- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Monitoring the Recruitment and Fusion of Autophagosomes to Phagosomes During the Clearance of Apoptotic Cells in the Nematode Caenorhabditis elegans
Published: Vol 12, Iss 22, Nov 20, 2022 DOI: 10.21769/BioProtoc.4554 Views: 2329
Reviewed by: Manish ChamoliMadhuja SamaddarAnupama Singh
Original research article
The authors used this protocol in:

Jan 2022
Advertisement





Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
During an animal's development, a large number of cells undergo apoptosis, a suicidal form of death. These cells are promptly phagocytosed by other cells and degraded inside phagosomes. The recognition, engulfment, and degradation of apoptotic cells is an evolutionarily conserved process occurring in all metazoans. Recently, we discovered a novel event in the nematode Caenorhabditis elegans: the double-membrane autophagosomes are recruited to the surface of phagosomes; subsequently, the outer membrane of an autophagosome fuses with the phagosomal membrane, allowing the inner vesicle to enter the phagosomal lumen and accumulate there over time. This event facilitates the degradation of the apoptotic cell inside the phagosome. During this study, we developed a real-time imaging protocol monitoring the recruitment and fusion of autophagosomes to phagosomes over two hours during embryonic development. This protocol uses a deconvolution-based microscopic imaging system with an optimized setting to minimize photodamage of the embryo during the recording period for high-resolution images. Furthermore, acid-resistant fluorescent reporters are chosen to label autophagosomes, allowing the inner vesicles of an autophagosome to remain visible after entering the acidic phagosomal lumen. The methods described here, which enable high sensitivity, quantitative measurement of each step of the dynamic incorporation in developing embryos, are novel since the incorporation of autophagosomes to phagosomes has not been reported previously. In addition to studying the degradation of apoptotic cells, this protocol can be applied to study the degradation of non-apoptotic cell cargos inside phagosomes, as well as the fusion between other types of intracellular organelles in living C. elegans embryos. Furthermore, its principle of detecting the membrane fusion event can be adapted to study the relationship between autophagosomes and phagosomes or other intracellular organelles in any biological system in which real-time imaging can be conducted.
Keywords: ApoptosisBackground
The phagocytic clearance of apoptotic cells plays an important role in eliminating potentially harmful subjects from the surrounding tissues, and in suppressing autoimmune and inflammatory responses that could be stimulated by the content of broken apoptotic cells. Whereas in simple organisms such as C. elegans multiple types of neighboring cells are capable of engulfing apoptotic cells, in more complex organisms such as mammals there are professional phagocytes that engulf dying cells. These include macrophages and dendritic cells in the immune system, and microglia cells and astrocytes in the brain (Yuan and Yankner, 2000; Poon et al., 2014). Phagocytosis and autophagy are two distinct lysosome-mediated degradation processes. Whereas phagocytosis refers to a cell-eat-cell event, autophagy traps protein aggregates, damaged organelles, and other components to be eliminated within a cell in the double-membrane autophagosomes. The formation of autophagosomes requires the organized, step-wise action of a set of proteins known as autophagy-related (ATG) proteins (Nakatogawa, 2020). Yeast ATG8, mammalian LC3 (microtubule-associated protein 1 light chain 3), and their orthologs in other organisms, such as the nematode C. elegans and the fruit fly Drosophila melanogaster, are attached to both the inner and outer membranes of an autophagosome and have been established as specific markers for autophagosomes (Schaaf et al., 2016). In addition, LC3-associated phagocytosis (LAP) vesicles, which are single-membrane, LC3-labeled vesicles, have been reported to facilitate phagocytosis in the mammalian system (Green et al., 2016; Martinez et al., 2016). In C. elegans embryos, by using electron microscopy, genetic analysis, and the real-time fluorescence microscopy protocol described here, we have discovered that the canonical double-membrane autophagosomes, but not the single-membrane LAP vesicles, fuse to phagosomes and facilitate the degradation of apoptotic cells (Peña-Ramos et al., 2022).
We tagged LGG-1 and LGG-2, two C. elegans homologs of mammalian LC3, with fluorescent reporters that are resistant or moderately resistant to acidic pHs, such as mCherry (pKa <4.5) or mNeonGreen (mNG, pKa = 5.1), respectively (Shaner et al., 2004, 2013; Shinoda et al., 2018). We specifically expressed these tagged reporters in engulfing cells by placing each fusion construct under the control of the ced-1 promoter (Pced-1), which is expressed in cell types that act as engulfing cells (Zhou et al., 2001; Lu et al., 2009). We monitored the enrichment of puncta labeled with LGG-1 or LGG-2 on phagosomes. C. elegans embryonic development follows a fixed lineage (Sulston et al., 1983); the identities of apoptotic cells, their engulfing cells, and the moments when the apoptosis events occur are all fixed from embryo to embryo (Sulston et al., 1983). Thus, by monitoring phagosomes containing apoptotic cells with specific identities in different genetic backgrounds, we can avoid variances linked to the different properties of particular engulfing cells. In this protocol, we monitor three phagosomes that contain apoptotic cells: C1, C2, and C3 (Figure 1A–1B). These are located on the ventral surface of an embryo, undergo apoptosis at around 330 min post first embryonic division, and each is immediately engulfed by one particular ventral hypodermal cell (Figure 1A). LGG-1+ or -2+ puncta are observed attaching to the surface of a phagosome as early as 2 min post phagosome formation (Figure 1B). The fluorescence signals elicited from the acid-resistant mCherry::LGG-reporters start to appear inside the phagosomal lumen at approximately 14 min post phagosome formation and continue to accumulate there over time until the phagosomal cargo is completely degraded in approximately 60 min. If the LGG+ vesicles were single-membrane, like an LAP vesicle, no membrane-attached LGG reporter would enter the phagosomal lumen because, as a result of membrane fusion, the reporter molecules would be retained on the phagosomal membrane (Figure 1C(a)). The observed entry of the LGG signal into the phagosomal lumen (Figure 1B) indicates that the LGG-labeled puncta represent the double-membrane vesicles, the autophagosomes (Figure 1C(b)).
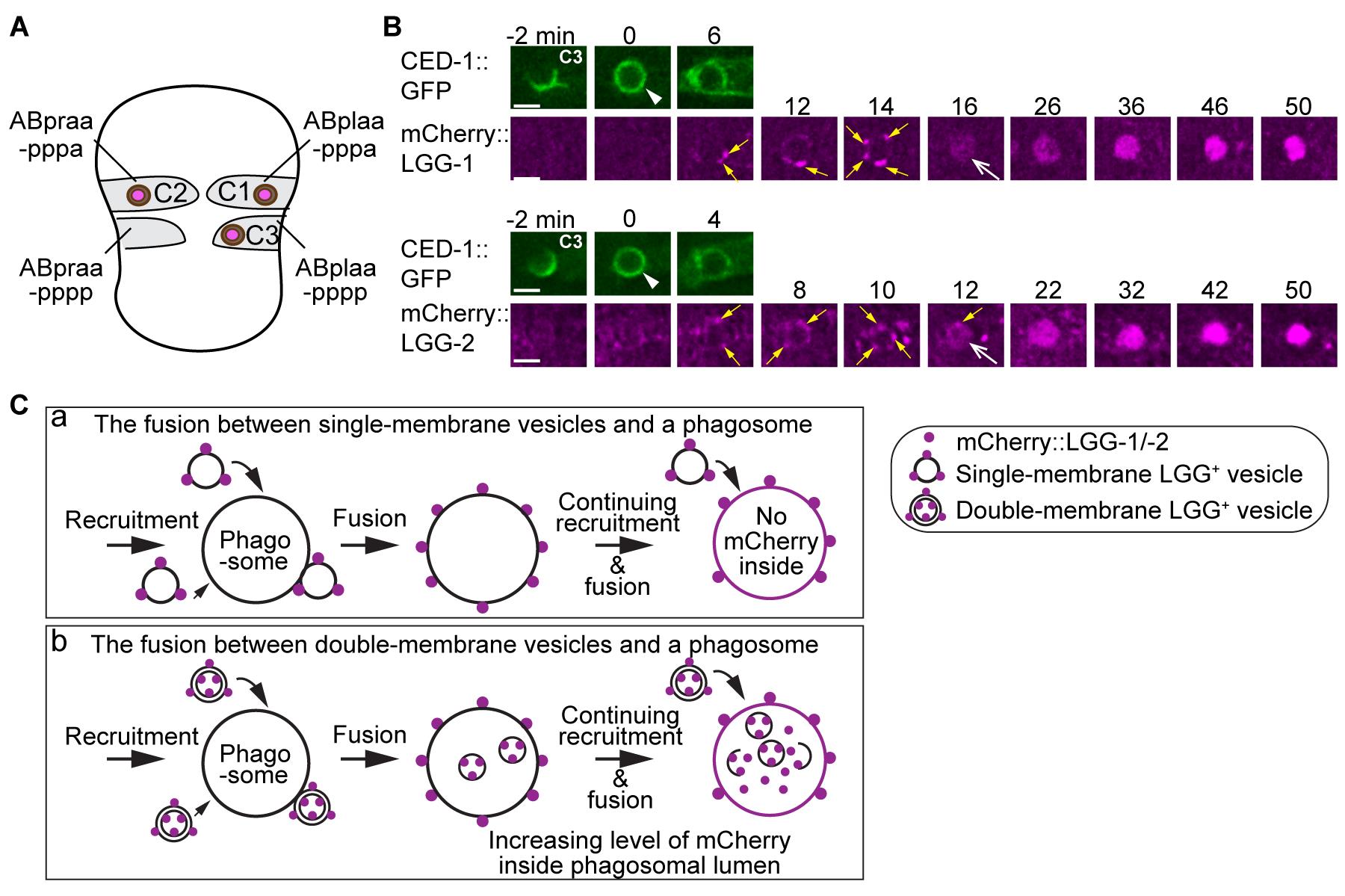
Figure 1. Vesicles labeled with mCherry::LGG-1 or ::LGG-2 are recruited to phagosome surfaces and subsequently fuse to phagosomes. (A) Diagram illustrating the three phagosomes that contain cell corpses, C1, C2, and C3, where we monitor the dynamic recruitment and fusion of autophagosomes, starting at approximately 330 min post first embryonic division. Both the positions of C1, C2, and C3 (purple dots) and the identities of their engulfing cells are shown. (B) Time-lapse images of the indicated reporters in C3 phagosomes in wild-type embryos. White arrowheads indicate the nascent phagosomes. Yellow arrows mark LGG-labeled puncta on the surface of phagosomes. White arrows indicate the moment when LGG signal can be observed in the phagosomal lumen. All reporters were expressed under the control of Pced-1. Scale bar = 2 µm. (C) Two diagrams illustrating the consequence of two fusion events: between single-membrane vesicles and a phagosome (a), and between double-membrane vesicles and a phagosome (b). The vesicles are first observed attaching to the phagosomal surfaces. After the fusion between the outer membrane of double-membrane vesicles and the phagosomal membrane, the mCherry::LGG-tagged inner membrane is released into the phagosomal lumen (b). The continuing incorporation of these vesicles to phagosomes increases the mCherry signal level in the phagosomal lumen over time (b). If the LGG-1 or LGG-2-labeled vesicles are of a single membrane, no fluorescence signal is expected to enter the phagosomal lumen (a).
This protocol, together with our genetic analysis, led us to discover that the signaling pathway led by the phagocytic receptor CED-1 promotes the recruitment of autophagosomes to phagosomes (Peña-Ramos et al., 2022); moreover, the subsequent fusion of autophagosomes with phagosomes requires the functions of the small GTPase RAB-7 and the HOPS complex (Peña-Ramos et al., 2022). Further analysis showed that autophagosomes provide apoptotic cell-degradation activities in addition to and independent of lysosomes (Peña-Ramos et al., 2022).
The principles of this protocol
Previously, methods for quantifying the efficiency of the interaction between highly dynamic intracellular organelles such as autophagosomes and phagosomes in real-time in C. elegans were not available. The protocol we describe here allows us to quantitatively measure the efficiencies of two separate events: (1) the recruitment and (2) the subsequent fusion of autophagosomes to the phagosomes in wild-type and mutant backgrounds. To evaluate the overall efficiency of the incorporation of autophagosomes into phagosomes, which is the end result of both the recruitment and fusion events, we measured the mCherry::LGG-1 and mCherry::LGG-2 signal intensities in the center of a phagosome over time, in a period starting from the formation of a phagosome (0 min) to 70 min afterward (Figure 2A). To evaluate the specific efficiency of recruitment, we measured the mCherry signal on the surface of a phagosome over time, starting from 0 min (Figure 2A). In wild-type embryos, the mCherry signal is detected evenly distributed in the phagosomal lumen starting at +14 min on average. In order to perform a meaningful comparison between the recruitment-defective mutants and wild-type samples, we choose to measure at the +12 min time point (Figure 2A).
In a mutant that is specifically defective in the fusion between autophagosomes and phagosomes, one will observe greatly reduced or absent mCherry signals inside the phagosomal lumen, with the mCherry signal accumulating on the surface of a phagosome over time (Figure 2B). On the other hand, in a mutant that is specifically defective in the recruitment of autophagosomes to phagosomes, at the +12 min, +50 min, and +70 min time points, very few mCherry+ puncta are observed on the phagosomal surfaces; in addition, no or very low mCherry signal is observed inside the phagosomal lumen (Figure 2C). If recruitment is partially defective whereas fusion is normal, the accumulation of the mCherry inside the phagosomal lumen will occur, albeit being delayed and/or at a lower intensity (Figure 2D). However, if recruitment is completely inactive, no autophagosome would be seen attached to phagosomes, further resulting in no mCherry signal inside the phagosomal lumen even if fusion is normal (Figure 2C). Such severe phenotype will make it impossible to determine whether the same mutant also suffers from a fusion defect.
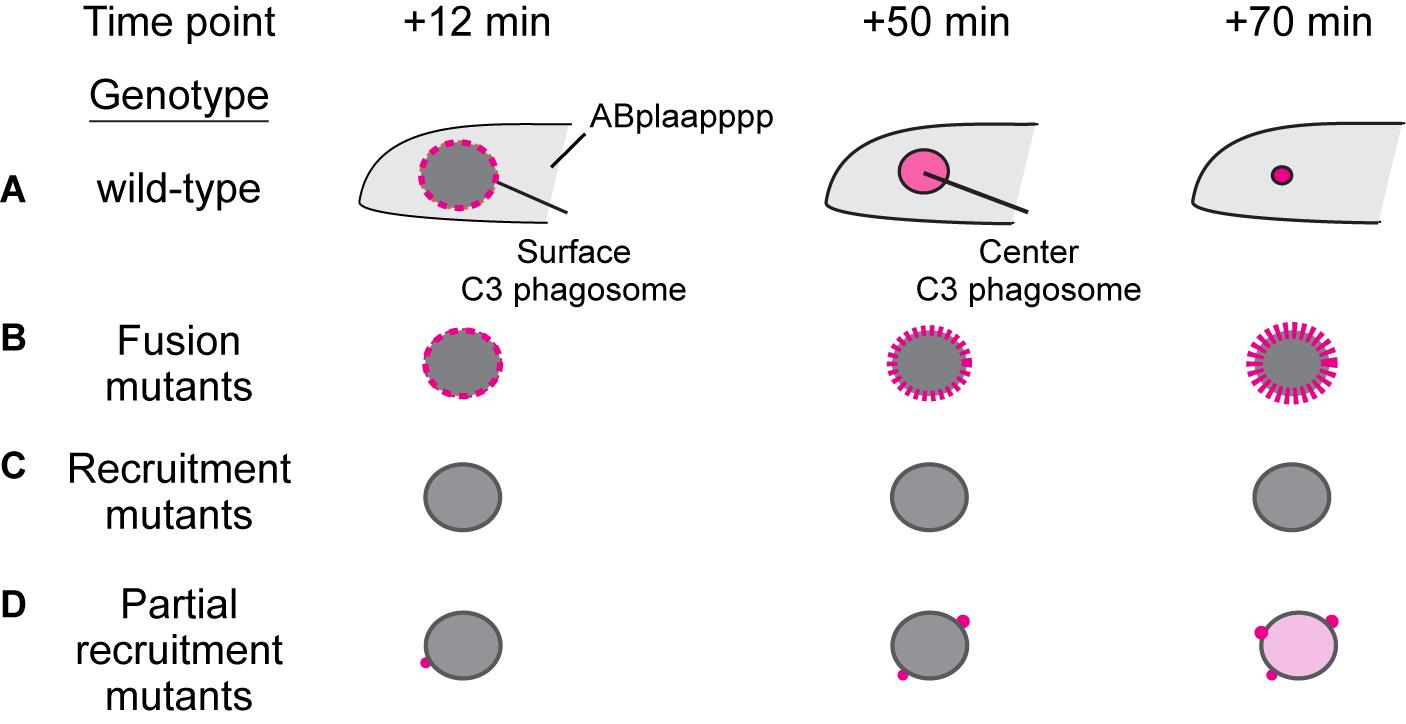
Figure 2. Recruitment and fusion of autophagosomes to phagosomes in wild-type and the mutants in which these events are impaired. Diagram illustrating the LGG::mCherry signal (in magenta) pattern in wild-type (A) and three kinds of mutants (B–D) on the surface and in the lumen of a C3 phagosome, 12, 50, and 70 min after phagosome formation. In (A), due to the efficient phagosomal degradation process, the size of a phagosome shrinks over time. In (B–D), the fusion and recruitment mutants are defective in phagosomal degradation, resulting in a much slower reduction of the phagosomal size.
Our protocol can be applied to characterize the fusion between autophagosomes and phagosomes that carry any kind of cargo, as well as between autophagosomes and other types of intracellular organelles in living C. elegans embryos, as long as the resolution is high enough to distinguish the surface of an organelle from its lumen. Furthermore, the principle of detecting the membrane fusion event between the double-membrane and single-membrane vesicles can be adapted to study the relationship between autophagosomes and phagosomes or between other intracellular organelles in any experimental systems that allows the expression of transgenic reporters and real-time microscopic recording of fluorescent signals.
Materials and Reagents
In vivo reporters
LGG-1 reporter construct: Pced-1mCherry::lgg-1 (for labeling autophagosomes)
LGG-2 reporter construct: Pced-1mCherry::lgg-2 (for labeling autophagosomes)
Pseudopod and phagosomal surface reporter construct: Pced-1ced-1::gfp (for labeling nascent phagosomes).
The Caenorhabditis elegans strains that carry both reporters
ZH2919, genotype: enIs82 [pUNC-76(+)(20 ng/μL), Pced-1ced-1::gfp (5 ng/mL), Pced-1mCherry::lgg-1 (5 ng/μL)] II; unc-76(e911) V
ZH2898, genotype: enIs83[pUNC-76(+)(20 ng/μL), Pced-1ced-1::gfp (5 ng/mL), Pced-1 mCherry::lgg-2 (5 ng/μL)] II; unc-76(e911) V
In these strains, the recruitment and subsequent fusion of autophagosomes to phagosomes can be monitored by recording the dynamic localization patterns of both the mCherry and GFP reporters.
Consumable reagents
Microscope slides (Premiere, catalog number: 9101)
Coverslips (22 × 22 mm) (Fisher Scientific, catalog number: 12-542B)
DeltaVision immersion oil, N = 1.516 (Cytiva, catalog number: 29162940)
Handmade worm pick, which is a platinum wire (Alfa Aesar, catalog number: 10287) mounted on a Pasteur pipette (Fisher Scientific, catalog number: 13-678-20B) (Figure 3)
High vacuum grease (Fisher Scientific, catalog number: 14-635)
Agarose (Fisher Scientific, catalog number: BP160-500)
KH2PO4 (MilliporeSigma, catalog number: PX1565)
Na2HPO4 (MilliporeSigma, catalog number: 567550)
NaCl (Fisher Scientific, catalog number: S671)
4% agarose solution (see Recipes)
M9 buffer (see Recipes)

Figure 3. A handmade worm pick
Equipment
Stereomicroscope (Nikon, catalog number: SMZ645)
DeltaVision Elite Deconvolution imaging system (GE Healthcare, Inc.), including AP1 F1- DeltaVision microscope (Olympus) equipped with 20×, 63×, and 100× Uplan Apo objectives, excitation/emission filter sets, fluorescent light source, differential interface contrast (DIC) microscopy accessories, motorized stage (X, Y, and Z-axis), and a Coolsnap HQ2 digital camera (Photometrics). For fluorescent imaging, two sets of fluorescence filters (Chroma Inc.) are used, including the GFP filter (excitation wavelength 475/28 nm; emission wavelength 525/50 nm) and the mCherry filter (excitation wavelength 575/25 nm; emission wavelength 632/60 nm). The DeltaVision microscope is kept in a room where the temperature is maintained at 20 °C.
Computer for image processing
Software
SoftWoRx 5.5 software (for the deconvolution and processing of images) (GE Healthcare, Inc.)
Microsoft Excel (Microsoft, Inc.)
Prism GraphPad (Dotmatics, Inc.)
Procedure
Mounting embryos on an agar pad
Melt 4% agarose solution in a microwave oven.
Dispense between 100 and 300 μL of the melted agarose solution in the center of the slide. Be cautious not to generate air bubbles. Immediately, flatten the drop by placing a glass slide perpendicular to the one holding the drop and press gently (Figure 4A). Let it stand for 1–2 min to allow the agarose to solidify.
Gently separate the two slides by sliding one against the other. Using a clean glass slide as a blade, trim the flattened agar pad into an approximately 12 × 12 mm square (Figure 4B).
Place 3 μL of M9 buffer at the center of the agar pad. The buffer is used to wash and maintain the embryos alive during the procedure.
Under the stereotype microscope, collect 50–80 embryos with the worm pick and transfer them to the drop of M9 buffer on the agarose pad. When transferring embryos from a Petri dish to the agar pad on the slide, carry as little bacteria as possible. If too many bacteria are transferred, they will consume oxygen and cause the embryos to die.
Gently squeeze a thin line of high vacuum grease around the agarose path (Figure 4C). Place a cover slip over the vacuum grease and press the corners gently to seal the slide (Figure 4D).
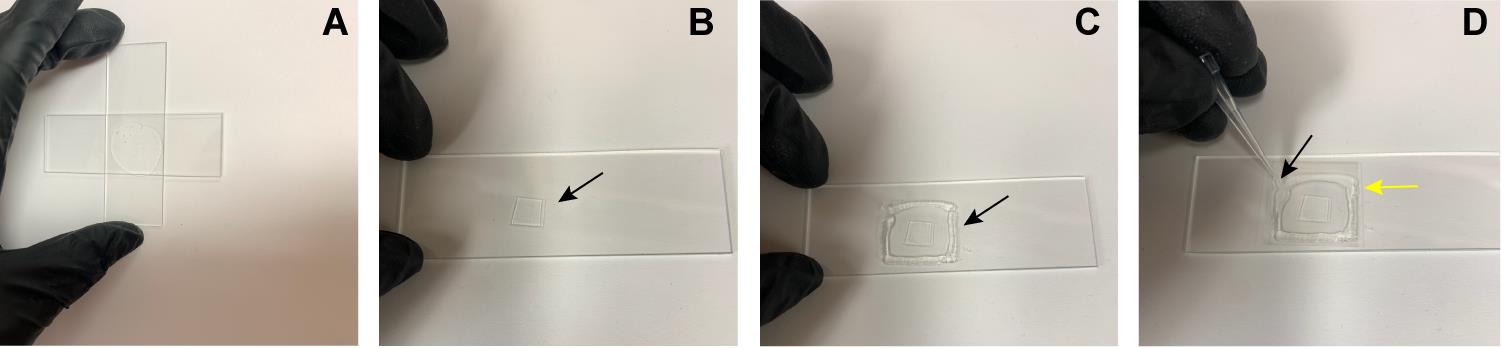
Figure 4. Construction of an agarose pad. (A) Agar flattened by two cover slips. (B) Agar pad cut into a square, marked by the black arrow. (C) High vacuum grease (black arrow) placed around the agar pad. (D) Agar pad covered with a glass cover slip (yellow arrow) by pressing gently pressing the corners with a piper tip (black arrow).Time-lapse recording of C1, C2, and C3 phagosomes
Set up microscope parameters: for DIC images, an exposure time of 0.2 s with a 10% neutral density filter is enough. The percentage of a neutral density filter represents the fraction of light that passes through the filter and illuminates the sample. For recording the mCherry::LGG-1/2 reporters, the exposure parameter for the mCherry channel is 0.1 s with 5% neutral density filter. For recording the engulfment marker ced-1::gfp, the parameter is 0.15 s with a 5% neutral density filter.
For optimal imaging, align the DIC light path according to the manufacturer's instructions.
Under the 20× objective, find embryos. Under the 100× objective, identify embryos at the age between 280 and 300 min post their first cell division and with their ventral side facing up (Figure 5). Mark these embryos utilizing the “point marking” and “position visiting” functions of the SoftWoRx software. For each strain, we image 15 embryos.
Define the position where the z-section recording should start. The serial z-section recording is performed from the embryo's ventral surface and proceeds to the center of the embryo. Set up 12–16 z-sections at 0.5 μm thickness for each section, which is sufficient to cover the entire ventral region.
Set up the time-lapse recording parameters. Start recording when the embryo is approximately 320 min post the first cell division; a moment later, the hypodermal cells will engulf apoptotic cells C1, C2, and C3 (Figure 1A). A clue hinting that the engulfment of C1, C2, and C3 is about to happen is the event when the middle part of the embryo slightly contracts towards its center, as shown in Figure 5. The recording time interval is set at every 2 mins for 60 to 120 min or until the embryos reach the 1.5-fold developmental stage, which is at 420 min post the first embryonic division.
Keep observing the images during the acquisition process. Adjust the starting focal plane if necessary. Abort the recording if the embryos slow down or arrest their development. Examples of time-lapse images of LGG+ vesicles are recruited to the phagosomal surfaces and subsequently fuse to phagosomes are shown in Figure 1B.
Figure 5. DIC image of a C. elegans embryo at 300 min post first cell division. Scale bar is 5 µm.
Data analysis
Measuring the signal intensity inside the phagosomal lumen
To measure the fluorescence signal intensity inside the phagosomal lumen over time in embryos expressing Pced-1mCherry::lgg-1 or -2, the boundary of a phagosome is identified at the 0 min time point when a nascent phagosome forms by the CED-1::GFP marker, which labels the surfaces of the extending pseudopods and nascent phagosomes. At each time point, the total mCherry::LGG-1 or -2 signal intensity within a fixed area (4 × 4 pixels) in the center of a phagosome (Intphagosome) is recorded (Figure 6B), as is the intensity of an area of the same size (4 × 4 pixels) outside the embryo as the background image intensity (Intbackground). The relative image intensity (RInt) at a particular time point (Tn) comparing to the start point (T0) is calculated as RIntTn = (Intphagosome - Intbackground)Tn / (Intphagosome - Intbackground)T0. The RIntTn value of 1.0 indicates no entry of LGG-1- or LGG-2-labeled autophagosomes into the phagosomal lumen. The data collected are plotted in a graph with the X-axis indicating the time points and RIntTn in the Y-axis. In addition, for each strain to be characterized, the RIntT50 (at the +50 min time point) of 15 phagosomes is measured. Standard statistical analysis is performed (Figure 6D). Whether the RIntT50 in a particular mutant strain is significantly different from that in the wild-type is determined by Student’s t-test.
Measuring the signal intensity on the surface of a phagosome
To evaluate the efficiency of recruitment of autophagosomes to phagosomes, the intensity of mCherry::LGG-1 or -2 is measured on the surfaces of phagosomes. First, the boundary of a phagosome at 0 min is identified by the CED-1::GFP reporter, which labels the surfaces of the extending pseudopods and nascent phagosomes. At a particular time point (Tn), the surface of a phagosome is outlined by two closed polygons (Figure 6A). The total signal intensities, as well as the areas of the polygons, are recorded. The unit signal intensity of the donut-shaped area between the two polygons is calculated as follows:
Unit Intensity (UIphagosome) = (Intensityexternal polygon - Intensityinternal polygon) / (Areaexternal polygon - Areainternal polygon). The Unit Background Intensity (UIbackground) was measured from a polygon outside the embryo and calculated as follows: UIbackground = Intensitybackground / Areabackground. At the time point Tn, the relative signal intensity on phagosomal surfaces (RsurfaceTn) = (UIphagosome - UIbackground)Tn / (UIphagosome - UIbackground)T0. The RsurfaceTn value 1.0 indicates no recruitment of the LGG-1- or LGG-2-labeled autophagosomes on phagosomal surfaces compared to the 0 min time point. The data collected are plotted in a graph with the X-axis indicating the time points and Y-axis indicating RsurfaceTn. In addition, for each strain to be characterized, the RsurfaceT12 (at the +12 min time point) of 15 phagosomes is measured (Figure 6C). Standard statistical analysis will be performed. Whether the RsurfaceT50 in a particular mutant strain is significantly different from that of the wild-type will be determined by the Student’s t-test.
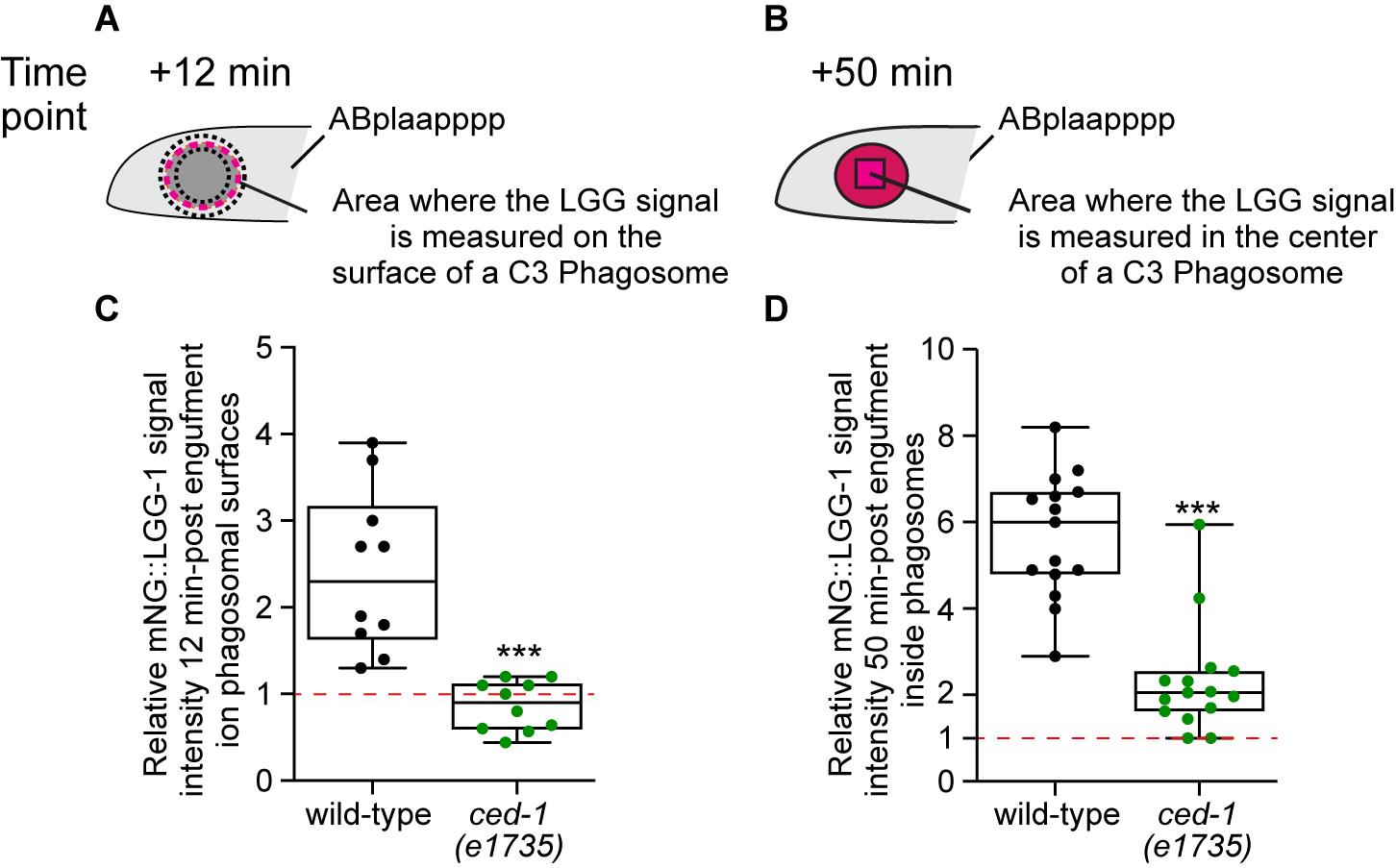
Figure 6. Two diagrams depicting the areas where the LGG signal intensity is measured in the C3 phagosome. (A) Diagram illustrating where the mCherry signal on the surface of a phagosome is measured over time. The area is delimited by the two black-dotted polygons. (B) Diagram illustrating where the relative mCherry signal in the center of a phagosome is measured over time. (C) Box and whisker plots of the relative mNeonGreen (mNG) signal intensities measured on the surfaces of ten C3 phagosomes 12 min post the formation of nascent phagosomes from wild-type and ced-1(e1735) mutant embryos. This graph is adapted from Peña-Ramos et al. (2022). (D) Box and whiskers plots of the relative mNG signal intensities measured in the center of fifteen C3 phagosomes 50 min post the formation of nascent phagosomes from wild-type and ced-1(e1735) mutant embryos. (C and D) Red dashed lines indicate the position of value 1.0, which represents no signal enrichment relative to the background signal. ***, p < 0.001, Student’s t-test of the ced-1(e1735) mutants compared to the wild-type value.
Notes
The reporter constructs are available upon request.
The strains mentioned in this protocol are available upon request.
Although this protocol specifically describes the usage of the DeltaVision Elite Deconvolution imaging system for the real-time recording of autophagosome recruitment and fusion events, it can be easily adapted to other real-time imaging systems such as the spinning disk confocal microscope. In addition, in C. elegans, programmed cell death events also occur in larvae and in the gonad of adult hermaphrodites in addition to embryos. Our protocol can also be applied to study the incorporation of autophagosomes into phagosomes in larvae and adults. Furthermore, besides C. elegans embryos, any experimental system in which real-time imaging can be conducted and that allows the expression of transgenic reporter constructs can adopt this protocol as well.
Recipes
4% agarose solution
Place 2.0 g of agarose in 50 mL of deionized water and heat up in a microwave until the agarose is completely dissolved.
After usage, cap the flask with aluminum foil and store it at room temperature.
To reuse the agar solution, melt the solidified solution for 35 s in the microwave.
M9 Buffer (1 L)
Dissolve 5.8 g of Na2HPO4 (40.9 mM), 0.5 g of NaCl (8.6 mM), 1.0 g of NH4Cl (18.7mM), and 3.0 g KH2PO4 (22.0 mM) in 400 mL of deionized water.
Bring the volume to 1 L and autoclave for 40 min.
Acknowledgments
This work is supported by NIH grant GM067648. This protocol is adapted from Peña-Ramos et al. (2022).
Competing interests
There are no conflicts of interest or competing interests.
References
- Green, D. R., Oguin, T. H. and Martinez, J. (2016). The clearance of dying cells: table for two. Cell Death Differ 23(6): 915-926.
- Lu, N., Yu, X., He, X. and Zhou, Z. (2009). Detecting apoptotic cells and monitoring their clearance in the nematode Caenorhabditis elegans. Methods Mol Biol 559: 357-370.
- Martinez, J., Cunha, L. D., Park, S., Yang, M., Lu, Q., Orchard, R., Li, Q. Z., Yan, M., Janke, L., Guy, C., et al. (2016). Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 533(7601): 115-119.
- Nakatogawa, H. (2020). Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol 21(8): 439-458.
- Peña-Ramos, O., Chiao, L., Liu, X., Yu, X., Yao, T., He, H. and Zhou, Z. (2022). Autophagosomes fuse to phagosomes and facilitate the degradation of apoptotic cells in Caenorhabditis elegans. Elife 11: e72466.
- Poon, I. K., Lucas, C. D., Rossi, A. G. and Ravichandran, K. S. (2014). Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol 14(3): 166-180.
- Schaaf, M. B., Keulers, T. G., Vooijs, M. A. and Rouschop, K. M. (2016). LC3/GABARAP family proteins: autophagy-(un)related functions. FASEB J 30(12): 3961-3978.
- Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N., Palmer, A. E. and Tsien, R. Y. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22(12): 1567-1572.
- Shaner, N. C., Lambert, G. G., Chammas, A., Ni, Y., Cranfill, P. J., Baird, M. A., Sell, B. R., Allen, J. R., Day, R. N., Israelsson, M., et al. (2013). A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods 10(5): 407-409.
- Shinoda, H., Ma, Y., Nakashima, R., Sakurai, K., Matsuda, T. and Nagai, T. (2018). Acid-Tolerant Monomeric GFP from Olindias formosa. Cell Chem Biol 25(3): 330-338 e337.
- Sulston, J. E., Schierenberg, E., White, J. G. and Thomson, J. N. (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol 100(1): 64-119.
- Yuan, J. and Yankner, B. A. (2000). Apoptosis in the nervous system. Nature 407(6805): 802-809.
- Zhou, Z., Hartwieg, E. and Horvitz, H. R. (2001). CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell 104(1): 43-56.
Article Information
Copyright
![]() Peña-Ramos and Zhou. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Peña-Ramos and Zhou. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Peña-Ramos, O. and Zhou, Z. (2022). Monitoring the Recruitment and Fusion of Autophagosomes to Phagosomes During the Clearance of Apoptotic Cells in the Nematode Caenorhabditis elegans. Bio-protocol 12(22): e4554. DOI: 10.21769/BioProtoc.4554.
- Pena-Ramos, O., Chiao, L., Liu, X., Yu, X., Yao, T., He, H. and Zhou, Z. (2022). Autophagosomes fuse to phagosomes and facilitate the degradation of apoptotic cells in Caenorhabditis elegans. Elife 11: e72466.
Category
Developmental Biology > Cell signaling > Apoptosis
Developmental Biology > Cell signaling
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.