- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Efficient Generation of Genome-wide Libraries for Protein–ligand Screens Using Gibson Assembly
Published: Vol 12, Iss 22, Nov 20, 2022 DOI: 10.21769/BioProtoc.4558 Views: 3678
Reviewed by: Kristin L. ShinglerEmmanuel Orta-ZavalzaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2023
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Genome-wide screens using yeast or phage displays are powerful tools for identifying protein–ligand interactions, including drug or vaccine targets, ligand receptors, or protein–protein interactions. However, assembling libraries for genome-wide screens can be challenging and often requires unbiased cloning of 105–107 DNA fragments for a complete representation of a eukaryote genome. A sub-optimal genomic library can miss key genomic sequences and thus result in biased screens. Here, we describe an efficient method to generate genome-wide libraries for yeast surface display using Gibson assembly. The protocol entails genome fragmentation, ligation of adapters, library cloning using Gibson assembly, library transformation, library DNA recovery, and a streamlined Oxford nanopore library sequencing procedure that covers the length of the cloned DNA fragments. We also describe a computational pipeline to analyze the library coverage of the genome and predict the proportion of expressed proteins. The method allows seamless library transfer among multiple vectors and can be easily adapted to any expression system.
Keywords: Genome-wide libraryBackground
Protein–ligand interactions are essential for nearly all cellular processes, including gene expression regulation, pathogen–host interactions, and the identification of drug or vaccine targets. Defining interacting partners can be challenging and usually relies on in vitro biochemical approaches or genetic manipulation of the organism of interest. For many non-model organisms, genetic manipulation tools are still scarce or of laborious application, thus limiting the approaches available for discovering protein–ligand partners or protein interaction networks.
Genome-wide libraries are powerful tools for exploratory studies in protein–ligand interactions (Bharucha and Kumar, 2007; Bidlingmaier and Liu, 2011). Once incorporated into an expression system, libraries can encode all possible polypeptides of an organism without bias associated with its life cycle stage or culture conditions. Generating a eukaryote genome-wide library requires cloning approximately 105–107 different DNA fragments. Hence, the method must be robust to ensure that all possible coding sequences are included and in frame with the proteins of the expression system. The most common methods for generating genomic libraries involve DNA fragmentation by enzymatic digestion or sonication, followed by cloning the DNA fragments into restriction enzyme–digested or T/A-based plasmid vectors. Adapters or barcode sequences are often added to the DNA fragments to help with library amplification and sequencing.
There are several expression systems for interaction screenings. Baker's yeast—Saccharomyces cerevisiae—has been widely used as an expression system due to its versatility for genetic manipulation, high levels of protein expression, inducible gene expression, and the presence of eukaryotic post-translational modifications (Boder and Wittrup, 1997; Bidlingmaier and Liu, 2011). Yeast surface display (YSD) is one of the most successful screening systems for discovering protein–ligand interactions (Gibson et al., 2009; Bidlingmaier and Liu, 2011; Gu et al., 2014; Danecek et al., 2021). In this system, library proteins are expressed in frame with proteins of an endogenous surface anchoring system (e.g., Aga1p-Aga2p) and then displayed in approximately 105 copies of a single protein per cell (Inoue et al., 1990). The yeast population can express a whole genomic library and is usually screened using fluorescence or magnetic-activated cell sorting to enrich yeast cells that react with the ligands. The library DNAs from the sorted yeasts are then sequenced to identify the coding sequences for the ligand–interacting partners. Hence, the YSD methodology coupled with a strategy to enrich or isolate the positive interactions can lead to the discovery of protein–ligand partners while overcoming the limitations of genetic manipulation of non-model organisms.
We present here an efficient method to construct genome-wide libraries. In our approach, the genomic DNA is fragmented to the average gene size, end-repaired, and A-tailed for the ligation of adapter sequences for PCR amplification (Figure 1). The fragments are PCR-amplified with a pair of primers that hybridizes with the adapters and contains a ~20 bp overlapping sequence to the receptor vector for Gibson assembly. The Gibson assembly entails an isothermal reaction to join DNA molecules using a ~20 bp overlapping sequence between DNA fragments (e.g., library fragments and plasmid vector). This reaction includes a 5' exonuclease that generates 3’ overhangs in the joining molecules. After DNA molecules annealing, a DNA polymerase fills existent gaps in the annealed strands, and a DNA ligase seals strand nicks completing the process (Kieke et al., 1997). Using the Gibson method, the amplicons originating from adapter-ligated genomic fragments are joined to a linearized vector in a highly efficient one-step library assembly. Our protocol was designed to facilitate library transfer between expression systems and streamline library sequencing by Oxford nanopore technology to speed up the screening process. The nanopore sequencing technology has the advantage of generating sequences covering the entire cloned fragment; however, the protocol can be adapted to other sequencing methods. Our protocol details the library assembly, optimized library sequencing, and a computational pipeline to analyze the library coverage of the genome and evaluate library diversity and completeness. We also include a computational tool that predicts the library polypeptides expressed in frame with proteins of the expression system.
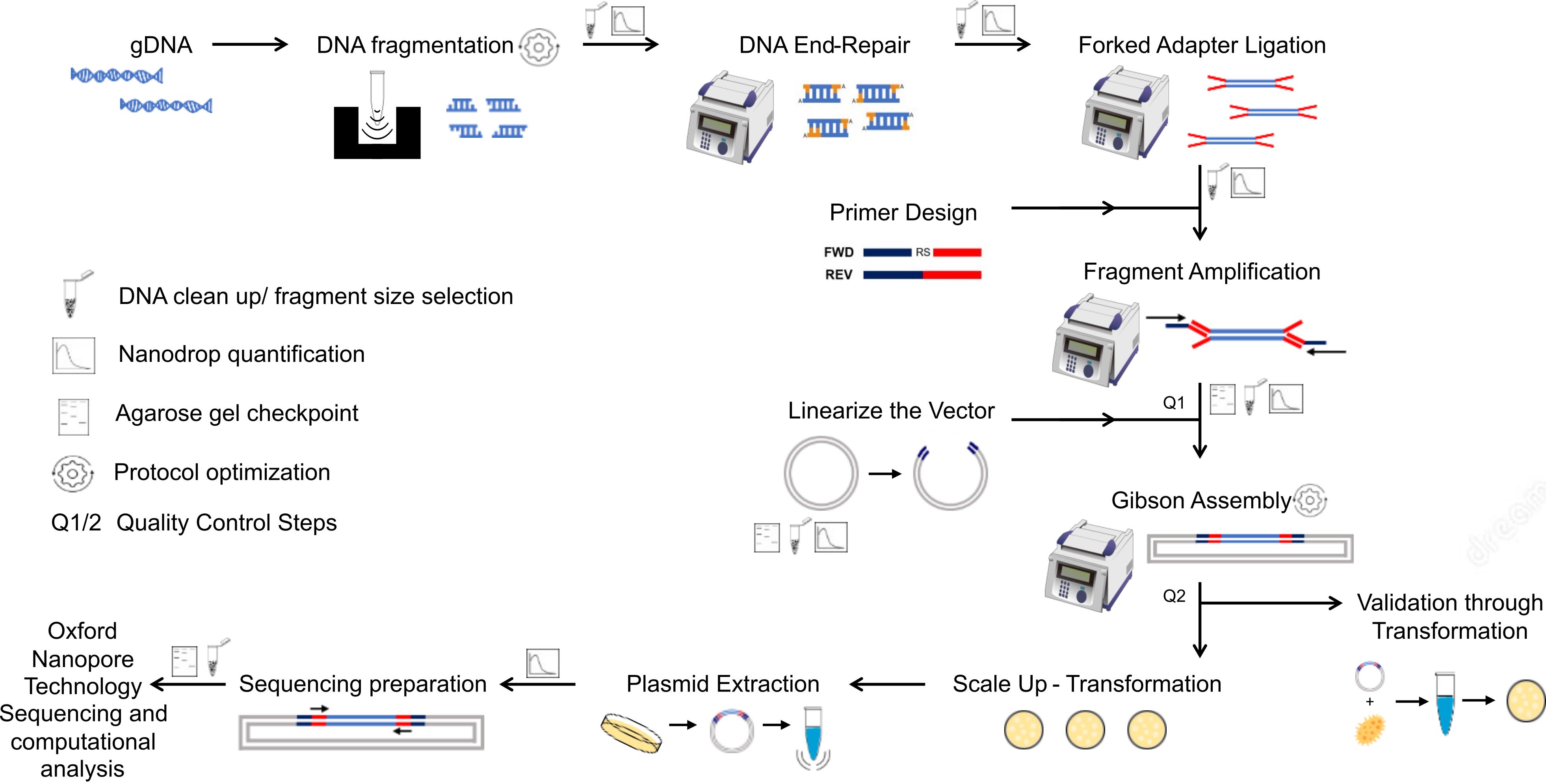
Figure 1. Diagram of genome-wide library preparation protocol and quality control steps
Materials and Reagents
Serological pipet, 10 mL (Drummond, catalog number: 6 000 010)
Labcon Eclipse refill universal-fit pipette tips with TubeGuard 0.1–10 µL (Labcon Eclipse, catalog number: 1036-260)
VWR refill pipette tips 100/200 µL (VWR, catalog number: 89079-476)
VWR refill pipette tips 1,000 µL (VWR, catalog number: 89079-470)
15 mL Centrifuge Tube, Bulk, Sterile, 25/pk, 500/cs (Montreal Biotech, catalog number: MBI601052C)
50 mL Centrifuge Tube, Bulk, Sterile, 25/pk, 500/cs (Montreal Biotech, catalog number: MBI602052C)
150 mm × 15 mm tissue culture-treated dishes (Fisher Scientific, catalog number: FB0875714)
Microtube-50 AFA fiber screw-cap vials (Covaris, catalog number: 520166)
Syringe Filter PES 0.22 µm 30 mm diameter, sterilized by Gamma irradiation (Ultident, catalog number: 229747)
NEB® 5-alpha F'Iq Competent Escherichia coli (High Efficiency) (New England Biolabs, catalog number: C2992H)
Mag-Bind® TotalPure NGS beads (Omega Bio-Tek, catalog number: M1378-01)
NEBNext® end-repair module (New England Biolabs, catalog number: E6050S)
Monarch® DNA elution buffer (New England Biolabs, catalog number: T1016L)
Ligation sequencing kit (Oxford Nanopore Technologies, catalog number: SQK-LSK110)
Native barcoding expansion 1-12 (Oxford Nanopore Technologies, catalog number: EXP-PBC001)
Blunt/TA ligase master mix (New England Biolabs, catalog number: M0367S)
NEBNext FFPE DNA repair mix (New England Biolabs, catalog number: M6630S)
NEBNext Quick Ligation Module – 20 rxs (New England Biolabs, catalog number: E6056S)
Taq DNA polymerase with ThermoPol buffer (New England Biolabs, catalog number: M0267S)
dNTPs mixture 10 mM (BioBasic, catalog number: DD0056)
Agarose (Bishop Canada Inc., catalog number: AGA002.250)
NEBuilder HiFi DNA assembly master mix (New England Biolabs, catalog number: E2621S)
Tryptone (BioBasic, catalog number: TG217(G211))
Yeast extract (BioBasic, catalog number: G0961)
Agar A (Biobasic, catalog number: FB0010)
Sodium chloride (Fisher Scientific, catalog number: S271-1)
Sodium Hydroxide (white pellets) (Fisher Scientific, catalog number: BP359-212)
Hydrochloric acid (Fisher Scientific, catalog number: A144-212)
Boric Acid (Omnipure, catalog number: 2710)
Ethylenediaminetetraacetic acid (EDTA) (Sigma Aldrich, catalog number: E9884-100G)
Manganesium chloride Hexahydrate (Fisher Scientific, BP214-500)
Calcium chloride dihydrate (Sigma Aldrich, catalog number: 223506-500G)
PIPES, Sesquisodium Sodium Salt (Fisher Scientific, catalog number: BP304-100)
Potasium hydroxide pellets (Fisher Scientific, catalog number: P250-500)
Trizma Base (AMRESCO, catalog number: 0497-5KG)
Ampicillin, trihydrate (BioBasic, catalog number: AB0064)
pUC-19 plasmid (NEB, catalog number: N3041S)
Covaris AFA-grade water (800 mL) (D-mark Biosciences, catalog number: Cov-520101)
Lambda DNA/Hind III plus marker (BioBasic, catalog number: M105R-2)
Ecostain (BioBasic, catalog number: DT81413)
Cell scraper (Cole-Parmer, catalog number: UZ01959-14)
EZ-10 spin column plasmid DNA miniprep kit (BioBasic, catalog number: BS414)
NucleoBond® Xtra maxi plus EF (Takara, catalog number: 740426.10)
pYD1 plasmid (Addgene, catalog number: 73447)
Restriction enzymes:
Bam HI (New England Biolabs, catalog number: R0136S)
Hind III (New England Biolabs, catalog number: R0104S)
Xho I (New England Biolabs, catalog number: R0146S)
NEBufferTM r3.1 (New England Biolabs, catalog number: B6003S)
Flow cell (R9.4.1) (Oxford Nanopore Technologies, catalog number: FLO-MIN106D)
Luria-Bertani (LB) medium with agar and ampicillin (see Recipes)
Ampicillin stock solution (100 mg/mL) (50 mL) (see Recipes)
Tris-Borate-EDTA buffer (10 L) (see Recipes)
10× TBE stock solution (1 L) (see Recipes)
SOB medium (1 L) (see Recipes)
Inoue solution (1 L) (see Recipes)
0.5 M PIPES buffer (100 mL) (see Recipes)
70% ethanol (see Recipes)
Equipment
Pipettes 0.2–2 µL (Gilson, catalog number: FA10001M), 2–20 µL (Gilson, catalog number: FA10003M), 20–200 µL (Gilson, catalog number: FA10005M), and 100–1,000 µL (Gilson, catalog number: FA10006M)
Portable Pipet-Aid® XP pipette controller (Drummond, catalog number: 4-000-101).
Focused-ultrasonicator (Covaris, M220, catalog number: 500295)
12-position magnetic rack for 1.5 mL tubes (Promega, catalog number: Z5342)
T100 thermal cycler (Bio-Rad, catalog number: 1861096)
Isotemp 215 water bath (Fisher, catalog number: 15-462-15)
Avanti® J-E centrifuge, 50 Hz, 200 V, 24 A (Beckman Coulter, catalog number: 369005)
Microcentrifuge 5418/5418R (Eppendorf, catalog number: EP5401000137)
Variable speed rocking platform shaker (VWR, catalog number: 75832-308)
NAPCO analog display automatic CO2 water-jacketed incubator (Napco, catalog number: 102219021)
Innova® 44 incubator shaker (New Brunswick, catalog number: M1282-0000)
Water system ultrapure (Millipore Synergy, catalog number: C9202)
Electrophoresis unit: Thermo Scientific Power Supply 400 mA 300 V (Fisher, catalog number: S65533Q)
NanoDrop ND-1000 (Thermo Fisher, catalog number: 2353-30-0010)
ChemiDocMP imaging system (BioRad, catalog number: 12003154)
Oxford nanopore MinION Mk1C sequencer (Oxford Nanopore Technologies, catalog number: MIN-101C)
Software
Minimap2 (Lemos Duarte et al., 2021) (https://github.com/lh3/minimap2)
Samtools (Li, 2018) (https://www.htslib.org/)
DeepTools (Liao et al., 2014) (https://deeptools.readthedocs.io/en/develop/index.html)
Libframe (This work) (https://github.com/cestari-lab/Libframe-tool)
Subread (Pointer et al., 2014) (http://subread.sourceforge.net/featureCounts.html)
Circlize (Ramirez et al., 2016) (https://jokergoo.github.io/circlize/)
Procedure
DNA fragmentation and size selection
Prepare three microtube-50 AFA fiber screw-cap vials, each containing 5 µg of genomic DNA in 55 µL of AFA-grade water. Sonicate using a focused-ultrasonicator for 3 s at 200 cycles per burst, 25 peak incidence power (W), and 10% duty factor at 7 °C, to obtain fragments of 0.5–3 kb (average gene range). Optimization of this step may be necessary depending on organism genome size, base composition, or DNA extraction method (see Note 1).
Combine the fragmented DNA from the three vials for a total of 165 µL in a 1.5 mL Eppendorf tube and analyze 30 µL in 1% agarose/TBE gel with 5 µL of ecostain. Load a well with 1 µg of non-fragmented DNA for comparison (optional). Run at 90 V for 30 min or two-thirds of the gel length and visualize the DNA in a ChemiDocMP imaging system. The fragmented DNA should migrate as a smear between 0.5–3 kb with peak intensity at 1.5 kb (Figure 4A). The peak intensity of fragments may vary depending on fragmentation conditions and genome composition.
Size select the DNA to obtain fragments above 500 bp by adding 95 µL of Mag-Bind® TotalPure NGS beads to 135 µL of fragmented DNA (ratio 0.7×, beads volume: fragmented DNA volume). Incubate for 10 min at room temperature with rotation at 50 rpm (see Note 2).
Quickly spin (approximately 10 s, 850 × g) the microfuge tube containing beads and DNA mixture, transfer the tube to a magnetic rack, and incubate it at room temperature until the beads separate from the solution (approximately 1 min). Pipette and discard the supernatant.
Slowly add 200 µL of 70% ethanol over the beads and incubate for 30 s or until beads separate from the solution. Collect and discard the ethanol. Repeat this wash step twice.
Air-dry the beads and mixture for 2 min at room temperature. Do not dry to the point of cracking the beads.
Add 55 µL of elution buffer to the beads, mix gently by flicking the tube, and incubate for 1 min at room temperature.
Transfer the Eppendorf tube to a magnetic rack to separate the beads from eluted DNA. Collect the supernatant, now containing the DNA, into a new 1.5 mL microtube tube. Discard the tube containing the beads.
Quantify the DNA concentration and purity using NanoDrop. Size selection should recover between 50% and 70% of the DNA. The ideal 260/280 and 260/230 absorbance ratios are 1.8 and 2.0, respectively.
Analyze 5 µL of recovered DNA in 1% agarose/TBE gel with 5 µL of ecostain, as described in step A2 to verify size selection and DNA recovery.
End-repair reaction and adapter ligation
Add 1 µg of DNA (from step A8) to a 0.2 mL tube and mix with 10 µL of NEBNext® end-repair buffer (10×) and 5 µL of end-repair enzyme mix. Add water for a final volume of 100 µL.
Incubate for 60 min at 20 °C in a thermocycler.
Clean up the end-repaired DNAs by adding 70 µL of Mag-Bind® TotalPure NGS beads (ratio of 0.7×, beads volume:end repair reaction volume). Follow the steps described in steps A3 to A8. Elute the DNA in 31 µL of elution buffer.
Quantify the DNA (1 µL of the sample) using NanoDrop. Expected DNA recovery is approximately 90%.
Add the remaining 30 µL of end-repaired DNA (approximately 1 µg) into a 0.2 mL tube. Add 20 µL of the Oxford nanopore barcode adapters (BCA reagent from the ligation sequencing kit) to the tube and 50 µL of 2× Blunt/TA ligase master mix. Pipette gently to mix the reaction.
Incubate at 25 °C for 2 h in a thermocycler.
Clean up the barcode adapter-ligated DNA by adding 70 µL of Mag-Bind® TotalPure NGS beads (ratio of 0.7×, beads volume:barcode adapter ligation reaction volume), as described in steps A3 to A8. Elute the DNA in 31 µL of elution buffer.
Quantify the DNA using NanoDrop. Expected DNA recovery is approximately 90% of the input material (step B5).
Primer design and library amplification
This step describes the design of primers and fragment amplification by PCR for library cloning into the expression vector using Gibson assembly. Figure 2 shows the primers' sequences and a diagram of the pYD1 plasmid used in this step. The primers should anneal to the barcode adapter ligated to the library fragments in step B5 (Figure 2A, sequences denoted by As) and have overhangs complementary to the expression vector (sequences indicated by Xs).
Each primer overhang sequence should be ~20 bp, and the primer melting temperature should be approximately 50 °C. If desired, add restriction enzyme sites in the primers to reconstitute any restriction sites in the plasmid. The restriction site used to linearize the vector will be lost if not reconstituted in the primer (Figure 2A).
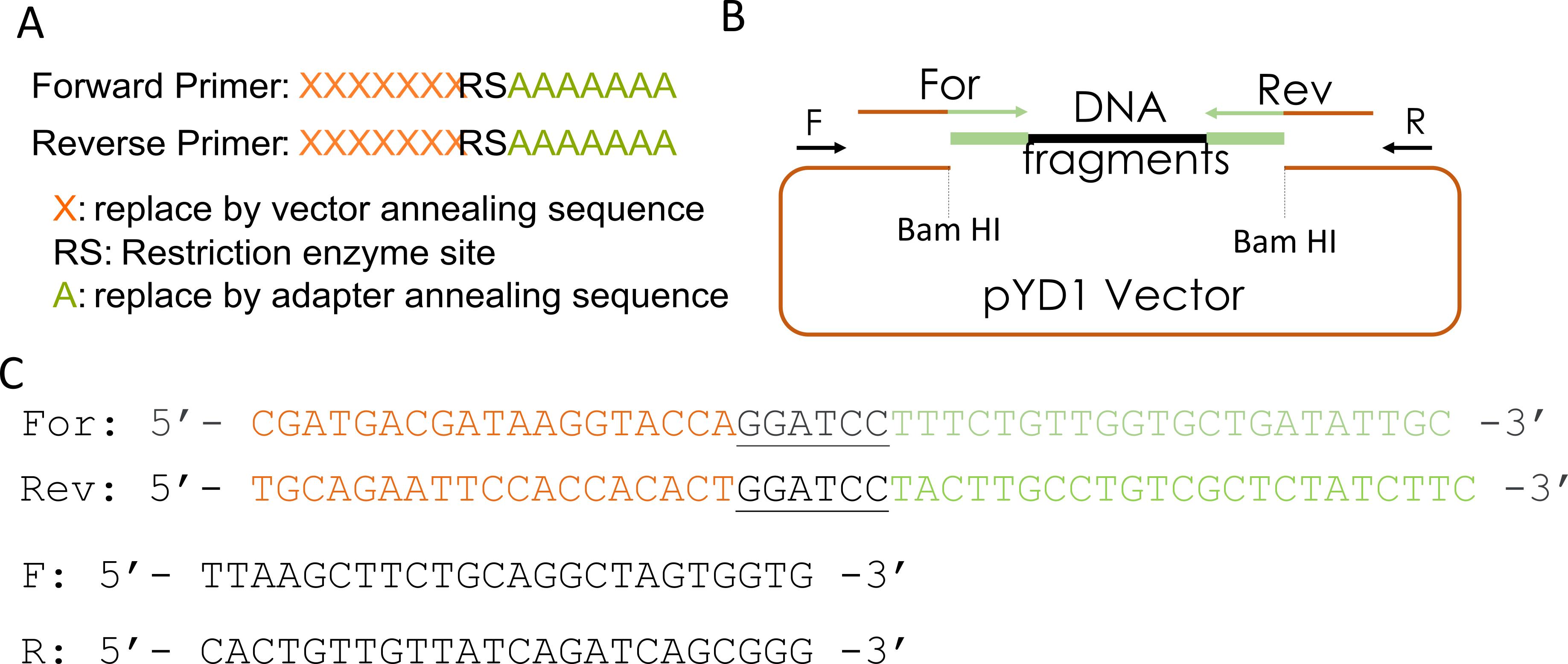
Figure 2. Design of primers for Gibson assembly. (A) Scheme of forward and reverse primers used for Gibson assembly. (B) Diagram of Bam HI-digested pYD1 vector, barcode adapter-ligated fragment (DNA fragments), and primers. (C) Forward (For) and reverse (Rev) primers used to amplify barcode adapter-ligated fragments for Gibson assembly (see pairing regions in B). The sequences in orange correspond to a ~20 bp vector sequence complementary to the pYD1 vector, the black letters are the Bam HI restriction sites, and the ~20 bp nanopore barcode adapter annealing sequences are in green. F and R: forward and reverse primers, respectively, that pair to the vector flanking the cloning site and are used to amplify cloned fragments by PCR for cloning validation or sequencing.
Prepare six PCR reactions, using the barcode adapter-ligated fragmented DNA as template, as follows: 10 µL of ThermoPol buffer 5×, 200 µM dNTPs, 500 nM primer mix (mix of forward and reverse primers), and 1.25 units of Taq DNA polymerase. Add the barcode adapter-ligated DNA (20 ng) and adjust the final volume to 50 µL with water. Amplify the reaction in a thermocycler with an initial denaturation at 95 °C for 30 s, then 14 cycles at 95 °C for 30 s, 49 °C for 30 s, and 68 °C for 3 min. Add a final extension of 5 min at 68 °C and store the reaction at 4 °C (see Note 3).
Combine the PCR reactions and purify the amplicons by adding 210 µL of Mag-Bind® TotalPure NGS beads to the 300 µL of reaction (ratio of 0.7×, beads volume: end repair reaction volume). Follow the procedure described in steps A3–A8. Elute the DNA in 31 µL of elution buffer.
Quantify the DNA using NanoDrop. Expected DNA recovery is approximately 80–140 ng/µL.
Linearize the vector
Digest 4 µg of pYD1 vector DNA by adding 3 µL of 10× reaction buffer r3.1, 2 µL (20 units) of Bam HI restriction enzyme, and water to 30 µL final volume. Incubate at 37 °C for 16 h.
Note: Do not dephosphorylate the vector, as it will interfere with the Gibson assembly reaction (see Note 4).
Purify the linearized DNA by adding 15 µL of Mag-Bind® TotalPure NGS beads (ratio of 0.5×, beads volume:digestion reaction volume). Follow the procedure described in steps A3 to A8. Elute the DNA in 31 µL of elution buffer.
Quantify the DNA using NanoDrop. Expected DNA recovery is approximately 90%.
Gibson assembly reaction and efficiency assessment
Add 10 µL of NEBuilder HiFi DNA assembly master mix to a 0.2 mL tube. Then add 50 ng (16 fmol) of digested vector (from step D3), 30 ng (32 fmol, see Note 5) of amplified fragments (from step C3), and water to a total of 20 µL reaction. The concentrations indicated result in a 1:2 ratio of vector:PCR fragments.
Incubate the Gibson assembly reaction for 1 h at 50 °C in a thermocycler.
Mix 1 µL of the Gibson assembly reaction with 50 µL of competent NEB 5-alpha F'Iq E. coli cells, defrosted in ice, in an ice-cold 15 mL Falcon tube. Incubate on ice for 30 min. We recommend performing additional transformations with 100 pg of pUC19 and 50 µL of competent cells to estimate the efficiency of the competent cells, and with 50 ng of the pYD1 vector as a control for no fragment insertion (to be used in step E9). See Notes 6 and 7.
Incubate the mix at 42 °C for 30 s, immediately transfer to ice, and incubate for 2 min.
Add 950 µL of SOB medium, incubate at 37 °C, and shake for 1 h.
Plate 500 µL of transformed cells into 150 × 15 mm Petri dishes containing LB medium with agar and ampicillin at 50 µg/mL for plasmid selection. For the pUC19 transformed cells, make serial dilutions (1:10, 1:100, and 1:1,000) and plate 500 µL into Petri dishes.
Incubate overnight at 37 °C.
Quantify the number of colonies obtained with the transformations to estimate the number of Gibson assembly reactions and transformations necessary for library assembly. Use the pUC19 transformation to estimate the efficiency of the competent cells. A total of 4,000–6,000 colonies per plate should be expected for the Gibson reaction transformation using competent cells with an efficiency of 109 CFU/µg of pUC19 DNA. See Note 8.
To check the efficiency of the Gibson assembly, select 10–20 colonies from the transformation for colony PCR to amplify the cloned fragments using primers pairing to the vector and flanking the library fragments (Figure 2B and 2C, primers F and R). In 0.2 mL tubes, prepare 22 PCR reactions using 4 µL of ThermoPol buffer 5×, 200 µM dNTPs, 500 nM primer mix (forward and reverse), and 1.25 units of Taq DNA polymerase in 20 µL. We recommend preparing a master PCR reaction mix, then splitting reactions into 21 tubes. Pick a colony with a 10 µL pipet tip and mix the colony with the reaction in the PCR tube. Prepare one reaction with a colony from pYD1-transformed (no library) bacteria as a control. Amplify the reaction with an initial denaturation at 95 °C for 2 min, then 35 cycles at 95 °C for 30 s, 54 °C for 30 s, and 68 °C for 3 min. Add a final extension of 5 min at 68 °C, and store the reaction at 4 °C.
Load 20 µL of the reaction to a 1% agarose/TBE gel with 5 µL ecostain, as described in step A2, to analyze the amplified DNA. The amplicons should appear as a single band with a size in the range of the selected fragments (0.5–3 kb). A higher than 90% efficiency in the assembly should be expected, i.e., 18 positive colonies out of 20. Otherwise, optimize the Gibson assembly reaction (step E1, see Note 9).
To check if fragments cloned are from the desired organism genome, pick ten colonies from the transformation to 15 mL cell culture tubes with 2 mL of LB medium and 50 µg/mL of ampicillin antibiotic for plasmid selection. Incubate at 37 °C, shaking at 225 rpm for 18–24 h.
Isolate the plasmid from colonies using a mini-prep kit and perform Sanger sequencing using primers pairing with the vector and flanking the library sequences (Figure 2C, primers F and R).
Perform sequence alignment with the basic local alignment search tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi) against the organism genome or the non-redundant database from the National Center for Biotechnology Information. The sequences should align with sequences from the organism's genome used for the initial DNA fragmentation.
Calculation of library size and large-scale transformation
Calculate the required number of cloned fragments (N) for each genome sequence to be represented in the library with a probability (P) of 99% using the Carbon and Clarke equation:
N=ln(1-P)/ln(1-F) , where F=i/g
F is the quotient between insert fragment size (i) and genome size (g) in bp. We recommend multiplying the final value by six to increase the chance that a fragment will be cloned in frame with proteins from the expression system, e.g., Aga2p in the pYD1 vector.
Divide the calculated number of required fragments (N) by the number of colonies obtained in step E8 to estimate the number of transformations and Gibson assembly required. One Gibson assembly reaction yields 20 transformations and results in 40 transformation plates. For a 44 Mb haploid genome (e.g., the protozoan Trypanosoma cruzi Sylvio X10/1 strain haploid genome) and 6-fold of the minimum calculated number of fragments at 1.5 kb mean size, expect to harvest approximately 811,000 colonies. Hence, 203 plates at 4,000 colonies each and approximately five Gibson assembly reactions.
Transform the Gibson assembly mix as described in steps E3–E7 to achieve the number of calculated fragments. Consider each colony a cloned fragment if assembly efficiency equals 100%.
Extraction of library plasmids DNA
Scrape all colonies from the plates and combine them in a 50 mL Falcon tube. Adjust the volume to 50 mL using LB medium with 50 µg/mL of ampicillin. If not extracting DNA immediately, keep the suspension at 4 °C for a maximum of one week.
Dilute the culture in LB with 50 µg/mL of ampicillin to obtain an OD between 1 to 2. Incubate at 37 °C for 3 h for cell recovery.
Isolate the plasmid DNAs using the NucleoBond® Xtra maxi plus EF according to the manufacturer's instructions.
Quantify the plasmid DNA on the NanoDrop. The expected DNA yield should be approximately 0.5–2 mg.
Digest 2 µg of plasmid DNA by mixing the DNA with 3 µL of 10× NEBufferTM r3.1 and 0.5 µL of Bam HI (5 units) at a final reaction volume of 30 μL. Incubate at 37 °C for 2–18 h. Other restriction enzymes that cut in the multiple cloning site, e.g., Hind III and Xho I, can also be used.
Load the 30 µL reaction to a 1% agarose/TBE gel with 5 µL of ecostain, as described in step A2, to analyze the digested DNAs. The gel should show a 5 kb band (digested pYD1) and a smear between 500 and 3,000 bp (digested library fragments) with a peak around 1.5 kb (Figure 4A).
Preparation of the library for Oxford nanopore DNA sequencing
Prepare six PCR reactions, each with 20 ng of plasmid, 10 µL of ThermoPol buffer 5×, 200 µM dNTPs, 400 nM barcode primers (included in the Native Barcoding Expansion 1-12 from Oxford Nanopore), 1.25 units of Taq DNA Polymerase, and water for a 50 µL final reaction volume. Run the PCR reactions in a thermocycler with an initial denaturation at 95 °C for 30 s, then 16 cycles at 95 °C for 30 s, 60°C for 30 s, and 68 °C for 3 min. Add a final extension of 5 min at 68 °C and store the reaction at 4 °C.
Mix the PCR reactions to obtain a total of 300 µL reactions. Add 210 µL of Mag-Bind® TotalPure NGS beads (0.7× ratio, beads volume:PCR reaction volume) and purify the DNA according to the steps described in steps A3 to A8. Elute the DNA in 61 µL of elution buffer.
Quantify the DNA using NanoDrop. The expected yield is approximately 30–60 ng/µL.
Add 1 µg of DNA to a 0.2 mL tube and mix with 3.5 µL of NEBNext® end-repair buffer and 3 µL of NEBNext end-repair enzyme mix, 3.5 µL NEBNext® FFPE DNA Repair Buffer, 3 µL NEBNext® FFPE DNA Repair mix, and 1 µL (approximately 10 ng) of CS DNA (control DNA, ~3 kb phage lambda DNA fragment). Add water for a final volume of 100 µL.
Incubate for 60 min at 20 °C in a thermocycler.
Clean up the end-repaired DNAs using Mag-Bind® TotalPure NGS beads with a ratio of 0.7× (beads volume:end repair reaction volume) by adding 70 µL of beads and follow the steps A3 to A8. Elute the DNA in 61 µL of elution buffer.
Quantify the DNA using NanoDrop. Expected DNA recovery is approximately 90%.
Add the end-repaired DNA (0.5–1 µg) in 60 µL volume to a 0.2 mL tube, then mix with 25 µL of Oxford nanopore ligation buffer (LNB), 10 µL of NEBNext Quick T4 DNA ligase, and 5 µL of Adapter mix (AMX) (LNB and AMX are reagents included in the Ligation sequencing kit from Oxford Nanopore). Mix by carefully pipetting the reaction up and down.
Incubate for 2 h at 20 °C in a thermocycler.
Add 40 µL of Mag-Bind® TotalPure NGS beads to the ligation reaction (0.4× ratio, beads volume:ligation reaction volume). Mix for 10 min with rotation at room temperature.
Quickly spin the sample for 10 s and separate the beads from the solution by incubation on a magnetic stand for 2 min at room temperature. Remove and discard the supernatant.
Add 250 µL of short fragmentation buffer and mix gently by flicking the tube.
Repeat steps H11 and H12. Then, separate the beads from the solution as described in step H11. Air-dry the beads for 30 s at room temperature. Do not over-dry.
Add 12 µL of Oxford nanopore elution buffer. Expect a yield of approximately 10–40 ng/µL, i.e., 200–1,500 fmol of DNA. Between 5 and 55 fmol of DNA are used for sequencing. See Note 5.
Load 5–55 fmol of the prepared library into the flow cell (R9.4.1) according to manufacturer’s instruction. Sequence the DNA following the manufacturer's instructions.
Data analysis
Computational analysis of sequenced library
The data used in these analyses were deposited in the Sequence Read Archive (SRA) with BioProject number PRJNA851089.
The Minknow software from the MinION Mk1C device creates sequencing report files that summarize the nanopore sequencing results. The .txt summary file can be used to extract information on sequence quality, reads' length, and barcodes. Sequences are produced in fast5 format and then basecalled using the Guppy tool through the MinION device during or after sequencing. For our experiments, we chose high accuracy basecalling through MinION, which generates the fastq files. The following steps describe the tools and scripts used for data analysis using fastq as input files in the pipeline. We also describe the scripts used for calculating genome mapping, visualizing mapped reads, and predicting the peptides produced by the libraries using the Libframe tool we developed. For a schematic summary of these steps and an illustration of the results, see Figures 3 and 4.
Note: Before running scripts, check the available computational resources, i.e., memory and processors. We recommend connecting to a server and submitting jobs via Bash scripts. The resources necessary for the job will vary according to the number and size of files and the tools used. The scripts below were submitted to a server (Compute Canada) using Linux Ubuntu in Windows Subsystem for Linux. A general file name is given for simplicity, e.g., "GenomeOfReference.fasta" for the reference genome or "reads-map_v1.sam" for the mapped output file.

Figure 3. Flow chart of the computation analysis stepsDecompress the fastq files and map the reads to the genome using minimap2. Mapping conditions can be changed to improve the data mapping to the genome. See minimap2 manual (https://github.com/lh3/minimap2).
#Before mapping, files in the *.fasta.tar.gz format must be decompressed using the bash command
tar xvzf yourdata.fastq.tar.gz -C pathdirectory
#This script aligns the reads obtained from the ONT sequencing to the reference genome, creating a .sam file.
module load minimap2/2.24
minimap2 -ax map-ont -k11 -m30 -w7 -I4G -t16 -2 GenomeOfReference.fasta \
/~/data/fastq/*.fastq.gz \
>/~/data/analysis/reads-map_v1.sam
Obtain the mapping statistics and convert .sam files to .bam files. Then, sort and index .bam files. The proportion of mapped reads will vary according to the data quality and assembly of the reference genome.
#Samtools commands extract statistics from the read mapping and will create the bam binary files for further processing.
module load samtools
samtools flagstat /~/data/analysis/reads-map_v1.sam > /~/data/analysis/reads-map_v1-flagstat.txt
samtools stat /~/data/analysis/reads-map_v1.sam > /~/data/analysis/reads-map_v1-stat_lib.txt
samtools view -S -b /~/data/analysis/reads-map_v1.sam > /~/data/analysis/reads-map_v1.bam
samtools sort /~/data/analysis/reads-map_v1.bam > /~/data/analysis/reads-map_v1_sorted.bam
samtools index /~/data/analysis/reads-map_v1_sorted.bam > /~/data/analysis/reads-map_v1_sorted.bam.bai
Perform coverage analysis to determine the read coverage distribution to the genome. Then, perform read counts to determine the number of reads mapped per gene.
#Using the DeepTools module to perform further analysis and generate visualizations.
module load python/3.8.2
#For these steps, a virtual environment was created
source ~/ENV/bin/activate
#plotCoverage generates a curve plot depicting the reads per base and the mean coverage of the genome.
plotCoverage -b /~/data/analysis/reads-map_v1_sorted.bam --labels reads-map_v1 --plotFileFormat pdf --outRawCounts Reads-V1_Lib.txt --numberOfProcessors 8
#Load required packages for readcount analysis (package subread). Read count was used to quantify the number of reads per gene.
module load gcc/7.3.0
module load StdEnv/2020
module load subread/2.0.3
#featureCounts counts mapped reads for genomic features such as genes, exons, promoters, etc. The script below will generate the number of read counts for each exon.
featureCounts -LMO -a GenomeOfReference.gtf -o reads-map_v1_counts.txt -F "exon" -g "gene_id" -s 0 -T4 reads-map_v1_sorted.bam
Process the data for visualization of library coverage and analysis of potential biases to genome regions.
#bamCoverage generates a coverage track by normalizing and binning the reads, producing a *.bw file. The file can be analyzed in a genome visualization tool (e.g., an integrated genome viewer such as https://igv.org/app/) for visual validation and to check for potential bias in library coverage.
bamCoverage -b reads-map_v1_sorted.bam -o reads-map_v1.bw --binSize 50 --normalizeUsing RPKM --extendReads 1000 --outFileFormat bigwig --numberOfProcessors 10
#computeMatrix creates a matrix of scores per region that is necessary as an intermediate for the visualization of the data as a heatmap.
computeMatrix scale-regions -b 500 -a 500 -m 3000 -R GenomeOfReference.gtf -S reads-map_v1.bw --skipZeros --sortRegions no --transcriptID 'exon’ –o matrix_reads-map_v1-sr1.gz
#plotHeatmap generates the heatmap to visualize gene coverage using the matrix file from computeMatrix.
plotHeatmap -m matrix_reads-map_v1-sr1.gz --outFileName reads-map_v1_heatmap.png --colorMap RdBu --whatToShow 'heatmap and colorbar' --zMin -4 --zMax 4
Generate graphs to visualize the read coverage over the genome using a circular plot. This step requires loading R installed in Linux. The same script can be used for the analysis in RStudio for Windows.
#Circlize package was used to generate the circular visualization of genome coverage. First, load R.
module load r
#Initiate R
R
#Install required packages
install.packages("circlize")
#Loading necessary libraries
library(dplyr)
library(circlize)
#Reading data from the plotCoverage output file (reads per base)
Lib_Cov.df = read.table('Reads-V1_Lib.txt', sep = "\t")
#Order the table according to read counts
Lib_Cov.ordered<-Lib_Cov.df[order(Lib_Cov.df$V4), ]
#Filter the data frame to have only the reads from complete chromosomes. This is a helpful step for partially sequenced or partially assembled reference genomes.
Lib_Cov.chrom <-dplyr::filter(Lib_Cov.ordered, grepl('CHR', V1)) %>%
#Converting the scale to log2 to improve visualization (optional)
dplyr::mutate(V4 = log2(V4))%>%
# Converting all cells that became –inf during log conversion to NA.
dplyr::mutate_if(is.numeric, list(~na_if(., -Inf)))
#Generate the circlize graph
#This first row inicializes the circular graph
circos.initializeWithIdeogram(Lib_Cov.chrom, plotType = NULL, circos.par(gap.degree = 8))
#This chunk generates the most outer ring with the chromosomes tracks
circos.track(ylim = c((0, 1), panel.fun = function(x, y) {
chr = CELL_META$sector.index #Uses the chromosome names as labels
xlim = CELL_META$xlim
ylim = CELL_META$ylim
circos.rect(xlim[1], 0, xlim[2], 1, col = "lightblue") #Changes the color of the chromosome rectangles
circos.text(mean(xlim), mean(ylim), chr, cex = 1.5, col = "white",
facing = "bending.inside", niceFacing = TRUE) #Changes the color of the text and the way it is oriented inside the rectangles.
}, track.height = 0.15, bg.border = NA)
#This chunk adds the counts track
circos.genomicTrack(Lib_Cov.chrom,
panel.fun = function(region, value, ...) {
circos.genomicPoints(region, value, type = "segment", lwd = 2,
col = "magenta", cex = 0.5, ...)
circos.yaxis("right", labels.cex = 0.4, col = "grey", labels.col = "darkgrey")
})
#This adds a text in the middle of the circle
text(0, 0, "Genome\ncoverage", cex = 1.5)
Determine the proportion of sequences in the library that are in frame with proteins in the expression system (e.g., Aga2p-Xpress in pYD1) and calculate their predicted peptide lengths and amino acid sequences.
Note: The Libframe tool used in this step was developed in Python and is used for pYD1. If using a different expression system, the code can be modified to replace the Xpress tag sequence with any sequence that should be in frame with the cloned fragments. The code is available at https://github.com/cestari-lab/Libframe-tool.
#The libframe script finds the Xpress tag sequence in the reads and translates library nucleotide sequences in-frame with the Xpress tag until a STOP codon is found.
#Load python and activate the environment
module load python/3.8.2
source ~/ENV/bin/activate
#Install biopython
pip install biopython
#Decompress the *.fasta.gz files into *.fasta files and concatenate all the multiple FASTA files.
gunzip *.fastq.gz
cat *.fastq > newfilename.fastq
#Execute libframe. It will output a text file with peptides' length and sequence. It takes three commands: 1) input fastq file; 2) name for the output file (.txt); and 3) the minimum length of a peptide. The Xpress tag has 8 aa (24 bases), plus linker (6 bases, 2 aa), and restriction site (Bam HI, 6 bases, 2 aa), resulting in the 12 aa sequence DLYDDDDKVPGS. Hence, we recommend the minimum value of 13 for a peptide.
python3 ./libframe.py path/to/fastq/file/.fastq path/to/output/file/.txt 13
#Example of results:
#Protein length is 127 aa and sequence is: DLYDDDDKVPGSTSHAPGRHGGRRIRLELHLDNFKLLPQLCSSVSADGSPAVPLQPVLPGIRQMLHHSVSIKCADARVARFLWPCPYALHCALLSITSEFAAACGSTIWISVVEFCEISSTVAAARV
#Protein length is 16 aa and sequence is: DLYDDDDKVPGSLCWC
#Protein length is 75 aa and sequence is: DLYDDDDKVPGSFLLVLILRRLLGCLTLIRAERHNRPQQGFVRRTAGVFHPSVATKSTCVFHFCISMYGRICGLL
#Protein length is 25 aa and sequence is: DLYDDDDKVPGSFVGADIAGDRAGK
#Protein length is 73 aa and sequence is: DLYDDDDKVPGSFLLVLILRRLHGCLTLCLRVHTLDTKQPEYPLLGGISRAPAHKGAALGGSFSSGCLHAEEV
Anticipated results
The protocol initiates with the sonication of genomic DNA using a Covaris M220 to obtain DNA fragments between approximately 0.5 and 3 kb. Optimization of sonication conditions may be necessary depending on the genome or sonicator used. We analyzed the sonicated genomic DNA on a 1% agarose/TBE gel to confirm that the DNA was fragmented in the expected length. Our fragmentation conditions resulted in 92% of the T. cruzi genome fragments ranging between 401 bp and 2,000 bp (Figure 4A and 4C). We then used magnetic beads to size select fragments above 500 bp (Figure 4A and B). We advise checking the manufacturer's instructions for size selection if another brand of magnetic beads is used. The size selected DNAs should migrate as a smear similar to the sonication range (Figure 4A) on a 1% agarose/TBE gel.
For library preparation steps, we found that end-repair and A-tailing of DNA fragment greatly improve subsequent ligations, even if PCRs are performed with a DNA polymerase that A-tails DNA. After each library preparation step (e.g., DNA end-repair, A-tailing, adapter ligation, PCR reactions, or vector linearization) DNAs were cleaned up with magnetic beads and the recovered DNA quantified using NanoDrop. The 260/280 and 260/230 ratios are ideally 1.8 and 2.0, respectively. Minor variations (approximately 10%) of these values are well tolerated in the assay. More considerable variations indicate potential contaminants that might affect subsequent reactions, especially the Gibson assembly. We found that DNAs purified using magnetic beads result in higher efficiency of Gibson assembly reaction than DNAs purified using silica-based columns and chaotropic buffers. To improve DNA purity, we recommend performing the DNA–beads washes with an ethanol volume equal to the reaction volume plus the beads volume, making sure to extract all the ethanol from the beads and air-drying the DNA before elution.
Before scaling up Gibson reactions, we recommend checking the efficiency of assembly. We performed PCR reactions with approximately 20 transformed colonies and analyzed the amplicons on a 1% agarose/TBE gel. Only proceed with the scale-up of library transformation if a minimum of 90% of the colonies show amplicons migrating in the expected size range (size of DNA fragments). We have often obtained 100% efficiency in the Gibson assembly reaction. To troubleshoot the Gibson assembly efficiency, we suggest performing ligations with various ratios of DNA fragments to vector, e.g., 1:2, 1:4, 1:6, and 1:8 (vector:DNA fragments). Most of our libraries worked well in the 1:2 to 1:4 ratio. It is also vital to Sanger sequence a few library clones before scaling up the library transformation to ensure that the library contains sequences from the desired organism.
After scaling up the transformations of the library, all colonies should be scraped and combined, and plasmids extracted. To ensure that the fragments were cloned, we performed plasmid DNA digestion to release the cloned fragments. Figure 4 shows results from our T. cruzi genome-wide library in the pYD1 vector. The library was analyzed on a 1% agarose/TBE gel and showed a 5 kb DNA band indicating digested pYD1 and a smear around 1.5 kb indicating digested library fragments (Figure 4A).
Our approach was designed for Oxford nanopore sequencing, as it produces long reads covering the entire sequence of the cloned fragment; however, the protocol can be adapted to other sequencing technologies. The extent of the sequencing data for analysis depends on the genome and library sizes. The T. cruzi Sylvio X10/1 strain has a haploid genome of 44 Mb. Our library included four million genomic fragments with an estimated mean length of 1.5 kb resulting in a 30-fold of the genome. In our example in Figure 4, we obtained approximately 8.7 Gb of DNA sequencing, resulting in approximately 8.2 million reads and 147× coverage of the genome (Figure 4D and 4E), which is sufficient for data analysis. After library sequencing and alignment of reads to the genome, we usually obtained 85%–99% genome mapping with minimap2. However, the percentage of genome mapped will vary according to the quality of the reference genome assembly. We recommend checking mapping per chromosome or contigs if the reference genome is not well assembled. We verified any potential bias in cloned fragments or the possible lack of sequences using a circular plot to visualize the reads mapped to the genome (Figure 4D). For a detailed analysis, we used a genome browser (e.g., integrated genome browser). We noted a high number of reads mapping to repetitive regions of large gene families, e.g., mucin or mucin-associated genes in T. cruzi (Figure 4D), which most likely reflects the quality of the genome assembly in these regions rather than bias in library cloning. We found that all the reference genome sequences were present in the library. We performed a plot heatmap analysis using DeepTools and confirmed that all gene sequences were covered by library reads (Figure 4F). Counting the number of reads per exon (featureCounts, package subread) may also help to determine how enriched a gene is in the library.
To evaluate the potential peptides generated by our library, we developed a tool that uses the nanopore reads (in fastq format) and searches for sequences in frame with the expression system, e.g., Aga2p and Xpress tag in the pYD1 vector. The Libframe tool predicts the library amino acid sequences and calculates their lengths using the available nanopore data. Using our data set, we found that approximately 90% of predicted peptides had between 21 and 160 amino acids (aa) in length (Figure 4G). The aa length reflects the size of library fragments and the presence of premature stop codons. In genomic libraries, not all DNA sequences are from coding genes, and fragments can be cloned in multiple frames and orientations. Hence, it is important to generate libraries that are at least 6-fold of the genome to ensure that all coding sequences are included (e.g., the T. cruzi library has 4 million fragment sequences and 30-fold of the genome).
Overall, our protocol details a step-by-step methodology to efficiently assemble genome-wide libraries into expression vectors for protein–ligand interaction studies.
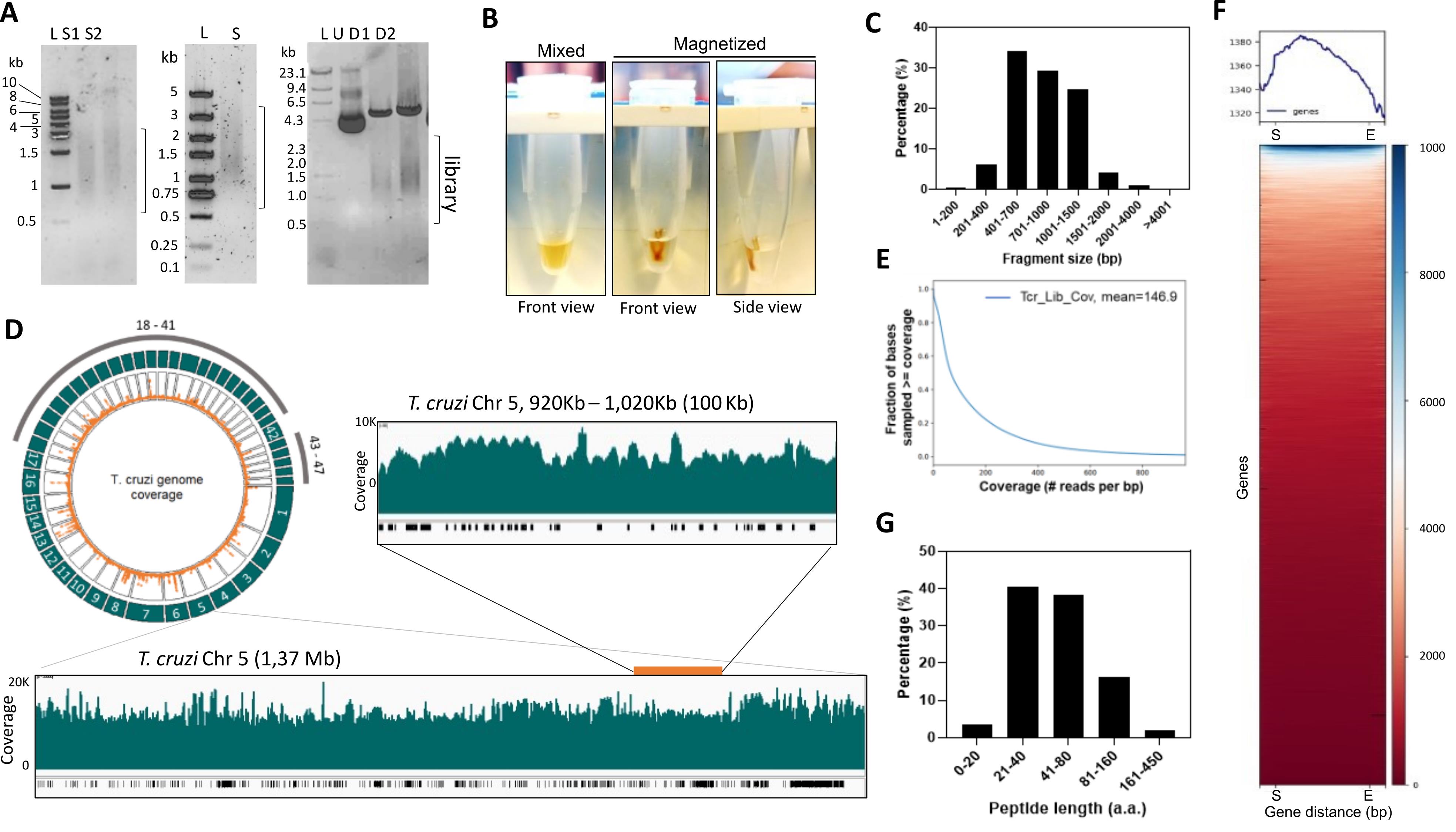
Figure 4. Analysis of T. cruzi genome-wide library for yeast surface display. (A) Left, genomic DNA sonicated using Covaris M220. DNA was resolved in 1% agarose/TBE gel. S1: incidence power 25, 2 duty factor, 200 cycles per burst, for 20 s; S2: incidence power of 10, duty factor 2, 700 cycles per burst, for 30 s. Middle, 1% agarose/TBE of sonicated genomic DNA size selected using magnetic beads to obtain fragments above 500 bp (S). Right, T. cruzi library cloned in pYD1 vector undigested (U) or digested (D1 and D2) with Hind III and Xho I restriction enzymes. D1: 1.0 µg of DNA; D2: 1.5 µg DNA. (B) Image of microcentrifuge tubes containing 100 µL of sonicated DNA sample and magnetic beads (0.7×) mixed before beads separation (mixed) and after separation in a magnetic rack (magnetization). Front and side views are indicated. (C) Analysis of fragment sequence length. Data were obtained from the Oxford nanopore sequencing summary file. (D) Circlize plot created using the Circlize R package shows the read coverage over the genome. Outer green tracks represent the chromosomes; inner orange dots represent mapped reads. Below is a snapshot of chromosome 5 (Chr 5) visualized using the integrated genome browser. The green plot shows read coverage, and the black bars below indicate genes. A 100 kb segment of the Chr 5 (inset above) is also shown. (E) Library coverage analysis using plotCoverage function (DeepTools) depicting read coverage per fraction of genome after mapping. (F) Heatmap of reads per gene generated by the computeMatrix and plotHeatmap functions. All genes were resized to the same length. A 0.5 kb from the start (S) and after the end of the gene (E) are shown. (G) Distribution of predicted library proteins in frame with proteins from the expression system, i.e., Aga2p and Xpress tag. Data were analyzed using the Libframe tool.
Notes
If using a standard probe sonicator, we recommend optimizing the sonication conditions starting with 50% frequency, 1,000 J, and three pulses of 5 s with 15 s rest between pulses. Perform the procedure in ice.
The user should refer to Omega Bio-Tek “Protocols and resources” section to select the ratio of beads during the size selection process according to the University of Oregon, Genomics & Cell Characterization Core Facility protocol (Weitzman, 2018).
We recommend first performing a PCR reaction with 35 cycles and analyzing the amplicons in an agarose gel to check the efficiency of amplification. It should result in a smear ranging from 0.5 to 3 kb (Figure 4A). Perform a gradient PCR to optimize annealing temperature if necessary. If no amplicons are observed, check for failed end-repair or adapter ligation reactions, and repeat section B. Once the amplification is confirmed, proceed with the multiple 14-cycle PCR amplification reactions, which should provide enough DNA to perform the next steps of the protocol. The number of PCR cycles should be kept between 14 and 16 to prevent bias in the library due to PCR amplification of selected sequences, or the introduction of PCR artifacts.
We recommend checking plasmid complete digestion before library assembly by performing agarose gel electrophoresis. Restriction enzyme–digested plasmids can be purified from agarose gel using silica-based columns; however, they should always be cleaned up using magnetic beads before Gibson assembly. We observed that the presence of impurities common to silica-based DNA purification methods with chaotropic buffers decreases the efficiency of the Gibson assembly.
Calculation of fmol based on average fragment size of 1.5 kb.
Chemical-competent Escherichia coli cultures were obtained using the low-temperature method (Ueda and Tanaka, 2000). We used the NEB® 5-alpha F'Iq E. coli strain for preparing the competent cells. Theefficiency should be 1 × 109–5 × 109 CFU/µg of pUC19 plasmid DNA.
Bacterial electroporation protocols usually produce highly efficient transformations using purified plasmids (1010 CFU/μg of pUC19 DNA). However, the electroporation of Gibson assembly reactions will result in lower efficiencies than chemical transformation. We noticed that the Gibson reaction components interfere with the electroporations. Hence, we recommend using chemical transformations. We also do not recommend using more than 1 µL of the Gibson assembly reaction for the chemical transformations.
Transformation efficiency (TE) is calculated as:
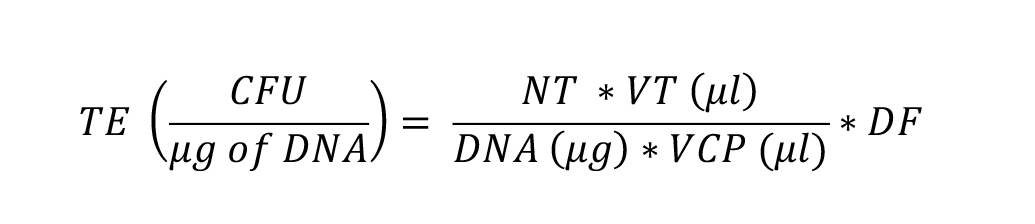
Whereas CFU is colony-forming units, NT is the number of transformants, VT (µL) is the volume of transformation in µL, DNA (µg) is the amount of DNA in µg added to the cells, VCP (µL) is the volume of cells plated in µL, and DF is the cell dilution factor in plating.
Poor quality DNAs can affect the Gibson assembly reaction. Ensure 260/280 and 260/230 ratios are in the recommended ranges. To troubleshoot the Gibson assembly efficiency, we recommend performing ligations with various ratios of fragments to vector, e.g., 1:2, 1:4, 1:6, and 1:8 (vector:DNA fragments).
Recipes
Luria-Bertani (LB) medium with agar and ampicillin
Prepare LB broth by mixing 10 g of Bacto-tryptone, 5 g of Bacto-yeast extract, and 5 g of NaCl; dilute to 900 mL of distilled water.
Adjust to pH 7.25 with 1 N NaOH or 1 N HCl as necessary, and then add distilled water to the final volume of 1 L.
Add 15 g Bacto-agar to the LB broth and autoclave immediately.
Wait to cool to approximately 40–50 °C and add 1 mL of ampicillin stock solution (100 mg/mL). Then, pour 40 mL into 150 mm × 15 mm Petri dishes.
Keep Petri dishes at 4 °C for a maximum of one month.
Ampicillin stock solution (100 mg/mL) (50 mL)
Dissolve 5 g of ampicillin in 45 mL of distilled water. Adjust the volume with distilled water to 50 mL.
Filter using a 0.2 µm syringe filter. Prepare 1 mL aliquots and keep them at -20 °C.
Tris-Borate-EDTA buffer (10 L)
Prepare TBE buffer by mixing 1 L 10× TBE stock solution (see below) with 9 L distilled water to a final volume of 10 L.
10× TBE stock solution (1 L)
Prepare TBE solution by mixing 108 g of Trizma base, 55 g of boric acid, and 8.2 g of EDTA to 800 mL of distilled water.
Adjust to pH 8.3 with 1 N NaOH or 1 N HCl as necessary.
Add distilled water to a final volume of 1 L.
Filter using a 0.2 µm syringe filter.
Keep at room temperature.
SOB medium (1 L)
Prepare SOB medium by mixing 5 g of NaCl, 20 g of tryptone, 5 g of yeast extract, and 2.5 mL of 1 M KCl, and dilute to 800 mL of distilled water.
Add distilled water to a final volume of 1 L.
Autoclave immediately.
Keep at 4 °C for up to six months.
Inoue solution (1 L)
86.62 mM MnCl2, 19.82 mM CaCl2, 250.84 mM KCl, 10 mM PIPES buffer
Prepare Inoue solution by mixing 10.9 g of MnCl2, 2.2 g of CaCl2, 18.7 g of KCl, and 20 mL of 0.5 M piperazine-1,2-bis [2-ethanesulfonic acid] (PIPES) buffer (see below). Add MilliQ water to final volume of 1 L and filter-sterilize using 0.2 µm filter.
0.5 M PIPES buffer (100 mL)
Dissolve 15.1 g of PIPES in 80 mL of MilliQ water by throwing in pellets of KOH until the solution clears up. Adjust to pH 6.7 with 1 N KOH or 1 N HCl.
Add MilliQ water to the final volume of 100 mL and aliquot in 10 mL tubes. Store at -20 °C.
70% ethanol
Mix 100 mL of MilliQ water with 368 mL of ethanol (95%).
Adjust the volume by adding MilliQ water to a final volume of 500 mL. Keep it at 4 °C.
Acknowledgments
This work was funded by the Canadian Institutes of Health Research (CIHR PJT-175222, to IC), the Natural Sciences and Engineering Research Council of Canada (NSERC RGPIN-2019-05271, to IC), the Canada Foundation for Innovation (JELF 258389, to IC), and by McGill University (130251, to IC). We thank Andressa Lira and Sahil Sanghi for their technical support at the early stages of this work. This research was enabled in part by computational resources provided by Calcul Quebec (https://www.calculquebec.ca/en/) and Compute Canada (www.computecanada.ca). This protocol is derived from the original research paper by Rhiannon Heslop et al. (2023).
Competing interests
The authors declare that they have no conflicts of interest with the contents of this article.
References
- Bharucha, N. and Kumar, A. (2007). Yeast genomics and drug target identification. Comb Chem High Throughput Screen 10(8): 618-634.
- Bidlingmaier, S. and Liu, B. (2011). Construction of yeast surface-displayed cDNA libraries. Methods Mol Biol 729: 199-210.
- Boder, E. T. and Wittrup, K. D. (1997). Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol 15(6): 553-557.
- Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., Whitwham, A., Keane, T., McCarthy, S. A., Davies, R. M., et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience 10(2).
- Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A., 3rd and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6(5): 343-345.
- Gu, Z., Gu, L., Eils, R., Schlesner, M. and Brors, B. (2014). circlize Implements and enhances circular visualization in R. Bioinformatics 30(19): 2811-2812.
- Inoue, H., Nojima, H. and Okayama, H. (1990). High efficiency transformation of Escherichia coli with plasmids. Gene 96(1): 23-28.
- Kieke, M. C., Cho, B. K., Boder, E. T., Kranz, D. M. and Wittrup, K. D. (1997). Isolation of anti-T cell receptor scFv mutants by yeast surface display. Protein Eng 10(11): 1303-1310.
- Lemos Duarte, M., Trimbake, N. A., Gupta, A., Tumanut, C., Fan, X., Woods, C., Ram, A., Gomes, I., Bobeck, E. N., Schechtman, D., et al. (2021). High-throughput screening and validation of antibodies against synaptic proteins to explore opioid signaling dynamics. Commun Biol 4(1): 238.
- Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34(18): 3094-3100.
- Liao, Y., Smyth, G. K. and Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7): 923-930.
- Pointer, K. B., Clark, P. A., Zorniak, M., Alrfaei, B. M. and Kuo, J. S. (2014). Glioblastoma cancer stem cells: Biomarker and therapeutic advances. Neurochem Int 71: 1-7.
- Ramirez, F., Ryan, D. P., Gruning, B., Bhardwaj, V., Kilpert, F., Richter, A. S., Heyne, S., Dundar, F. and Manke, T. (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44(W1): W160-165.
- Smith, A. M., Ammar, R., Nislow, C. and Giaever, G. (2010). A survey of yeast genomic assays for drug and target discovery. Pharmacol Ther 127(2): 156-164.
- Ueda, M. and Tanaka, A. (2000). Genetic immobilization of proteins on the yeast cell surface. Biotechnol Adv 18(2): 121-140.
- Weitzman, M. (2018). Evaluation of Omega Mag-Bind® TotalPure NGS Beads for DNA Size Selection. Genomics & Cell Characterization Core Facility, University of Oregon Technical Note.
- Heslop, R., Gao, M., Brito Lira, A., Sternlieb, T., Loock, M., Sanghi, S. R. and Cestari, I. (2023). Genome-Wide Libraries for Protozoan Pathogen Drug Target Screening Using Yeast Surface Display. ACS Infect. Dis: e2c00568.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sternlieb, T., Loock, M., Gao, M. and Cestari, I. (2022). Efficient Generation of Genome-wide Libraries for Protein–ligand Screens Using Gibson Assembly. Bio-protocol 12(22): e4558. DOI: 10.21769/BioProtoc.4558.
Category
Systems Biology > Interactome > Protein-ligand interaction
Microbiology > Heterologous expression system > Saccharomyces cerevisiae
Drug Discovery > Drug Design
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.