (1988). The bacteriophage T4 insertion/substitution vector system. A method for introducing site-specific mutations into the virus chromosome. J Biol Chem 263(23): 11336-11347.
- Suttle, C. A. (2007). Marine viruses--major players in the global ecosystem. Nat Rev Microbiol 5(10): 801-812.
- Weber-Dabrowska, B. Mulczyk, M. and Górski, A. (2001). Bacteriophage therapy for infections in cancer patients. Clin Appl Immunol Rev 1(3-4): 131-134.
- Yanisch-Perron, C., Vieira, J. and Messing, J. (1985). Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33(1): 103-119.
- Yosef, I., Kiro, R., Molshanski-Mor, S., Edgar, R. and Qimron, U. (2014). Different approaches for using bacteriophages against antibiotic-resistant bacteria. Bacteriophage 4(1): e28491.
- Yosef, I., Manor, M., Kiro, R. and Qimron, U. (2015). Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc Natl Acad Sci U S A 112(23): 7267-7272.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Manor, M. and Qimron, U. (2017). Selection of Genetically Modified Bacteriophages Using the CRISPR-Cas System. Bio-protocol 7(15): e2431. DOI: 10.21769/BioProtoc.2431.
Category
Microbiology > Microbial genetics > DNA > Chromosomal
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.