- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A High Resolution Short Interfering RNA (siRNA) Detection Method from Virus-infected Plants


Published: Vol 3, Iss 20, Oct 20, 2013 DOI: 10.21769/BioProtoc.940 Views: 13390
Reviewed by: Tie Liu
Original research article
The authors used this protocol in:

Apr 2013
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







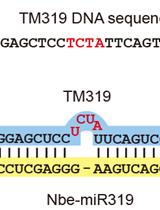
Abstract
Plant viruses are strong inducers as well as targets of RNA silencing. In plants RNA silencing acts as a natural defense mechanism against viral infection and is associated with accumulation of virus-specific small interfering RNAs (siRNAs). The continuing discoveries, increasing awareness and interest in the regulatory roles of non-coding small RNAs have raised the need for methods that can reliably detect and quantitate the expression levels of small RNAs. Northern blot analysis of small RNAs involving the separation of RNA molecules using polyacrylamide gel electrophoresis (PAGE) has remained a popular and valuable analytical method to validate small RNAs. Northern blot analysis consist of resolving RNAs by gel electrophoresis, followed by transferring and fixing to nylon membranes as well as detecting by hybridization using radioactive probes. The following protocol provides a method for isolation and detection of small RNAs from virus-infected plants and was successfully used in Panwar et al. (2013a), Panwar et al. (2013b).
Keywords: RNA SilencingMaterials and Reagents
- Virus-infected plant tissue
- TRIzol reagent (Invitrogen)
- Chloroform (Sigma-Aldrich)
- Isopropanol (Sigma-Aldrich)
- Ethyl alcohol (EtOH)
- Diethylpyrocarbonate (DEPC) (Sigma-Aldrich)
- Hybond NX Neutral Membrane (Amersham biosciences, catalog number: RPN203T )
- 40% Acrylamide/N’N’-bis-methylene-acrylamide (19:1) (Life Technologies, Ambion®)
- Tetramethylethylenediamine (EDTA) (Sigma-Aldrich)
- Urea (Sigma-Aldrich)
- Hyperfilm TM MP (Amersham biosciences, catalog number: 28-9068-45 )
- Ethidium bromide (Sigma-Aldrich)
- Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) (Sigma-Aldrich, catalog number: 39391 )
- Ammonium persulphate (APS)
- Tetramethylethylenediamine (TEMED) (Sigma-Aldrich)
- Tris base (AMRESCO)
- Boric acid (Fisher Scientific)
- Formamide (Sigma-Aldrich)
- Bromophenol blue (Sigma-Aldrich)
- Xylene cyanol (Sigma-Aldrich)
- 3 MM Whatman filter paper
- 1-methylimidazole (Sigma-Aldrich)
- Hydrochloric acid (HCl)
- Sodium dodecyl sulphate (SDS) (Fisher Scientific)
- 20x Saline sodium citrate (SSC) buffer (Sigma-Aldrich)
- ULTRAhyb-Oligo buffer (Life Technologies, Ambion®, catalog number: AM8669 )
- Megaprime DNA Labeling System (General Electric Company, model: RPN1604 )
- Liquid nitrogen
- RNase-free water
- Small gels (size of regular protein gels 1.5 mm thick) (Bio-Rad, Mini-Protean® Cell)
- Neutral Hybond nitrocellulose membrane
- RNase ZAP (Life Technologies, catalog number: AM9784 )
- DEPC treated water (see Recipes)
- 10x Tris-Borate-EDTA (TBE) buffer (see Recipes)
- 10% APS (see Recipes)
- 15% polyacrylamide gel (see Recipes)
- 2x loading buffer (see Recipes)
Equipment
- Pestle and mortars
- Semi dry electroblotter (Bio-Rad)
- Protean II vertical gel system (Bio-Rad)
- Nanodrop spectrophotometer
- Centrifuge
- Hybridization oven
- UV transilluminator
- Microcentrifuge tube
- Electrophoresis apparatus
- X-ray film
Procedure
- RNA isolation
- Using mortar and pestle grind 50-100 mg of virus-infected plant tissue in liquid nitrogen. Immediately add 1 ml of TRIzol reagent and continue grinding until a uniform paste is formed. Pipet the homogenized material in to a sterile microcentrifuge tube and incubate for 5 min at 25-30 °C to permit complete dissociation of nucleoprotein complexes.
- Add 200 μl of chloroform per ml of TRIzol reagent used for homogenization of sample tissue. Shake the samples vigorously by inverting the microcentrifuge tubes several times by hand and incubate at 20-30 °C for 2-3 min.
- Centrifuge the samples at no more than 12,000 x g for 15 min at room temperature (20-25 °C).
- Following centrifugation the mixture will separate in a lower red phenol-chloroform phase, an interphase, and a colorless upper aqueous phase.
- Without disturbing the interphase, carefully pipette out the aqueous phase and transfer it to a new sterile microcentrifuge tube.
- Precipitate the RNA from the aqueous phase by adding 0.5 ml of isopropanol per ml of TRIzol reagent used.
- Incubate sample at 20-30 °C for 10 min. Recover the total RNA by centrifuging the sample at no more than 12,000 x g for 10 min at 2-8 °C. RNA will form a gel like pellet at the bottom and side of the microcentrifuge tube.
- Without disturbing the pellet remove the supernatant. Wash the RNA pellet by slowly adding 1 ml of 75% ethanol down the side of the tube.
- Mix the sample briefly by vortexing and centrifuge at no more than 7,500 x g for 5 min at 2-8 °C.
- Discard the supernatant carefully without disturbing the pellet. Vacuum or air dry the pellet for 10-15 min and resuspend in 50 μl RNase-free water (commercially available or DEPC-treated sterile water). The volume is adjustable but higher concentrations are preferable since this will allow lower volumes to be loaded on the gel. Do not overdry the pellet otherwise it will be hard to dissolve. Quantify the concentration of RNA by measuring the absorbance at 260 nm using a spectrophotometer (e.g., Nanodrop) and store at -20 °C until ready to use. RNA will be stable for months at these conditions.
- Using mortar and pestle grind 50-100 mg of virus-infected plant tissue in liquid nitrogen. Immediately add 1 ml of TRIzol reagent and continue grinding until a uniform paste is formed. Pipet the homogenized material in to a sterile microcentrifuge tube and incubate for 5 min at 25-30 °C to permit complete dissociation of nucleoprotein complexes.
- 15% Polyacrylamide gel electrophoresis (PAGE)
Small gels (size of regular protein gels 1.5 mm thick) are good to detect the expression of small RNAs. However, to analyze length variants of individual small RNAs (e.g. 20 nt, 21 nt, 22 nt), you need to run longer gels. Before assembling the electrophoresis system, clean all equipment’s thoroughly for any RNase contamination using RNase ZAP. Avoid nuclease contamination throughout the procedure by using sterile solutions and RNase free plasticware.
- Assemble gel casting chamber following manufacturer instructions and handcast 15% polyacrylamide gel. Let the gel polymerize for at least 30 min to 1 h at room temperature (20-25 °C). Setup the electrophoresis module as recommended by the manufacturer. Rinse the wells thoroughly with running buffer (0.5x TBE) using a syringe to remove any traces of urea and pre run gel with tracking dye for nearly 15 min before loading the samples.
- While pre-running the gel, prepare RNA samples. Ideally, for a good siRNAs resolution, 35-40 μg of total RNA is recommended for loading (based on the purity and concentration of the RNA). Accordingly, adjust the volume of each sample to be loaded (according to the least concentrated sample) using DEPC-treated water. Add equal volumes of 2x loading dye to the samples. Mix well by gentle tapping and spin down briefly.
- Cap the tubes and denature RNA samples by heating at 90-100 °C for 15 min. Spin down the samples briefly followed by snap cooling on ice for 5 min.
- Load the samples and the siRNA size marker and run the gel at 200 V for nearly an hour.
Note: The amount of voltage and duration of the run for the gel depends on the types of power supply and gel-electrophoresis system used. Stop the run when the bromophenol blue dye has migrated to the end of the gel. Do not overrun.
- Assemble gel casting chamber following manufacturer instructions and handcast 15% polyacrylamide gel. Let the gel polymerize for at least 30 min to 1 h at room temperature (20-25 °C). Setup the electrophoresis module as recommended by the manufacturer. Rinse the wells thoroughly with running buffer (0.5x TBE) using a syringe to remove any traces of urea and pre run gel with tracking dye for nearly 15 min before loading the samples.
- Semi-dry electroblotting of siRNAs onto nylon membrane
- Carefully dismantle the electrophoresis apparatus without breaking the gel. Cut the lower portion of the gel containing both tracking dye (xylene cyanol and bromophenol blue) bands. On a 15% polyacrylamide gel, the bromophenol blue dye front runs near 10 nt long RNA. The upper portion of the gel which includes tRNA and 5S RNA is stained with ethidium bromide solution (0.25 μg/ml ethidium bromide in 0.5x TBE buffer) for 10-20 min. Inspect the integrity and equal loading of RNA in the gel using a 360 nm UV transilluminator. The sharpness and clarity of tRNA bands is a good indicator of RNA quality and integrity (Figure 1).
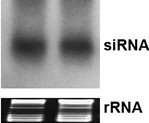
Figure 1. Northern blot revealing small interfering RNA molecules of 23 nucleotides (upper panel). Ethidium bromide-stained rRNA served as loading controls in the gel prior to RNA transfer (lower panel).
- To prepare a transfer sandwich, cut 3 MM Whatman filter paper (six pieces) and neutral Hybond nitrocellulose membrane to the size of the gel. Soak the membrane and Whatman paper in 0.5x TBE buffer until they are wet completely. Complete wetting of the membrane is important to insure proper binding of nucleic acids. Assemble the sandwich as follows.
- Place three pieces of Whatman paper on the anode plate of electroblotter. Make sure to roll out any air bubble that may inhibit transfer.
- Transfer the membrane on top of the filter papers and roll out any air bubble formed between the membrane and filer paper. After electrophoresis, equilibrate the gel in transfer buffer (0.5x TBE) for 5 to 10 min. Equilibration facilitates the removal of electrophoresis buffer salts and detergents. Carefully place the equilibrated gel over the membrane, aligning the stack as perfect as possible.
- Place the remaining three pieces of Whatman paper saturated in running buffer on the stack and roll out any trapped air.
- Slightly wet the stack with transfer buffer (0.5x TBE) and place the anode plate on the top without disturbing the gel-nitrocellulose stack and secure firmly.
Note: It is important to exclude excess buffer and air bubbles trapped in the filter paper and membrane when setting up the transfer.
- Set the power supply and run the transfer unit for 30 min at 10 V and 200 mAmp (constant current settings). Carefully disassemble the transfer unit and remove the filter paper and gel on top of the nylon membrane. Mark the orientation of the gel slots on the membrane with a pencil on RNA transfer side.
- Carefully dismantle the electrophoresis apparatus without breaking the gel. Cut the lower portion of the gel containing both tracking dye (xylene cyanol and bromophenol blue) bands. On a 15% polyacrylamide gel, the bromophenol blue dye front runs near 10 nt long RNA. The upper portion of the gel which includes tRNA and 5S RNA is stained with ethidium bromide solution (0.25 μg/ml ethidium bromide in 0.5x TBE buffer) for 10-20 min. Inspect the integrity and equal loading of RNA in the gel using a 360 nm UV transilluminator. The sharpness and clarity of tRNA bands is a good indicator of RNA quality and integrity (Figure 1).
- Chemical cross linking
Cross linking of the RNA to the membrane frequently improves the sensitivity of northern blots. RNA can be immobilized to the membrane using conventional methods such as exposure to standard dose of UV (120 mJ/cm2) or by baking in an oven (80 °C for 30 min). Both of these methods can be used successfully, however using the EDC-based cross-linking of small RNAs to membrane greatly improves the signal resolution of small RNAs by hybridization (Pall and Hamilton, 2008).- Immediately prior to use, prepare a solution of 0.16 M EDC solution in 0.13 M 1-methylimidazole and adjust the pH of the solution to 8 with HCl.
- Cut a single sheet of 3 MM Whatman filter paper slightly bigger than the size of the membrane and saturate it with the EDC solution.
- The membrane is placed on the EDC-saturated 3 MM paper with the side on to which RNA was transferred facing up. Roll out any air bubbles trapped between the membrane and filter paper.
- Cover the tray holding the membrane and filter paper with saran wrap and incubate at 60 °C for between 1 to 2 h.
- Remove the membrane and rinse with RNase free water to remove any residual EDC solution.
- The membrane can be dried and stored at -20 °C after removal of residual EDC without compromising the sensitivity of siRNA detection, or be used immediately for hybridization.
- Immediately prior to use, prepare a solution of 0.16 M EDC solution in 0.13 M 1-methylimidazole and adjust the pH of the solution to 8 with HCl.
- Hybridization
Finally, the membranes with fixed RNAs are incubated with specific, radiolabelled probes. For this, follow general prehybridization and hybridization procedures.
- Carry out prehybridization using ULTRAhyb-Oligo buffer for 30 min to one hour at 42 °C with gentle agitation.
Note: Probe signal strength obtained for small RNAs may vary depending on the composition of the hybridization buffer used.
- End-label probes with 32P; a number of kits are commercially available but we used the Megaprime DNA labeling system and followed the manufacturer’s instructions. Hybridize the membrane with the prepared 32P-end-labelled oligonucleotide probes. The probes must be labeled at high specific activity (≥ 108 cpm/μg template).
- Hybridize the membrane overnight (14-16 h) at 38 to 42 °C with gentle agitation.
- Posthybridization, the membrane is washed twice with a low stringency buffer solution (2x SSC, 0.5% SDS) for 10 min each, and once using a high stringency buffer solution (0.1x SSC, 0.1% SDS) for 5 min at 42 °C. If necessary, washing time can be increased under more stringent conditions (e.g., when high background levels are seen).
- Expose the membrane to X-ray film at -80 °C for signal visualization. Adjust the exposure time depending on signal intensity.
Note: It is extremely important that all steps involving radioactive material are followed under appropriate safety guidelines.
- Carry out prehybridization using ULTRAhyb-Oligo buffer for 30 min to one hour at 42 °C with gentle agitation.
Recipes
- DEPC treated water
Dissolve 1 ml of DEPC in 1 L of distilled water with continuous stirring and autoclave
- 10x TBE (1 liter)
108 g Tris Base
55 g Boric acid
40 ml EDTA (0.5 M, pH 8.0)
Bring volume to 1 liter with DEPC-treated water, mix by stirring and autoclave
- 10% APS
Mix 1 g APS in 10 ml of RNase free water and filter sterilize
- 15% polyacrylamide gel
21 g urea
5 ml 10x TBE
18.8 ml 40% Acrylamide/Bis-acrylamide (19:1)
Bring volume to 50 ml with RNase free water and vortex well until urea is dissolved
When ready to pour gel add by swirling briefly
250 μl 10% Ammonium persulfate
50 μl TEMED
Note: TEMED should be added last. Mix the solution well, immediately pour gel smoothly without creating any air bubbles and let it polymerize. This recipe is sufficient to fill four mini gel cassettes. Amount may be adjusted depending on the application. Gel can be stored in 4 °C degree fridge after tightly wrapping in saran wrap with comb still inserted. Do not freeze gels.
- 2x loading buffer (50 ml)
47.5 ml 95% Formamide
2 ml EDTA (20 mM, pH 8.0)
0.025 g Bromophenol blue
0.025 g Xylene cyanol
Acknowledgments
This protocol was adapted from the protocol described by Pall and Hamilton (2008). We are grateful for discussions with and technical advice from Mrs. Melanie Walker and Dr. Hélène Sanfaҫon.
References
- Pall, G. S. and Hamilton, A. J. (2008). Improved northern blot method for enhanced detection of small RNA. Nat Protoc 3(6): 1077-1084.
- Panwar, V., McCallum, B. and Bakkeren, G. (2013a). Host-induced gene silencing of wheat leaf rust fungus Puccinia triticina pathogenicity genes mediated by the Barley stripe mosaic virus. Plant Mol Biol 81(6): 595-608.
- Panwar, V., McCallum, B. and Bakkeren, G. (2013b). Endogenous silencing of Puccinia triticina pathogenicity genes through in planta-expressed sequences leads to the suppression of rust diseases on wheat. Plant J 73: 521-532.
Article Information
Copyright
© 2013 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Panwar, V. and Bakkeren, G. (2013). A High Resolution Short Interfering RNA (siRNA) Detection Method from Virus-infected Plants. Bio-protocol 3(20): e940. DOI: 10.21769/BioProtoc.940.
Category
Plant Science > Plant molecular biology > RNA > RNA interference
Plant Science > Plant immunity > Perception and signaling
Molecular Biology > RNA > RNA interference
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.