- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Preparation of Homogenous Actin Filaments for Dynamic and Electrophoretic Light Scattering Measurements
(*contributed equally to this work) Published: Vol 12, Iss 22, Nov 20, 2022 DOI: 10.21769/BioProtoc.4553 Views: 2065
Reviewed by: Giusy TornilloAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Actin filaments are essential for various biological activities in eukaryotic cellular processes. Available in vitro experimental data on these systems often lack details and information on sample preparation protocols and experimental techniques, leading to unreproducible results. Additionally, different experimental techniques and polymerization buffers provide different, sometimes contradictory results on the properties of these systems, making it substantially difficult to gather meaningful data and conclusive information from them. This article presents a robust, accurate, detailed polymerization protocol to prepare high-quality actin filament samples for light scattering experiments. It has been shown to provide unicity and consistency in preparing stable, dispersed, aggregates-free, homogenous actin filament samples that could benefit many other scientific research groups currently working in the field. To develop the protocol, we used conventional actin buffers in physiological conditions. However, it can easily be adapted to prepare samples using other buffers and biological fluids. This protocol yielded reproducible results on essential actin filament parameters such as the translational diffusion coefficient and electrophoretic mobility. Overall, suitable modifications of the proposed experimental method could generate accurate, reproducible light scattering results on other highly charged anionic filaments commonly found in biological cells (e.g., microtubules, DNAs, RNAs, or filamentous viruses).
Graphical abstract:
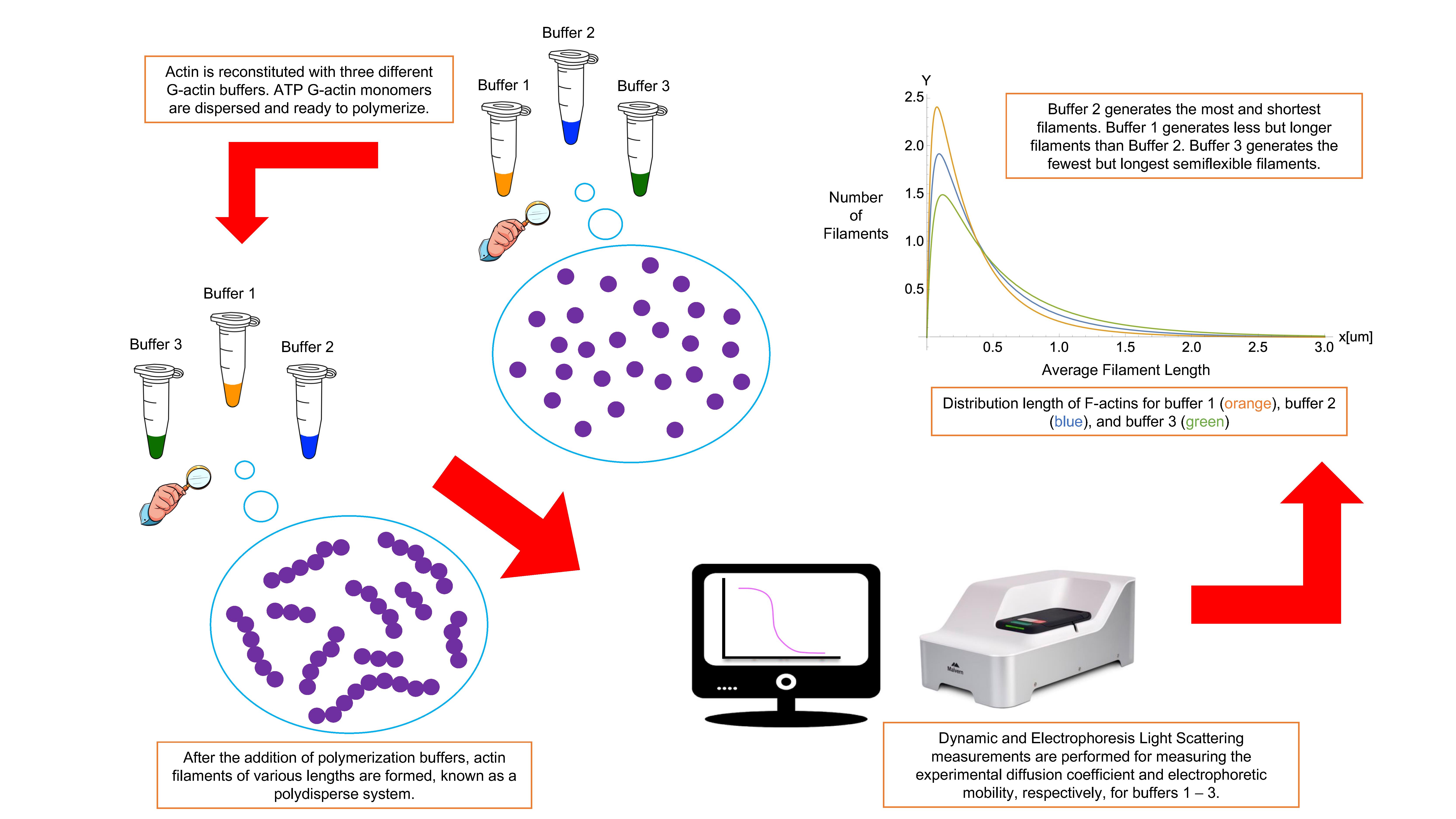
Background
Actin filaments (F-actin) are highly charged double-stranded semiflexible polyelectrolytes formed by the polymerization of G-actin subunits. Due to their hydrodynamic, mechanical, and electrostatic properties, these cytoskeleton filaments have the extraordinary ability to dynamically change conformations in response to alterations in G-actin concentration and type of crosslinker/binding proteins, as well as electrolyte concentration. These cytoskeleton properties are crucial for eukaryotic cells to achieve specific biological functions in different cellular compartments, which may vary depending on the cell type and location conditions. While a substantial amount of research has been done in the biochemistry and biophysics fields (Lanni and Ware, 1984; Janmey et al., 1986, 1994; Hou et al., 1990; Käs et al., 1996; Xu et al., 1998; Bonet et al., 2000; Niranjan et al., 2001; Y. H. Wang and Narayan, 2008; Crevenna et al., 2013; Del Rocio Cantero et al., 2020), the underlying principles and molecular mechanisms that support the hydrodynamic, electrostatic, and stability properties of these filaments remain elusive (Lanni and Ware, 1984; F. Wang et al., 1989; Hou et al., 1990; Käs et al., 1996; Bonet et al., 2000).
Modern dynamic light scattering (DLS) and electrophoretic light scattering (ELS) instruments are the most accurate, non-invasive, experimental techniques to characterize hydrodynamic and electrostatic properties of polydisperse charged biomolecules even at low concentrations and using small sample volumes. These instruments use advanced technology and multi-functional software to measure the hydrodynamic size, polydispersity, dispersion stability and aggregation, biomolecular charge, and zeta potential of particles and polymers immersed in aqueous biological environments. To assure high accuracy and reproducibility of the results, it is essential to use high-quality samples, accurate instruments, robust software, and optimized measurement protocols.
This article presents a complete, accurate, detailed polymerization protocol to prepare high-quality actin filament samples for light scattering experiments. Obtaining actin filament samples that are stable, dispersed, clean from aggregates and impurities, and homogenous could benefit many other scientific research groups in the field. We describe the polymerization procedure for three typical actin buffers in physiological conditions (Janmey et al., 1986, 1994; F. Wang et al., 1989). Nevertheless, this protocol can easily be adapted to prepare samples using other buffers and biological fluids. Additionally, we provide a unique methodology based on fundamental biostatistical tools (McDonald, 2009) and a measurement protocol with optimized configuration to perform DLS and ELS experiments on actin filament samples at critical G-actin concentrations. We measured actin filament’s translational diffusion coefficient and electrophoretic mobility using the same sample and experimental conditions for both DLS and ELS experiments to assure unicity and consistency in the results (Alva et al., 2022). A fitting approach was adequate to characterize other filaments’ essential properties, such as asymmetric length distribution, effective electric charge, hydrodynamic size, effective diameter, dispersion stability, aggregation, and semi-flexibility (Kroy and Frey, 1997; Tassieri et al., 2008; Alva et al., 2022). Additionally, the comparison of the light scattering results obtained for different polymerization buffers elucidated the impact of their chemical composition, reducing agents, pH values, and ionic strengths on the hydrodynamic, electrostatic, and stability properties of these actin structures. This kind of comparison and characterization provides a deeper understanding of how actin filaments behave in various cellular environments and conditions. Overall, suitable modifications of the proposed experimental method might allow other scientific groups to obtain accurate and reproducible light scattering results on other highly charged anionic polyelectrolyte filaments, commonly found in biological cells (e.g., microtubules, DNA, RNA, or filamentous viruses), having hydrodynamic, electrostatic, and stability properties resembling those characterizing actin filaments (Janmey et al., 2014).
Materials and Reagents
2 mL glass vial (Agilent, catalog number: 20097845)
Polypropylene centrifuge tube, 15 and 50 mL (Corning, catalog number: 430791)
Polypropylene microfuge tube, 11 × 38 mm, 1.5 mL (Beckman Coulter, catalog number: 9080511)
12 mm square polystyrene cell cuvettes (Malvern Panalytical, number: DTS0012)
2 mL cryogenic vials (Corning, catalog number: 30721070)
Business source stainless steel scissors (Fiskars, catalog number: 01-004250J)
Optifit pre-sterilized tips, 10–1,000 μL (Sartorius, catalog number: PR151159)
Optifit pre-sterilized tips, 5–350 μL (Sartorius, catalog number: PR149531)
Quartz spectrophotometer cell rectangular, stopper, 1 mm (Starna Cells, catalog number: 21Q1)
Actin protein (>99% pure): rabbit skeletal muscle; store at 4 °C (Cytoskeleton, catalog number: AKL99)
Water for molecular biology (Millipore, catalog number: H20MB1001)
Calcium chloride, dried, powder, 97% (CaCl2) (Alfa Aesar, catalog number: 10219230)
Magnesium chloride, anhydrous, 99% (MgCl2) (Alfa Aesar, catalog number: W07D102)
Potassium chloride (KCl) (VWR, BDH Chemicals, catalog number: 18J2256059)
Tris base (Fisher Bioreagents, catalog number: BP152-500)
Beta-mercaptoethanol, molecular biology grade (BME) (Millipore, catalog number: 3192024)
Adenosine triphosphate (ATP), 100 mM stock (Cytoskeleton, catalog number: BSA04)
DL-Dithiothreitol (DTT), molecular grade (Promega Corporation, catalog number: 0000383224)
Sodium hydroxide, pellets, 97%, A.C.S. reagent (NaOH) (Sigma-Aldrich, catalog number: 1310-73-2)
HCl volumetric standard, 0.1 N solution in water (Sigma-Aldrich, catalog number: 7647-01-0)
Precision red protein assay reagent (Cytoskeleton, catalog number: ADV02)
5 3/4” pipets (Fisherbrand, Fisher Scientific, catalog number: 13-678-20A)
70% v/v denatured ethanol solution (Fisher Bioreagents, catalog number: 216731)
Orion buffers pH 4.01, pH 7.00, and pH 10.00 (Thermo Scientific, catalog numbers: YX1, YW1, YX1)
Universal dip cell kit (Malvern Panalytical, catalog number: ZEN1002)
50 mM CaCl2 (see Recipes)
50 mM MgCl2 (see Recipes)
1.0 M KCl (see Recipes)
102.24 mM KCl (see Recipes)
G-actin buffer 1 (pH 7.80) (see Recipes)
G-actin buffer 2 (pH 7.66) (see Recipes)
G-actin buffer 3 (pH 8.23) (see Recipes)
Polymerization buffer 1 (pH 7.56) (see Recipes)
Polymerization buffer 2 (pH 7.64) (see Recipes)
Polymerization buffer 3 (pH 8.07) (see Recipes)
Electrolyte buffer 1 (pH 7.72), buffer 2 (pH 7.66), and buffer 3 (pH 8.06) (see Recipes)
Equipment
Analytical Explorer Pro-Scale (Ohaus Industrial Scales, model: PX84)
Pipette+ (Sartorius, Andrew Alliance Stand+)
Smart electronic pipettes: 5–350 μL, 10–1,000 μL, 5–10 mL (Sartorius, Andrew Alliance Stand+)
Vortex mixer with standard tube head, 120V (Corning LSE, The Lab Depot, Ref:6775)
Magnetic stirrer RT Basic-12 (Thermo Scientific, catalog number: 88880007)
Orion Star A211 pH meter, accuracy: ± 0.002 (Thermo Scientific, catalog number: X56954)
Allegra 64R benchtop centrifuge machine (Beckman Coulter, product number: 367585) with a F1202 Rotor, 30,000 rpm (Beckman Coulter, catalog number: 19U3138)
Zetasizer ULTRA, accuracy MW: ± 10% typical, temperature accuracy: 0.1 °C (Malvern Panalytical, model: ZSU5700)
Cary 100 UV-Vis spectrophotometer, accuracy: ~2% (Agilent Technologies, product number: 10069000)
Software
ZS Xplorer software (Malvern Panalytical, https://www.malvernpanalytical.com/en/products/product-range/zetasizer-range/zetasizer-advance-range/zetasizer-ultra)
Microsoft Word 2021 (Microsoft, https://www.microsoft.com/)
Cary WinUV Kinetics (Agilent Technologies, https://www.bioprocessonline.com/doc/cary-winuv-software-0001)
Cary WinUV Scan (Agilent Technologies, https://www.bioprocessonline.com/doc/cary-winuv-software-0001)
Procedure
ATP reconstitution
Briefly centrifuge to collect the white powder at the bottom of the storage tube.
Add 1 mL of cold 100 mM Tris base at pH 7.5 to reconstitute the ATP at 100 mM.
Aliquot the ATP into experiment-sized amounts as needed.
Snap frozen the ATP using liquid nitrogen.
Store at or below -20 °C.
Notes:
The lyophilized ATP (desiccated to <10% humidity) is stable for six months at 4 °C.
The ATP is stable for six months if stored at or below -20 °C.

Figure 1. Experimental instrumentation and timeline. (A) Daily timeline. (B) Workplace area and instrumentation. (C) Electronic Pipette+ OneLab, which reduces errors and assures consistency across many tests. (D) The protein solution is transferred to the cell cuvette and placed into the Zetasizer ULTRA holder to begin the diffusion coefficient measurements. (E) Subsequently, the dip cell (ZEN1002) is introduced into the Zetasizer ULTRA holder to perform ELS experiments and obtain the electrophoretic mobility, as well as the phase and frequency shift data plots.
Protein reconstitution
Notes:
Muscle actin is a liable protein; it should be handled with care.
To avoid repeated thawing cycles, consider specific experimental sized amounts.
Some of the steps considered in this section are recommended by the producer (Cytoskeleton, Inc). In addition to those, we considered the addition of mixing steps (B3 and B5), due to the rapid agglomeration/aggregation of the actin as the buffer is continuously added. Thus, we aim to achieve an aggregate-free solution by considering the following: as actin should be handled with care, the vortex speed is set at low to medium speed (4/10) for a short amount of time. Pipetting should also be carefully handled to avoid high mechanical stress in the protein solution.
This step is performed on Day 3 (Figure 1).
The aliquots containing the protein and G-actin buffers are stable for six months at -80 °C.
Transfer the protein powder into a 3 mL glass vial.
Add 100 μL of ultra-pure water into the glass vial containing the protein powder to reconstitute at 10 mg/mL G-actin density.
Vortex the solution for 30–40 s at low to medium speed (4/10) to dissolve the protein powder as much as possible.
Add 2.4 mL of G-actin buffer (buffers 1, 2, or 3—Recipes section B) to the glass vial containing the protein solution.
Vortex one more time for 35–45 s at low to medium speed (4/10) to dissolve the aggregates as much as possible. If aggregates are still present, pipette and mix the solution to achieve a homogeneous protein solution.
Aliquot the solution into experimental sized amounts according to the number of experiments needed. Transfer the small-size solutions into cryotube vials. We recommend aliquoting the protein solutions into multiples of 250 μL (1×250 μL, 2×250 μL, 3×250 μL, …).
Once the wanted protein solutions are aliquoted, snap-freeze the cryotube vials with liquid nitrogen and immediately store at -80 °C.
Actin polymerization
Notes:
ATP and DTT are initially stored at -20 °C. When ready to prepare the solutions, thaw both solutions. Immediately after use, snap-freeze them and store them back in the freezer.
Following the manufacturer’s recommendation, after the 2 h centrifuge process, remove the top 90% (198 μL) of the supernatant from each microcentrifuge tube by following step C11. The amount of translucent pellet left in the microcentrifuge will be 22 μL.
This step is performed on Day 4 (Figure 1).
A timeline for the actin polymerization steps can be seen in Figure 2.
Extract one of the cryotube vials containing 1×250 μL of protein solution from the -80 °C freezer.
Wait 5 min for de-frosting at room temperature.
Incubate the vial on ice for 1 h to depolymerize actin oligomers that form during storage.
Extract 200 μL from the cryotube vial and transfer to a 1.5 mL polypropylene microcentrifuge tube.
Add 20 μL of polymerization buffer (1/10th volume) (buffers 1, 2, or 3—Recipes section C) to the microcentrifuge tube containing the 200 μL protein solution to start polymerization.
Vortex the solution for 40–50 s at low speed (3–4/10).
Incubate the protein solution at room temperature for 1 h.
Turn on the centrifuge 10–15 min before the previous step is finalized. Set up the centrifuge at 50,000 × g for 2 h at 4 °C.
Note: The acceleration and deceleration should both be set at very low (2/10) speed.
Centrifuge the protein solutions for 2 h.
Carefully remove the tubes containing the actin protein solutions from the centrifuge and place them on ice.
Set the 5–350 μL Pipette+ in titration mode at very low speed (1/10) and extract the supernatant from the solution in the following manner, to avoid any possible stress in the solution that could lead to the breakage of the actin filaments: 100 μL, 50 μL, and 48 μL.
Using the same Pipette+ specifications as in the previous step, add 978 μL of electrolyte buffer (buffers 1, 2, or 3—Recipes section D) to the actin protein pellet (22 μL).
Store the solution at 4 °C and leave it overnight.
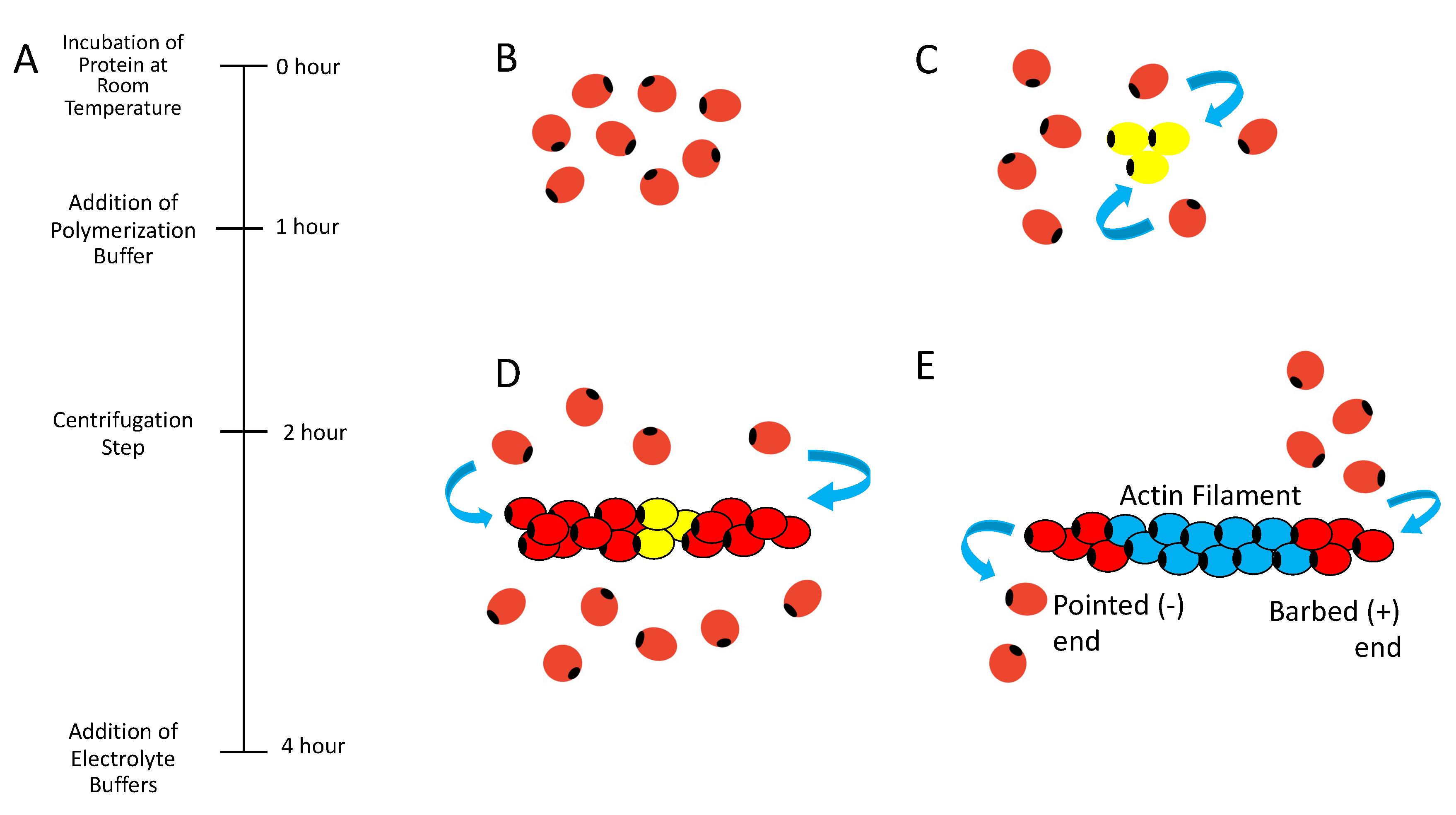
Figure 2. Activation, nucleation, elongation, and annealing of actin filaments. (A) Hourly polymerization timeline of F-actin, starting with the incubation of actin protein at room temperature and the addition of the polymerization buffer. After one hour, the centrifugation process begins to separate the supernatant from the pellet (F-actins), and finally, the electrolyte buffer is added. (B) G-actin monomers undergo a polymerization process that starts with the activation. In this step, Mg2+, K+, and Ca2+ bind to G-actin monomers reducing its electrostatic repulsion between monomers and inducing a structural change. (C) Subsequently, the nucleation begins, where G-actin monomers form stable nucleus due to the presence of ATP- and ADP-actin, supporting the addition of more monomers. (D) During the elongation stage, monomers are rapidly added to the nucleus through both ends of the filament (pointed and barbed end). (E) Finally, in the annealing step there is an association and dissociation of G-actin monomers at both ends of the filament.UV-Vis spectrophotometer absorbance
Notes:
Turn on the UV-Vis spectrophotometer 10–15 min before use.
Determine the protein concentration in the supernatant with the precision red protein assay reagent. The goal is to obtain the protein concentration in the pellet and after adding the electrolyte buffers. For more information, see the Data analysis > Protein Concentration Analysis.
Pipette 1 mL of precision red protein assay reagent into a 1.5 mL disposable microfuge tube.
Add 10 μL of the supernatant protein solution obtained in step C11 and mix by inverting.
Incubate at room temperature for 1 min.
Transfer the solution into the quartz cuvette.
Set the Scan Controls at 0.100 s for Ave Time, 0.12 nm for Data Interval, and 100 nm/min for Scan Rate.
Blank the spectrophotometer on precision red protein assay reagent at 580–620 nm and read the absorbance of the protein sample at 600 nm.
Dynamic light scattering
A measurement consists of several averaged normalized intensity autocorrelation decays, called runs, each lasting 10 s. The number of runs can be determined automatically by the software, based on the scattered light intensity, which depends on the concentration and size of the macromolecule. Alternatively, it can be entered manually by the user. For F-actins, the number of runs is typically five (see more details under Data analysis).
Instrument start-up
Turn on the computer.
Turn on the Zetasizer ULTRA by pressing the power switch located on the back of the instrument.
The status light on the Zetasizer ULTRA will initially be red.
Open the ZS Xplorer software.
The status light on the Zetasizer ULTRA will become green shortly after.
Wait 10–15 min for the instrument to stabilize.
Standard operating procedure (SOP)
Select Analyze > select Explorer > select Project Explorer ‘+’ sign > type the name of the new project.
Select Measure (see Figure 3)
Name: Type a sample name for this measurement.
Cell: Select DTS0012
Material: Select Protein
Dispersant: Select Water
Project: Select the project created in step 2a.
Measurement type: Select Size and 5 runs
Measurement
Temperature: Select 25 °C
Return to default temperature: Select Yes
Equilibration time (s): Type 180
Data Processing
Analysis Model: Select General Purpose
Advanced Settings
Angle of Detection: Select Back scatter
Positioning Method: Automatically selected as Measure at a fixed position
Cell Position: Automatically selected as 5.50
Attenuation: Select Automatic
Measurement Process: Select Automatic
Use pause after sub runs: Select No
Optical Filter: Select No filter
Pause between repeats (s): Type 0
Sample preparation for measuring translation diffusion coefficient and correlation functions:
Remove the 1 mL aliquots of polymerized F-actins (buffers 1, 2, or 3—Recipes section B) from the 4 °C fridge and bring to room temperature for 10–15 min.
In the meantime, clean and sterilize the scissors with water and ethanol. Cut Optifit tips up to a diameter of 3–5 mm from the tip. This action will greatly reduce the breakage of filaments when transferring the aliquot into a cell cuvette.
The cell cuvette must be clean (air pressure) and free of scratches since this could lead to erroneous measurements.
Set the Pipette+ (5–350 μL) in titration mode at low speed (1–2/10) to extract the protein solution into the cell cuvette. Titrate the protein at a 45° angle mode. The cell should be filled slowly at a 45° angle to avoid creating air bubbles.
Do not fully cap the cell. Fully cap only one side of the cell. This action will also reduce the presence of air bubbles during measurements.
Open the cell area lid by pressing the button in front of the lid. Push the cell cuvette into the cell holder until it stops. The small triangle at the top of the cell indicates the front of the cell, which should be facing towards the user. Close the cell area lid.
During the measurements (every two sets of five runs), open the lid, remove the cell cuvette from the cell holder, and mix the protein solution 2–3 times using a cut tip of 3–5 mm in diameter. Immediately after, place the cell cuvette back in the cell holder and proceed to the next set of experiments. This action will minimize sedimentation.
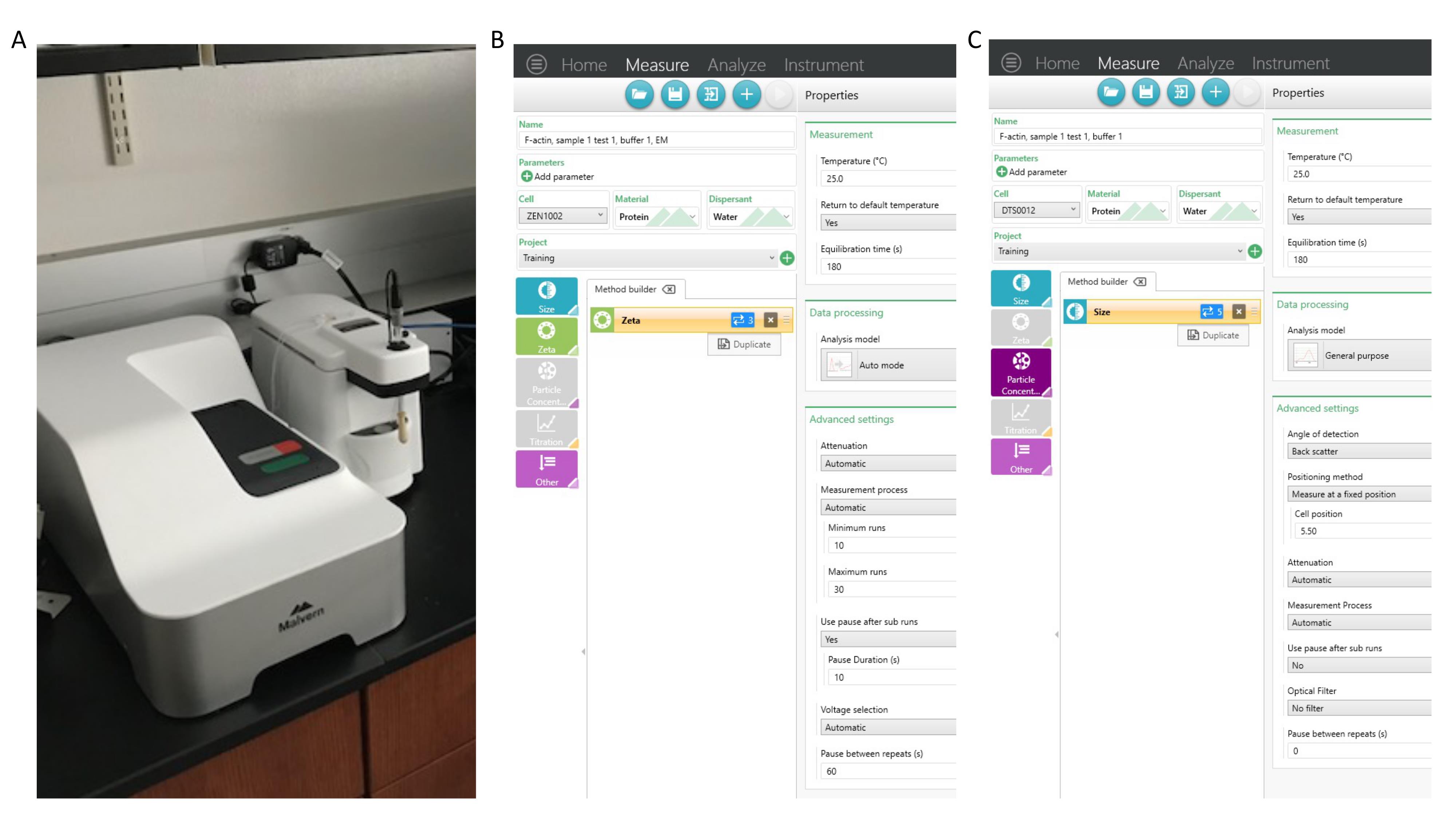
Figure 3. Zetasizer ULTRA. (A) The DLS and ELS Zetasizer ULTRA instrument uses a 633 nm He-Ne laser. It houses a single cuvette and measures light scattered at 173°, 90°, and 17°. It is interfaced to a computer running Windows 10 and controlled with the Zetasizer software. (B) SOP window for ELS measurements. The settings shown here are also presented under the ELS SOP (step I1). (C) SOP window for DLS measurements. The settings shown here are also presented under DLS SOP (step H2).
DLS measurements
When the project and specifications are set and the sample is loaded, begin the measurements. Click the Play bottom icon located by properties selection, and the initial green light of the Zetasizer ULTRA will turn blue.
During DLS measurements, select Multi-view to display the following three windows: 1) “g2(t)-1” versus decay time “t,” which is updated as the run continues (see Figure 5); 2) the intensity fluctuations described in Background; and 3) the intensity distribution.
As a series of runs accumulates into a measurement, select the Analyze tab and locate the Project Name to get all the data generated by this measurement.
After each run is over, the distribution function is calculated, and many data options are available such as “Intensity vs. Size,” “Intensity vs. Volume,” and “Correlogram.” This process is repeated five times, obtaining five similar correlation functions.
After the measurement is complete, the blue light will return to green, meaning that the Zetasizer ULTRA is ready for the next set of measurements.
Electrophoresis light scattering
A measurement consists of several averaged fast field reversal phase shift, called runs, each lasting 1.4 s. The number of runs can be determined automatically by the software, based on the scattered light intensity, which depends on the concentration and size of the macromolecule. Alternatively, it can be entered manually by the user. For F-actins, the number of runs is typically three (see more details under Data analysis).
Standard operating procedure (SOP)
Select Analyze > select Explorer > select Project Explorer ‘+’ sign > type the name of the new project.
Select Measure (see Figure 3)
Name: Type a sample name for this measurement.
Cell: Select ZEN1002
Material: Select Protein
Dispersant: Select Water
Project: Select the project created in step 1a.
Measurement type: Select Zeta and 3 runs
Measurement
Temperature: Select 25 °C
Return to default temperature: Select Yes
Equilibration time (s): Type 180
Data Processing
Analysis Model: Select Auto mode
Advanced Settings
Attenuation: Select Automatic
Measurement Process: Select Automatic with minimum runs set at 10 and maximum runs set at 30
Use pause after sub runs: Select Yes and a pause duration (s) set at 10
Voltage selection: Select Automatic
Pause between repeats (s): Type 60
Sample preparation for measuring electrophoretic mobility, frequency, and phase shifts:
Once the DLS measurements are over, remove the protein solution from the cell holder and proceed to insert the dip cell (ZEN1002).
Before inserting the dip cell, its electrodes must be cleaned with ethanol and water. After every measurement, the electrodes must be cleaned to reduce contamination across samples.
The dip cell must be placed into the sample cuvette. Place the ZEN1002 cell into the cell cuvette containing the protein solution. Ensure that the small triangle at the top of the cell still faces the front of the instrument, as indicated in section I. Make sure that the sample does not overflow the cuvette when the ZEN1002 is fully inserted.
Holding the base of the ZEN1002 cell cap and the top of the cuvette simultaneously, push the cell into the cell holder until it stops.
Open the cell area lid by pressing the bottom in front of the lid. Simultaneously holding the base of the ZEN1002 cell cap and the top of the cuvette, push the cell into the cell holder until it stops. Close the cell area lid.
During the ELS measurements, check the dip cell electrodes and sample for any potential contamination. Failing to clean the electrodes after each run could lead to cross-contamination between samples.
ELS measurements
When the project and specifications are set and the sample is loaded, begin the measurements. Click the Play bottom icon located by properties selection, and the initial green light of the Zetasizer ULTRA will turn blue.
During ELS measurements, select Multi-view to display the following three windows: 1) “PALS: phase(rad) vs t”, which is updated as the run continues (see Figure 6); 2) the intensity fluctuations described in Background; and 3) the frequency shift.
As a series of runs accumulates into a measurement, select the Analyze tab and locate the ‘Project Name’ to get all the data generated by this measurement.
After each run is over, the fast field reversal (FFR) of the phase analysis will be available along with other data options such as the frequency shift.
After the measurement is complete, the blue light will return to green meaning that the Zetasizer ULTRA is ready for the next set of measurements.
Shutdown
Exit ZS Xplorer software (File > Exit).
Turn off the Zetasizer equipment.
Shut down computer.
Data analysis
Detailed information on data analyses already appears in the “Materials and Methods” section of the original research article (Alva et al., 2022).
DLS analysis
Note: The Backscatter angle technique significantly reduces the presence of dust in the signal-to-noise ratio correlation plots. However, a wavering behavior can appear in the correlogram’s baseline due to multiple exponential decays and an increase in the number of fluctuations. In that case, it may lead to aggregation and sedimentation of the sample. Thus, we recommend following Note 12.
Once the set of measurements is completed, select the Analyze tab and select the appropriate project name where all the measurements have been saved.
Select the Size tab. Under the data options, select the multiple parameters available for each run. For instance, select Size > Z-average, PDI, translational diffusion coefficient, derived mean count rate, or any other size parameter according to the user’s needs (see Figure 4).

Figure 4. Table properties. (A) Full display of available parameters and properties that can be selected according to the user’s needs for DLS and ELS. (B) Selected and recommended parameters for DLS measurements. (C). Selected and recommended parameters for ELS measurements.Once the chosen parameters are displayed, select the correlogram as one of the data plots to be displayed for each run. The correlogram is an important data function for analyzing and determining the quality of the sample and measurements. The correlograms must follow an experimental criterium to minimize errors and assure reproducibility.
The correlogram plot’s intercepts must be under 1 and within a range in the low polydispersity index (0.4 < PDI < 0.8), indicating a good quality in the polydispersity samples.
The resulting derived count rate should be higher than 100 kpcs, the minimum value required to obtain suitable measurements.
The correlogram plot should also present a smooth and uniform single exponential decay. However, the correlogram data plots may show multiple exponential decays within a single run due to the sample’s aggregates, sedimentation, and dust. In that case, the data is disregarded. This is also considered under the experimental criterium to reduce error and increase reproducibility (see Figure 5).
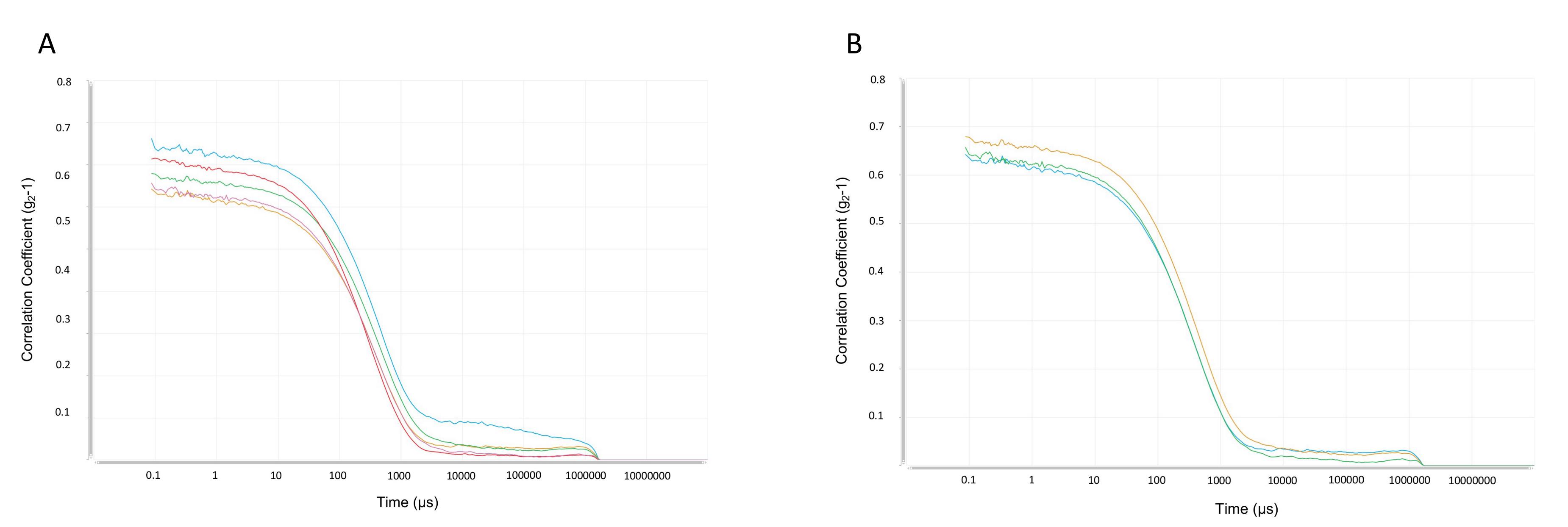
Figure 5. Correlation coefficient. Illustrative example of the correlation data function [g2-1 vs. time (μs)] plots obtained from size measurements. (A) Data measured from five consecutive, independent experiments. (B) The three correlation data functions selected for data analysis after two measurements (2/5) are disregarded according to the experimental criterium to increase accuracy.
ELS analysis
Once the set of measurements is completed, select the Analyze tab and the appropriate project name where all the measurements have been saved.
Select the Zeta tab. Under the data options, select the multiple parameters available for each run. For instance, select Zeta > zeta potential, electrophoretic mobility, quality factor, conductivity, or any other electrophoretic parameter according to the user’s needs (Figure 4).
Once the chosen parameters are displayed, select the phase plot and frequency shift data plots to be displayed for each run. The fast field reversal (FFR) phase plot is an important data function to determine the quality of the sample and measurements. Three independent experiments were tested for each actin filament sample to reduce statistical errors in electrophoretic mobility values.
The quality factor is a parameter that derives from the phase analysis plot during the FFR stage of the measurement; it must be higher than 1 to be considered as good quality data.
The frequency shift plots should have very low traces of noise since this is another quality data check-up. Also, the phase FFR plot should have a sinusoidal behavior, which also implies a good quality data (see Figure 6).
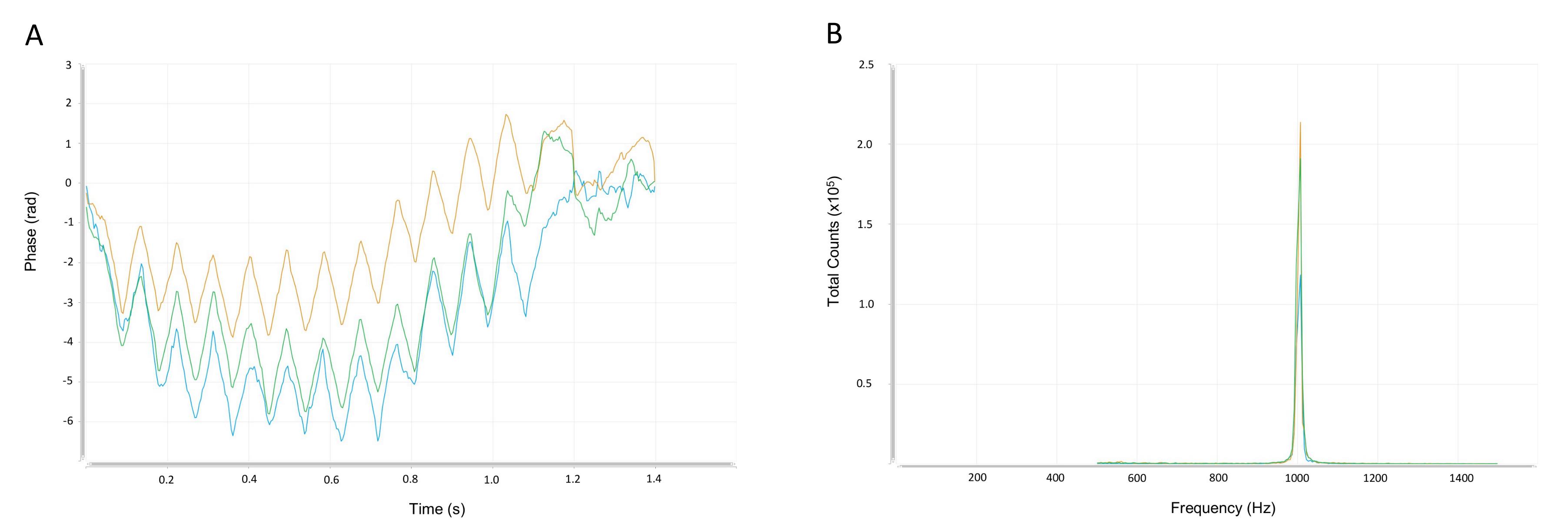
Figure 6. Phase and frequency shifts. Illustrative example of phase and frequency shift plots obtained from electrophoretic measurements. Three independent experiments are measured on the phase (A) and frequency shifts (B) to reduce statistical errors in the electrophoretic mobility values. An evidence of good data quality is displayed in the frequency shift plot, since there are no traces of noise and the curves match very well.
Protein concentration analysis
Note: We performed the calculation on the pellet concentration to confirm that the samples used in our experiments were in the dilute regime, where the light scattering measurements correspond to single actin filament properties. This condition is achieved when the pellet concentration value “c” = 1.37 μM is lower than 1/<L>3 *NA, where <L> and NA are the average filament length and Avogadro’s number, respectively. Using a fitting approach (Alva et al., 2022, Table 8), we obtained <L> ~0.5 μm and 1/<L>3 *NA ~13.3 μM. Thus, our experiments were performed in the dilute regime.
Calculation of initial concentration (Co)
When reconstituting the actin protein by adding 100 μL of ultra-pure water, the formulation is the following:
C(concentration)= (Mass of Protein Powder (mg))/Volume=10 mg/mL
Adding 2.4 mL of G-actin buffer will change the concentration of protein to 0.4 mg/mL:
C=(1 mg)/(0.1 ml+2.4 ml)=0.4 mg/mL
From each tube, we extract 200 μL and add 20 μL of polymerization buffer. Thus, the resulting initial concentration of protein for experimental measurements is the following:
Co=(0.08 mg)/(0.2 mL+0.02 mL)=0.3636 mg/mL
Calculation of mass supernatant concentration (MSN) (Figure 7)
Note: In this stage, the spectrophotometric absorbance range is 0.009–0.012. This range comes from the multiple independent measurements performed for all buffers. If the absorbance value is outside this range, it is possible that a percentage of G-actins have not polymerized into filaments. In other words, the centrifugation was incomplete and/or some excess protein concentration is found in the supernatant.
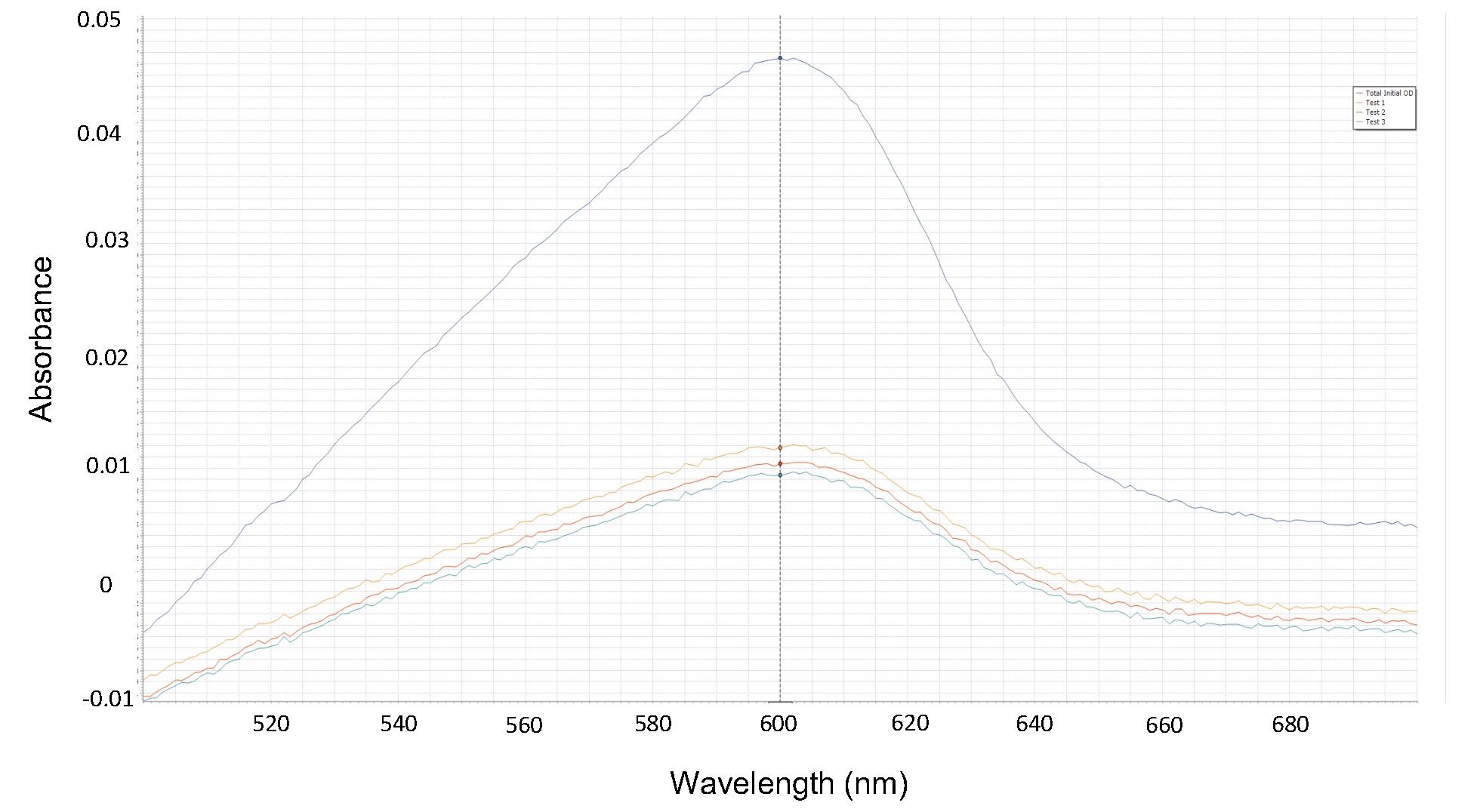
Figure 7. Absorbance. Illustrative example of an absorbance data plot obtained from spectrophotometric measurements. The absorbance curves for the total initial protein concentration (black line) and three independent experiments for the supernatant were measured at 600 nm. The average absorbance value is used to determine the mass of the supernatant in step C2 of Data analysis.The average of three independent absorbance values obtained for the supernatant at 600 nm is 0.01055.
Calculate the supernatant concentration based on 1.00 OD600 nm = 100 μg protein per mL reagent per cm and multiply this protein concentration by 10 to achieve the protein concentration in μg/mL of the original protein solution.
The supernatant concentration is 0.1055 mg/mL in 198 μL. Thus, the following equation is applied to solve for MSN:
MSN=Supernatant Concentration *Supernatant Volume=0.0209 mg
Calculation of pellet concentration (F-actin) (CP)
First, the difference between the initial mass concentration (0.08 mg) and the mass of the supernatant concentration (0.0209 mg) can be used to obtain the mass of the pellet, MP, as follows:
MP=Mass Initial Concentration-Mass Supernatant Concentration=0.0591 mg
Furthermore, once the mass of the pellet (0.0591 mg) is known, the pellet concentration is easily predicted in 22 μL (volume pellet):
CP=(Mass Pellet)/(Volume Pellet)=2.687 mg/mL
Last, to find the molarity (M) of the pellet, we use the following equation where the pellet concentration (mg/mL) is divided by the molecular weight of actin (43 KDa):
Pellet Molarity (MP)= (Pellet Concentration(mg/mL))/(Actin Molecular Weight (Da))=1.37 μM
Sample quality control
Note: The following steps/comments are intended for new experimenters to correlate the quality of their samples to light scattering measurement data plots.
The correlation function is an excellent indicative plot to ensure the system is free from aggregation/sedimentation. As indicated in the DLS Analysis, if multiple exponential decays and oscillated baselines are observed, it will most likely represent the sample’s aggregation and/or sedimentation.
A good practice to ensure homogeneity in the sample is to mix and vortex the solution prior to any DLS and ELS measurement to avoid the tendency to sedimentation and ensure good quality data.
Knowing the protein concentrations in the dilute regime is highly recommended to ensure dispersion. If increased protein concentration is achieved, longer filaments and higher-order structures of actin may be formed, leading to different results in the DLS measurements from those presented in our article (Alva et al., 2022). From the ELS experiments point of view, high zeta potential values are correlated with high repulsive forces between polymers, leading to a disperse system. If increased polyelectrolyte concentration is achieved, zeta potential values may decrease, and the dispersion may be weakened.
Notes
It is important to have good laboratory practices that include working in very clean conditions. To minimize any source of contamination, tightly close tubes, vials, and samples. Make sure the cuvettes and vials are clean and dust-free to prevent the presence of contaminants (see Figure 1).
The usage of a 15 mL conical tube was vital since it facilitated measuring and adjusting the pH of the buffers (1–3). If the tube is smaller, the pH meter probe will not be able to fit; also, there will not be enough solution to cover the tip of the probe leading to inaccurate pH readings.
When adjusting the pH (G-actin and polymerization buffers) use glass pipets to carefully add the base and acid solutions drop by drop into the buffers. The addition of too many drops may increase or decrease the pH significantly, so use caution. It is important to mix well by vortexing the solutions after adding these drops; failing to do so will lead to inaccurate pH readings.
To begin the protein reconstitution, the 1 mg G-actin protein powder needs to be extracted. The powder is usually frozen and stuck to the bottom of the vial. We suggest vortexing the protein vial at a low to medium speed (5/10) for 30–45 s to smooth the protein powder and allow easy extraction when the protein is still dry.
After adding the G-actin buffer to the 10 mg/mL G-actin density, the protein will agglomerate. Mix the solution well by vortexing and pipetting to lose the agglomerates. These structures are visible, so mix the solution well to create and maintain a homogeneous solution.
G-actin proteins may concentrate at the solution’s surface. If so, mix the solution well before aliquoting into experimental samples.
When cutting the Optifit tips using sterilized scissors, have a reference mark on the tips to make this process easier and more consistent. The tips need to have a diameter of approximately 3–5 mm to reduce the breakage of the filaments.
During the centrifugation step, we used a 50,000 × g spin for 2 h to ensure that short to long filaments pellet. If a faster ultracentrifugation (50,000 × g < speed < 100,000 × g) is available, we recommend a duration of 1.5–2 h. If the ultracentrifugation is able to reach 100,000 × g, we highly recommend a duration of 1 h.
We recommend imaging actin filaments by fluorescent labeling of phalloidin and/or similar imaging techniques such as transmission electron microscopy, to ensure the quality of actin final preparation and formation (see Figure 8).

Figure 8. Micrograph image of actin filaments. Illustrative micrograph image of actin filaments using the JEOL 1400 transmission electron microscopy. During the TEM sample preparations, the samples underwent a series of steps using 2% uranyl acetate on a formvar-coated grid. The samples were washed 2–3 times with distilled water.WARNING: If the actin filaments or other polymers are exposed to high electrolyte salt concentrations (ionic strength), there is a high possibility that new order structures (bundles and networks) and aggregates are formed in solution. We recommend caution on the type and salt concentrations that are used in solution that could induce bundles for actin filaments (Tang and Janmey, 1996) or other polymers.
Examples of different actin preparations:
At actin concentrations higher than the critical, the ATP-G-actin increases in solution leading to a significant increase in the elongation rates (Crevenna et al., 2013) and the formation of longer and more semiflexible filaments. As the length of filaments grows, the breakage of filaments may be higher. On the other hand, longer filaments generate smaller diffusion coefficients and higher electrophoretic mobility values.
The actin filament elongation kinetics depends on the pH solution. At pH higher than 7, shorter filaments may be formed due to decreased elongation rates (Crevenna et al., 2013). Thus, a higher diffusion coefficient and smaller electrophoretic mobility values would be expected at higher pH level solutions.
The polymerization rate depends on the type of divalent cation bound to actin. Thus, if Mg2+ is used as part of the polymerization buffer (instead of Ca2+), it could lead to changes in the mechanical and flexibility properties of actin filaments (Steinmetz et al., 1997). These changes may affect the hydrodynamic and electrostatic properties of actin filaments. Thus, changes in the diffusion and electrophoretic mobility values may be expected when using different metal ions in the polymerization buffers.
High concentrations of Ca2+ and Mg2+ in the electrolyte solution may attenuate the repulsive inter-filament interaction and the bundling formation of actin filaments (Castaneda et al., 2018). Thus, the actin preparation under different divalent concentrations may generate actin bundles with different bending, diameter, and length, and consequently, hydrodynamic and electrostatic properties.
Capping proteins, namely gelsolin, regulate the assembly and length of actin filaments (Warshavsky et al., 2022). For instance, the lower the ratio between G-actin and gelsolin concentration, the shorter the average filament length, the higher the diffusion coefficient, and the smaller the electrophoretic mobility values.
To avoid sedimentation and/or aggregation of protein during light scattering experiments, gently pipette 2–3 times the solution in between runs using the 3–5 mm cut tip.
If the sample has a higher protein and/or salt concentration than proposed in this protocol, we highly recommend considering the viscosity parameter by adding this value in the ZS Xplorer software under Measure > Dispersant.
The attenuation factor in the DLS instrument uses multiple positions (1–11) to control the beam intensity from 100% to 0.0003%. This feature was set to automatic to allow the ZS Xplorer software to determine the best possible attenuator. For F-actins, the attenuation factor usually varies between 10 and 11.
The refractive index and absorption of the material have no bearing on the Z-average, polydispersity, and intensity. We selected protein as the material being measured, consisting of a refractive index of 1.450 and an absorption of 0.001. Similarly, the dispersant’s refractive index was set to 1.33, and viscosity to 0.8872 mPa·s. These values may vary according to different materials and/or electrolytes being measured.
The correlation functions were measured at the back-scattering angle (173°), where the incident beam does not have to travel through the entire sample. The effect of multiple scattering and dust is greatly reduced.
During DLS experiments, the measurement duration was automatically determined from the detected count rate. The lower the count rate, the longer the measurement duration and the higher the noise.
Since F-actins form a polydisperse system of unknown size distribution, the general purpose analysis model using the CONTIN algorithm was selected during the DLS measurements.
The ZS Xplorer software measures the sample electrical conductivity of the system when set as automatic. It adjusts the cell voltage to keep a low current flowing, close to 5 ms/cm, in the sample. If this is not considered, the sample temperature may increase near the electrodes, inducing bubble formation and sample degradation, leading to inaccurate results.
The fast field reversal (FFR) of the phase analysis light scattering was selected since the mobility measured during this period is due to the electrophoresis of the particles only. It is not affected by electro-osmosis associated with the soft field reversal (SFR).
This protocol can also be applied for other manufacturer’s instruments such as Dynamics Mobius from Wyatt Technology and NanoBrook Series from Brookhaven Instruments Corporation. Like ULTRA Zetasizer, these instruments can perform DLS and ELS measurements. The software setup for both instruments is somewhat similar, except the data analysis. We recommend the multimodal analysis for size measurements, specifically for polydisperse systems.
The Mobius and NanoBrook Series instruments use different volumes and cuvettes for size and zeta potential measurements. We highly recommend following their websites and manuals for usage of their cuvettes.
A time delay between measurements helped to reduce sample heating when long run measurements raised the temperature of the cell to approximately 50 °C, allowing the sample to recover 25 °C between consecutive measurements, reducing critical sample degradation and avoiding increasing mobility with sequential measurements.
Standard operating protocol, data analysis (Parker and Lollar, 2021), and analysis of biomolecular preparations to detect aggregation of proteins, glycoproteins, protein-protein complexes, and others, can be found elsewhere (Stetefeld et al., 2016).
Recipes
Stock buffers
50 mM CaCl2
Reagent Final concentration Amount CaCl2 50 mM 55.50 mg H2O ultra-pure n/a 10 mL Total 50 mM 10 mL 50 mM MgCl2
Reagent Final concentration Amount MgCl2 50 mM 47.61 mg H2O ultra-pure n/a 10 mL Total 50 mM 10 mL 1.0 M KCl
Reagent Final concentration Amount KCl 1.0 M 745.5 mg H2O ultra-pure n/a 10 mL Total 1.0 M 10 mL 102.24 mM KCl
Reagent Final concentration Amount KCl 102.24 mM 76.22 mg H2O ultra-pure n/a 10 mL Total 102.24 mM 10 mL Notes:
1). To maintain the stock buffers biologically active, they need to be remade on a weekly basis.
2). This step is performed on Day 1 (see Figure 1).
G-actin buffers
G-actin buffer 1 (pH 7.80)
Reagent Final concentration Amount Tris base (100 mM, pH 7.6) 2 mM 100 μL H2O ultra-pure n/a 4,845 μL CaCl2 (50 mM) 0.2 mM 20 μL ATP (100 mM) 0.5 mM 25 μL DTT (100 mM) 0.2 mM 10 μL Total n/a 5 mL G-actin buffer 2 (pH 7.66)
Reagent Final concentration Amount Tris base (100 mM, pH 7.6) 2 mM 100 μL H2O ultra-pure n/a 4,860 μL CaCl2 (50 mM) 0.2 mM 20 μL ATP (100 mM) 0.2 mM 10 μL BME (50 mM) 0.1 mM 10 μL Total n/a 5 mL G-actin buffer 3 (pH 8.23)
Reagent Final concentration Amount Tris base (100 mM, pH 7.6) 2 mM 100 μL H2O ultra-pure n/a 4,830 μL CaCl2 (50 mM) 0.2 mM 20 μL ATP (100 mM) 0.5 mM 25 μL DTT (100 mM) 0.5 mM 25 μL Total n/a 5 mL Notes:
1). Calibrate the pH meter at three-point calibration using Orion buffers pH 4.01, 7.00, and 10.00 before its usage.
2). To increase/decrease the pH, add drops of a base (0.74 M NaOH) or acid (0.1 N HCl) solution, respectively. Add these drops carefully and constantly check the pH, as it can rapidly change.
3). This step is performed on Day 2 (Figure 1).
4). Have ice ready in a cooler, as all G-actin buffers must be done on ice.
Polymerization buffers
Polymerization buffer 1 (pH 7.56)
Reagent Final concentration Amount H2O ultra-pure n/a 4,050 μL MgCl2 (50 mM) 2 mM 200 μL KCl (1.0 M) 150 mM 750 μL Total n/a 5 mL Polymerization buffer 2 (pH 7.64)
Reagent Final concentration Amount H2O ultra-pure n/a 4,050 μL MgCl2 (50 mM) 2 mM 200 μL KCl (1.0 M) 150 mM 750 μL Total n/a 5 mL Polymerization buffer 3 (pH 8.07)
Reagent Final concentration Amount H2O ultra-pure n/a 4,550 μL MgCl2 (50 mM) 2 mM 200 μL KCl (1.0 M) 50 mM 250 μL Total n/a 5 mL Notes:
1). This step is also performed on Day 2 (Figure 1).
2). Have ice ready in a cooler, as all polymerization buffers must be done on ice.
Electrolyte buffers
Electrolyte buffer 1 (pH 7.72), buffer 2 (pH 7.66), buffer 3 (pH 8.06)
Reagent Final concentration Amount H2O ultra-pure n/a 10 mL KCl 102.24 mM 76.22 mg Total n/a 10 mL
Acknowledgments
This work was supported by NIH grant 1SC1GM127187-04. We thank Drs. Carrie Schindler, Anna Morfesis, Ronald Soriano, and Matthew Brown from Malvern Panalytical Instruments Inc. for their assistance and advice in the configuration of the light scattering instrument and data analysis. We also thank Drs. Brian Hoover and Lee Toni from Cytoskeleton Inc. for their assistance and advice in the sample’s preparation and protein concentration analysis. This protocol was derived from previous work (F. Wang et al., 1989; Janmey et al., 1994, 1986).
Competing interests
The authors declare no conflict of interest.
Ethics
There are no human subjects or animal studies described in this protocol.
References
- Alva, E., George, A., Brancaleon, L. and Marucho, M. (2022). Hydrodynamic and Polyelectrolyte Properties of Actin Filaments: Theory and Experiments. Polymers 14(12): 2438.
- Bonet, C., Ternent, D., Maciver, S. K. and Mozo-Villarias, A. (2000). Rapid formation and high diffusibility of actin-cofilin cofilaments at low pH. Eur J Biochem 267(11): 3378-3384.
- Crevenna, A. H., Naredi-Rainer, N., Schonichen, A., Dzubiella, J., Barber, D. L., Lamb, D. C. and Wedlich-Soldner, R. (2013). Electrostatics control actin filament nucleation and elongation kinetics. J Biol Chem 288(17): 12102-12113.
- Castaneda, N., Zheng, T., Rivera-Jacquez, H. J., Lee, H. J., Hyun, J., Balaeff, A., Huo, Q. and Kang, H. (2018). Cations Modulate Actin Bundle Mechanics, Assembly Dynamics, and Structure. J Phys Chem B 122(14): 3826-3835.
- Del Rocio Cantero, M., Gutierrez, B. C. and Cantiello, H. F. (2020). Actin filaments modulate electrical activity of brain microtubule protein two-dimensional sheets. Cytoskeleton (Hoboken) 77(3-4): 167-177.
- Hou, L., Lanni, F. and Luby-Phelps, K. (1990). Tracer diffusion in F-actin and Ficoll mixtures. Toward a model for cytoplasm. Biophys J 58(1): 31-43.
- Janmey, P. A., Hvidt, S., Kas, J., Lerche, D., Maggs, A., Sackmann, E., Schliwa, M. and Stossel, T. P. (1994). The mechanical properties of actin gels. Elastic modulus and filament motions. J Biol Chem 269(51): 32503-32513.
- Janmey, P. A., Peetermans, J., Zaner, K. S., Stossel, T. P. and Tanaka, T. (1986). Structure and mobility of actin filaments as measured by quasielastic light scattering, viscometry, and electron microscopy. J Biol Chem 261(18): 8357-8362.
- Janmey, P. A., Slochower, D. R., Wang, Y.-H., Wen, Q. and Cēbers, A. (2014). Polyelectrolyte properties of filamentous biopolymers and their consequences in biological fluids. Soft Matter 10(10): 1439-1449.
- Käs, J., Strey, H., Tang, J. X., Finger, D., Ezzell, R., Sackmann, E. and Janmey, P. A. (1996). F-actin, a model polymer for semiflexible chains in dilute, semidilute, and liquid crystalline solutions. Biophysical Journal 70(2): 609-625.
- Kroy, K. and Frey, E. (1997). Dynamic scattering from solutions of semiflexible polymers. Physical Review E 55(3): 3092-3101.
- Lanni, F. and Ware, B. R. (1984). Detection and characterization of actin monomers, oligomers, and filaments in solution by measurement of fluorescence photobleaching recovery. Biophys J 46(1): 97-110.
- McDonald, J. H. (2009). Handbook of Biological Statistics. Sparky House Publishing. Baltimore, MD.
- Niranjan, P. S., Forbes, J. G., Greer, S. C., Dudowicz, J., Freed, K. F. and Douglas, J. F. (2001). Thermodynamic regulation of actin polymerization. J Chem Phys 114(24): 10573-10576.
- Parker, E. T. and Lollar, P. (2021). Conformation of the von Willebrand factor/factor VIII complex in quasi-static flow. J Biol Chem 296: 100420.
- Pollard, T. D. (1986). Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. J Cell Biol 103(6 Pt 2): 2747-2754.
- Steinmetz, M. O., Goldie, K. N. and Aebi, U. (1997). A correlative analysis of actin filament assembly, structure, and dynamics. J Cell Biol 138(3): 559-574.
- Stetefeld, J., McKenna, S. A. and Patel, T. R. (2016). Dynamic light scattering: a practical guide and applications in biomedical sciences. Biophys Rev 8(4): 409-427.
- Tang, J. X. and Janmey, P. A. (1996). The Polyelectrolyte Nature of F-actin and the Mechanism of Actin Bundle Formation (∗). J Biol Chem 271(15): 8556-8563.
- Tassieri, M., Evans, R. M., Barbu-Tudoran, L., Trinick, J. and Waigh, T. A. (2008). The self-assembly, elasticity, and dynamics of cardiac thin filaments. Biophys J 94(6): 2170-2178.
- Wang, F., Sampogna, R. V. and Ware, B. R. (1989). pH dependence of actin self-assembly. Biophys J 55(2): 293-298.
- Wang, Y. H. and Narayan, M. (2008). pH dependence of the isomerase activity of protein disulfide isomerase: insights into its functional relevance. Protein J 27(3): 181-185.
- Warshavsky, V., and Marucho M. (2022). Theory of Weakly Polydisperse Cytoskeleton Filaments. Polymers 14(10): 2042.
- Xu, J., Schwarz, W. H., Kas, J. A., Stossel, T. P., Janmey, P. A. and Pollard, T. D. (1998). Mechanical properties of actin filament networks depend on preparation, polymerization conditions, and storage of actin monomers. Biophys J 74(5): 2731-2740.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Alva, E., George, A., Brancaleon, L. and Marucho, M. (2022). In vitro Preparation of Homogenous Actin Filaments for Dynamic and Electrophoretic Light Scattering Measurements. Bio-protocol 12(22): e4553. DOI: 10.21769/BioProtoc.4553.
Category
Neuroscience > Neuroanatomy and circuitry
Biological Sciences > Biological techniques
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.