- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Staphylococcus aureus 30S Ribosomal Subunit Purification and Its Biochemical and Cryo-EM Analysis
Published: Vol 12, Iss 20, Oct 20, 2022 DOI: 10.21769/BioProtoc.4532 Views: 3015
Reviewed by: Alba BlesaKristin L. ShinglerNathan Rhys James
Original research article
The authors used this protocol in:

Nov 2021
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The ribosome is a complex cellular machinery whose solved structure allowed for an incredible leap in structural biology research. Different ions bind to the ribosome, stabilizing inter-subunit interfaces and structurally linking rRNAs, proteins, and ligands. Besides cations such as K+ and Mg2+, polyamines are known to stabilize the folding of RNA and overall structure. The bacterial ribosome is composed of a small (30S) subunit containing the decoding center and a large (50S) subunit devoted to peptide bond formation. We have previously shown that the small ribosomal subunit of Staphylococcus aureus is sensitive to changes in ionic conditions and polyamines concentration. In particular, its decoding center, where mRNA codons and tRNA anticodons interact, is prone to structural deformations in the absence of spermidine. Here, we report a detailed protocol for the purification of the intact and functional 30S, achieved through specific ionic conditions and the addition of spermidine. Using this protocol, we obtained the cryo-electron microscopy (cryo-EM) structure of the 30S–mRNA complex from S. aureus at 3.6 Å resolution. The 30S–mRNA complex formation was verified by a toeprinting assay. In this article, we also include a description of toeprinting and cryo-EM protocols. The described protocols can be further used to study the process of translation regulation.
Graphical abstract:
Background
Protein synthesis is performed by the ribosome, which is composed of two dynamic subunits. These are made by RNAs and proteins that interact with several ligands and adopt several conformations during the translation process (Frank et al., 2007; Agirrezabala et al., 2008; Zhang et al., 2009). Structural and functional studies have revealed that ions impact the stability and dynamics of the ribosome (Nierhaus, 2014; Akanuma, 2021). Optimal ions and polyamine concentrations are necessary for maintaining both structural integrity and intrinsic flexibility. The small subunit (30S), responsible for the translation initiation process and the mRNA decoding, is particularly structurally dynamic and, thus, more challenging for structural studies. This subunit is composed of two main structural domains, the head and the body, which undergo continuous rearrangement (swiveling and nodding, respectively), heavily influencing the position of the mRNA in the decoding channel (Fahnestock et al., 1977; Hussain et al., 2016; Belinite et al., 2018). For this reason, specific ionic conditions have been used to stabilize the position of the mRNA for structural studies. Ribosome structure and dynamics are especially affected by the presence of cations, notably Mg2+, Zn2+, K+, NH4+, and polyamines (Gordon and Lipmann 1967; Weiss et al., 1973; Stahli and Noll 1977; Kim et al., 2007; Wohlgemuth et al., 2010; Nierhaus, 2014; Rozov et al., 2019). For instance, recent studies have demonstrated that different magnesium concentrations drastically affect the conformation of the 30S, and high temperatures help induce some conformational changes in Escherichia coli (Jahagirdar et al., 2020). In addition, spermidine has been shown to stabilize the active conformation of Staphylococcus aureus 30S by interacting with rRNA (Belinite et al., 2021); indeed, the correct folding of the decoding center at the top of the helix 44 (h44) in the 30S body can be achieved by maintaining a certain concentration of spermidine along the 30S purification process (Belinite et al., 2021). Spermidine and spermine are polyamines, organic polycations present in all cell types (Cohen and Lichtenstein, 1960; Stevens et al., 1969; Weiss and Morris, 1970; Cohen, 1971; Hardy and Turnock, 1971; Turnock and Birch, 1973), including in S. aureus (Herbst et al., 1958; Hamana and Satake 1995; Rosenthal and Dubin, 1962). These have been shown to associate with the ribosome (Cohen and Lichtenstein, 1960) and contribute to both the efficiency and fidelity of protein synthesis (Echandi and Algranati, 1975; Igarashi et al., 1980; Igarashi and Kashiwagi, 2018; McMurry and Algranati, 2018). They are known to stimulate in vitro translation (Hershko et al., 1961; Martin and Ames, 1962; Takeda, 1969; Igarashi et al., 1973; Konecki et al., 1975; Hetrick et al., 2010), lessening the Mg2+ requirement (Rheinberger and Nierhaus, 1987; Bartetzko and Nierhaus, 1988).
In this study, we describe in detail the protocols for the efficient 30S purification from S. aureus and the complex preparation with a natural S. aureus mRNA, spa mRNA, which encodes the virulence factor protein A. The proper conformation of the h44, full accommodation of the mRNA in the channel, and reduced flexibility of certain parts of the 30S were achieved by maintaining spermidine at a proper concentration throughout the whole process, starting from the 30S subunit isolation. The 30S subunit was obtained by separating the 70S ribosome under dissociative conditions and analyzed for high-molecular-weight contaminants and rRNA integrity. Activity of the isolated 30S was tested using a toeprinting assay (Fechter et al., 2009), while its structure was obtained by cryo-electron microscopy (cryo-EM). This technique has also been used to test different buffer conditions, thanks to the development of methods to rapidly get multiple structures (Cressey and Callaway, 2017; Nakane et al., 2020). The final S. aureus 30S–mRNA structure was obtained with focused refinement of the head and body at 3.6 and 3.4 Å resolution, respectively. Our previous work demonstrated the requirement of spermidine for the rRNA stabilization and its use in the cryo-EM studies (Belinite et al., 2021). Here, we show that the 30S subunits are stable in these conditions, confirming that spermidine not only contributes to their structural integrity, but also assures their required flexibility. Hence, protocols described in this article can be used for the future investigations of S. aureus functional complexes.
Materials and Reagents
Cells
Staphylococcus aureus RN6390 strain (Khusainov et al., 2016)
70S ribosome purification
Polycarbonate 50 mL centrifuge tubes (NalgeneTM, catalog number: 3117-0500)
Open-top thinwall ultra-clear tube, 38.5 mL (Beckman Coulter, catalog number: 344058)
Blood agar plates (BDTM Columbia agar with 5% sheep blood, catalog number: PA-254005.06)
Brain–heart infusion broth (BHI, Sigma-Aldrich, catalog number: 53286)
Lysostaphin (Sigma-Aldrich, catalog number: L7386)
DNase I (Roche, catalog number: 4716728001)
Protease inhibitor cocktail without ethylenediaminetetraacetic acid (EDTA, Thermo ScientificTM, catalog number: 78429)
30% PEG 20000 (Hampton Research, HR2-609)
Buffer A (see Recipes)
Buffer B (see Recipes)
5× buffer E (see Recipes)
Dissociation buffer (see Recipes)
30S subunit purification
Open-top thinwall ultra-clear tube, 13.2 mL (Beckman Coulter, catalog number: 344059)
Amicon ultra-15 centrifugal filter unit (Merck Millipore, catalog number: UFC910008)
Liquid nitrogen
30S storage buffer (see Recipes)
mRNA purification
RNaseZap (Thermo ScientificTM, catalog number: AM9780)
NucleoSpin Plasmid, mini kit for plasmid DNA (Macherey-Nagel, catalog number: 740588.50)
NucleoSpin Gel and PCR Clean-up, mini kit for gel extraction and PCR clean-up (Macherey-Nagel, catalog number: 740609.50)
BamHI enzyme (NEB, catalog number: R0136S)
CutSmart® buffer (NEB, catalog number: B7204S)
MEGAscript kit with DNase turbo (Ambion, catalog number: AM1333)
ROTI® phenol/chloroform/isoamyl alcohol, 250 mL, pH 7.5–8.0 (Carl Roth, catalog number: A156.1)
ROTI® Aqua-P/C/I, 250 mL, pH 4.5–5.0 (Carl Roth, catalog number: X985.1)
3 M sodium acetate
Buffer ELU (see Recipes)
30S and mRNA quality analysis
PageRulerTM protein ladder (Thermo ScientificTM, catalog numbers: 26616 and 26619)
InstantBlue® coomassie protein stain (Abcam, catalog number: ab119211)
ROTI® aqua-phenol, 250 mL, pH 4.5–5.0 (Carl Roth, catalog number: A980.1)
Chloroform (Thermo ScientificTM, catalog number: AC158210010)
Agarose (SeaKemTM, catalog number: 11590717)
1 M potassium thiocyanate (Sigma, catalog number: P3011)
Ethidium bromide (Thermo ScientificTM, catalog number: 15585011)
100% formamide (Sigma, catalog number: F9037)
RiboRuler low-range RNA ladder (Thermo ScientificTM, catalog number: SM1831)
4% stacking gel (see Recipes)
15% resolving gel (see Recipes)
4× blue buffer (see Recipes)
10× running buffer (see Recipes)
6% polyacrylamide gel (see Recipes)
10× TBE (see Recipes)
Toeprinting assay
10 μM Toe primer (5′ CCTGCAGGTCGACTC 3′)
10× PNK buffer (Thermo ScientificTM, catalog number: EK 0032)
10 U/μL T4 PNK (Thermo ScientificTM, catalog number: EK 0032)
[γ-32P]-ATP (370 MBq/mL, 10 mCi/mL, 185 MBq)
Micro bio-spin chromatography columns (Bio-Rad, catalog number: 732-6200)
20 U/μL AMV reverse transcriptase, isolated from Avian Myeloblastosis Virus (QBIOgene, catalog number: EMAMV200)
10× AMV buffer (QBIOgene, catalog number: EMAMV200)
10 mM dATP, dCTP, dGTP, and dTTP (Roche, catalog numbers: 11051440001, 11051458001, 11051466001, and 11051482001)
0.3 M Na acetate (Sigma, catalog number: S2889)
Phenol/chloroform, pH 4.5–5.0 (Carl Roth, catalog number: X985.1)
Chloroform/isoamyl alcohol (Carl Roth, catalog number: X984.1)
100% cold ethanol
80% cold ethanol
8 M urea (Sigma, catalog number: U5128)
0.1% bromophenol blue (Sigma, catalog number: B0126)
0.1% xylene cyanol (Sigma, catalog number: X4126)
100 µM ddATP, ddGTP, ddCTP, and ddTTP (Roche, catalog number: 3732738001)
3 M KOH (Sigma, catalog number: 221473)
1 M tris-HCl, pH 8.0 (Sigma, catalog number: T1503)
10% SDS (Bio-Rad, catalog number: 1610416)
Acetic acid (Sigma, catalog number: A6283)
10× toeprinting buffer (see Recipes)
30S–mRNA complex preparation for cryo-EM
Liquid ethane
Liquid nitrogen
Quantifoil R2/2300-mesh holey carbon grids
Equipment
70S ribosome purification
2 L flasks
Microbiological incubator (Thermo ScientificTM, catalog number: 50125882 or equivalent)
Cell culture shaker at 30 °C
Class II biosafety cabinet
Floor centrifuges (Beckman, Avant J20 XP and/or Avanti J-E or equivalent)
JA-25.50 rotor (Beckman, catalog number: 363055)
JLA-16.250 rotor (Beckman, catalog number: 363930)
Ultracentrifuge (Beckman, OPTIMA XE90 or equivalent)
Type 45 Ti rotor (Beckman, catalog number: 339160)
SW28 rotor (Beckman, catalog number: 342204)
Gradient MasterTM base unit (Biocomp, catalog number: B108-2)
Gradient MasterTM device (BioComp Instruments, Fredericton, Canada)
Fraction collector (Bio-Rad, model: 2110)
NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA or equivalent)
30S subunit purification
Ultracentrifuge (Beckman, OPTIMA XE90 or equivalent)
SW41 Ti swinging-bucket rotor (Beckman, catalog number: 331362)
Floor centrifuges (Beckman, Avanti J-E or equivalent)
JLA-16.250 rotor (Beckman, catalog number: 363930)
mRNA purification
Cell culture shaker at 37 °C
Benchtop centrifuge (Eppendorf MiniSpin® Plus personal microcentrifuge with 12-place rotor or equivalent)
Eppendorf thermomixer R, dry block heating and cooling shaker or equivalent
30S and mRNA quality analysis
Mini-PROTEAN® Tetra Handcast systems (Bio-Rad or equivalent)
PowerPacTM basic power supply (Bio-Rad or equivalent)
Wide mini-sub cell GT cell (Bio-Rad or equivalent)
GelDoc Go Gel imaging system (Bio-Rad)
Toeprinting assay
Benchtop centrifuge (Eppendorf MiniSpin® Plus personal microcentrifuge with 12-place rotor or equivalent)
Phosphor imaging or autoradiography (Typhoon or equivalent)
30S–mRNA complex preparation cryo-EM data acquisition
Thermo Scientific Vitrobot Mark IV
Talos Arctica equipped with Falcon III direct electron detector (Thermo Fisher)
Microscope control PC with installed EPU software
Software
MotionCor2 (https://emcore.ucsf.edu/ucsf-software/)
Gctf (https://www2.mrc-lmb.cam.ac.uk/research/locally-developed-software/zhang-software/)
Relion 3.0 (https://www3.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page/)
ResMap (http://resmap.sourceforge.net/)
Chimera (https://www.cgl.ucsf.edu/chimera/)
Procedure
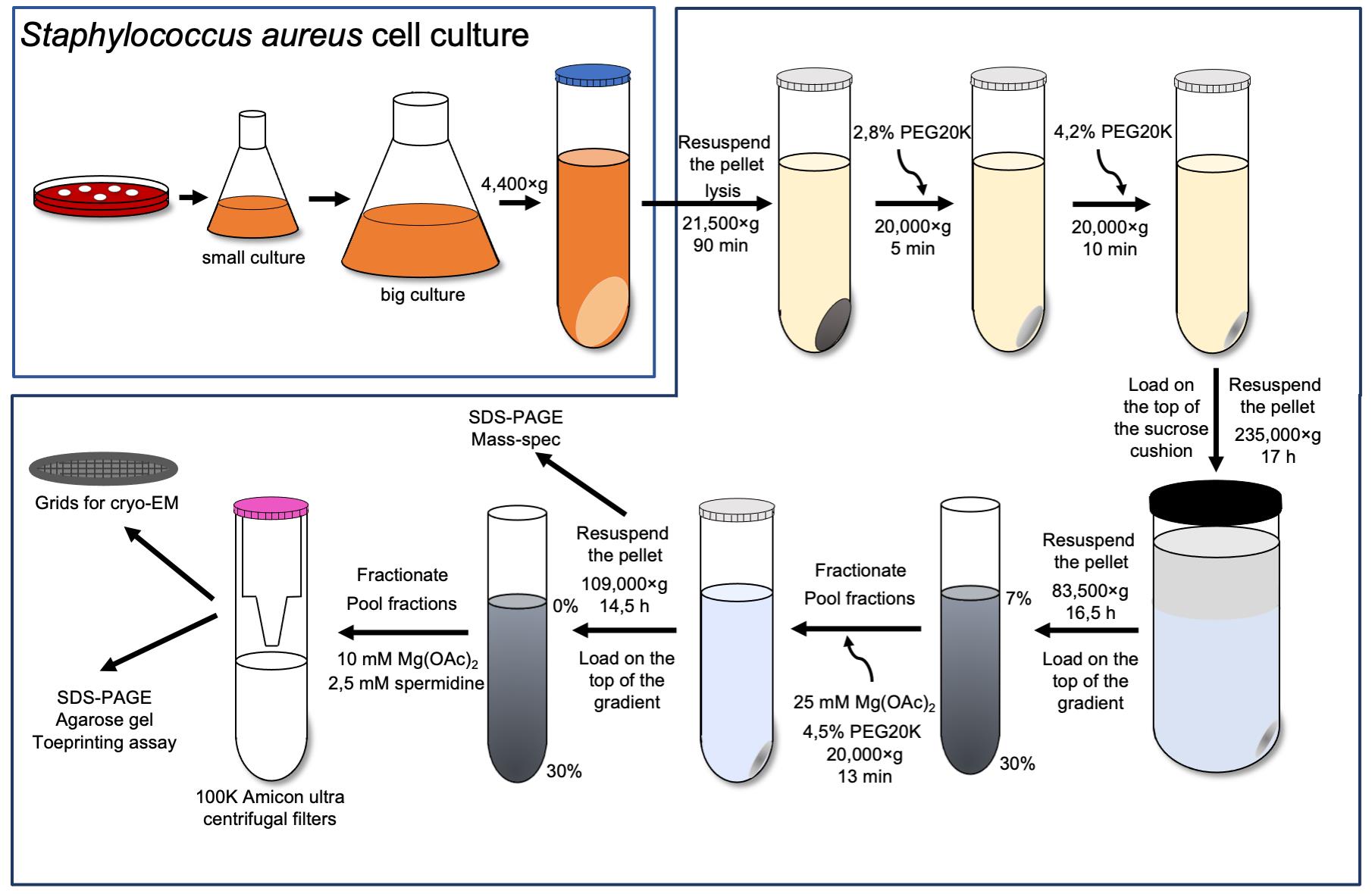
70S ribosome purification
Streak Staphylococcus aureus’ RN6390 cells on a blood agar plate and grow at 37 °C for 20 h.
Inoculate a single colony in 50 mL of sterile BHI medium in a 250 mL flask and grow for 17 h at 37 °C and 180 rpm.
Add 5 mL of overnight culture to four 2 L flasks, each filled with 500 mL of sterile BHI medium, and grow for 3 h at 37 °C and 180 rpm until OD600 = 1.
Harvest cells by centrifugation at 4,400 × g for 15 min at 4 °C.
Resuspend the pellet in 50 mL of buffer A.
Collect the pellet by centrifugation at 4,400 × g for 15 min.
Repeat the previous steps (5–6) two times.
Weigh cells and keep at -80 °C.
Note: The typical yield is 2.8 g of cells from 2 L of culture.
Thaw the cells (2.8 g) on ice for 15–20 min and resuspend in 6.25 mL of buffer A supplemented with 0.7 mg lysostaphin, 1× protease inhibitor cocktail, 1 mM EDTA pH 7.5, and 6 µl DNase I per 1 gram of cell weight. Incubate for 1 h at 37 °C.
Spin down the lysate at 21,500 × g (JA-25.50 rotor) for 90 min at 4 °C.
Adjust the PEG20K concentration in the collected supernatant to 2.8% and leave the reaction on ice under agitation for 5 min.
Collect the supernatant by centrifugation at 20,000 × g for 5 min (JA-25.50 rotor).
Increase the PEG20K concentration to 4.2% and leave the reaction on ice under agitation for an additional 10 min.
Collect the supernatant by centrifugation at 20,000 × g for 10 min (JA-25.50 rotor).
Resuspend each pellet in 17.5 mL buffer A with 1 mM EDTA, lay on a 25 mL sucrose cushion (buffer B), and spin at 235,000 × g for 17 h at 4 °C (Type 45 Ti rotor).
Rinse the walls of each tube with 5 mL of buffer E to remove excess sucrose and resuspend the pellet in 1 mL of buffer E.
Lay the obtained sample on twelve 7%–30% sucrose gradients and spin at 83,000 × g for 16.5 h (SW28 rotor).
Fractionate sucrose gradients (Figure 1A-I), pool fraction of 70S, and adjust the magnesium acetate to 25 mM and PEG20K to 4.5%. Leave on ice under agitation for 10 min.
Pellet down the ribosomes by centrifugation at 20,000 × g for 13 min (JA-25.50 rotor) (Khusainov et al., 2016).
Wash the tube walls with the dissociation buffer to remove the excess of sucrose and PEG. Resuspend the pellet in 1.5 mL of dissociation buffer.
Note: The usual yield is 14 mg of 70S with at least 95% purity.
Check the purity of 70S by 15% polyacrylamide gel (Figure 1A-II) and by mass-spectrometry analysis (Figure 1A-III).
30S subunit purification
Load 14 mg of 70S in the dissociation buffer directly on 0%–30% sucrose gradients equilibrated in the same buffer and spin at 109,000 × g for 14.5 h (SW28 rotor).
Fractionate the sucrose gradients (Figure 1B-I) and adjust the magnesium acetate of pooled fractions to 10 mM and spermidine to 2.5 mM.
Concentrate the 30S sample to 2–3 mg/mL in the Amicon ultra centrifugal filter.
Note: By using two filters in parallel, it takes 1 h to concentrate the sample.
Make aliquots, flash freeze them in liquid nitrogen, and store at -80 °C.
Note: The volume of the aliquots should not be less than 3 μL to prevent evaporation of the sample. Moreover, the size of the aliquots should be small (3–5 μL), as a large amount of sample is not required for subsequent experiments.
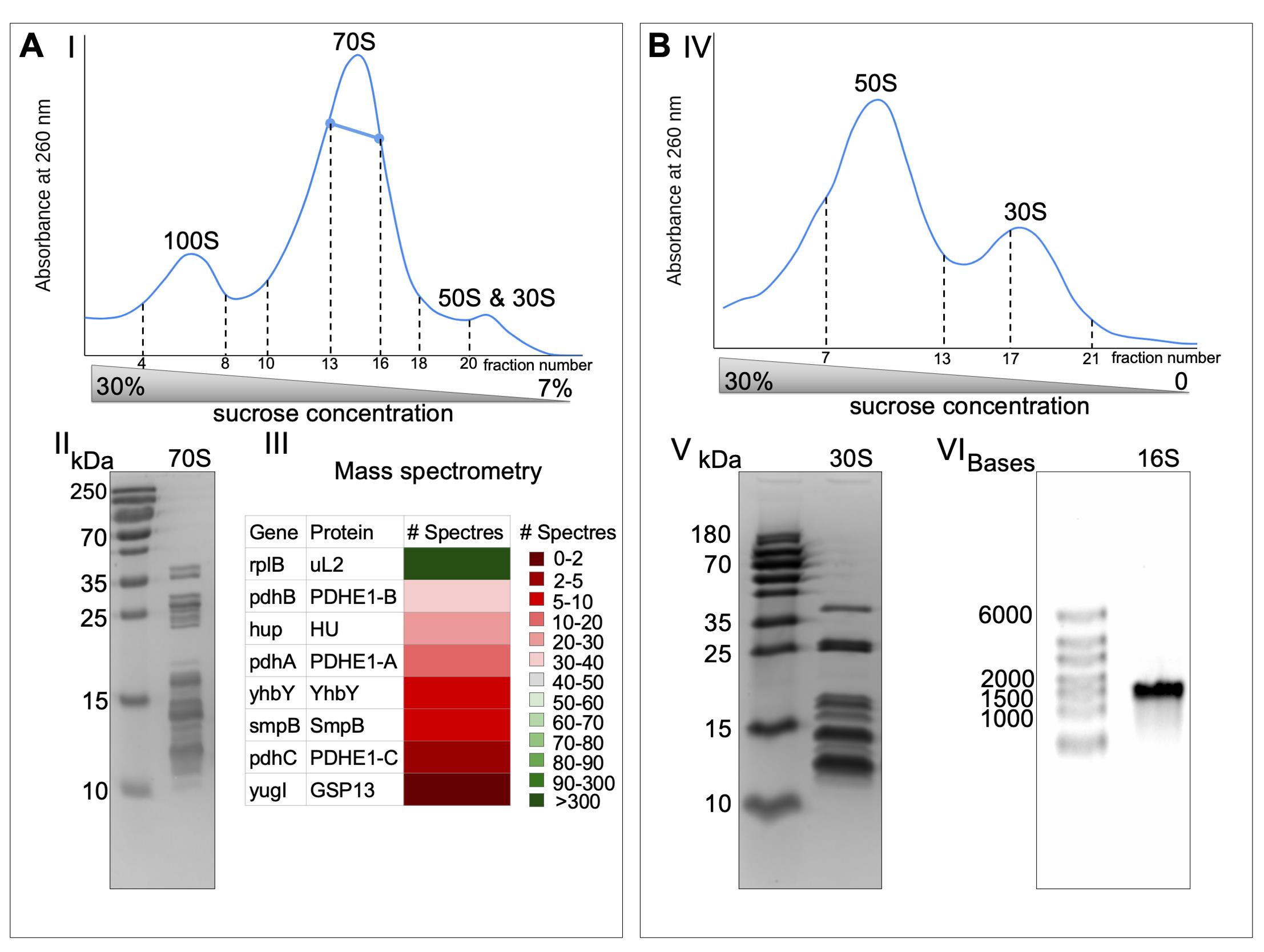
Figure 1. The 30S ribosomal subunits purification. The 30S (B) were obtained from 70S (A) by running through sucrose gradients twice (I and IV, 35 mL is the total volume of sucrose gradients). The bold blue line represents the fraction used for further experiments. Both 70S and 30S fractions were run on SDS-PAGE to check the purity of the samples (II and V). 70S ribosome was sent to mass spectrometry analysis to assess the sample purity (III). 16S was obtained from 30S by phenol/chloroform extraction and run on agarose gel in denaturing conditions to represent its integrity (VI).
spa mRNA purification
The plasmid used in this study was previously described in Benito et al. (2000) and Huntzinger et al. (2005). The pUT7 plasmid contains a T7 promoter for T7 in vitro transcription and ampicillin resistance. spa mRNA sequence with a strong Shine–Dalgarno AGGGGG (underlined) and UUG start codon (bold) is shown below:
ACAAAUACAUACAGGGGGUAUUAAUUUGAAAAAGAAAAACAUUUAUUCAAUUCGUAAACUAGGUGUAGGUAUUGCAUCUGUAACUUUAGGUACAUUACUUAUAUCUGGUGGCGUAACACCUGCUGCAAAUGCUGCGCAACACGAUGAAGCUCAACAAAAUGCUUUUUAUCAAGUCUUAAAUAUGCCUAACUUAAACGCUGGGAUCCUCUAGAGUCGACCUGCAGG
Note: Before starting to work with the mRNA, wipe the bench and pipettes with a liquid that removes RNase contamination (e.g., RNaseZap).
Transform XL1 Escherichia coli cells with the pUT7-spa plasmid.
Grow cells in 250 mL of LB medium with 100 μg/mL ampicillin at 37 °C for 16 h.
Purify the plasmid using a NucleoSpin Plasmid mini kit and linearize it (up to 50 μg of plasmid per reaction) with BamHI enzyme in a 1× CutSmart buffer for 2 h at 37 °C.
Note: The usual yield is 10 μg of plasmid per one reaction. One linearization reaction is enough to proceed with the experiments.
Extract the linearized plasmid DNA from 1% agarose gel using a NucleoSpin Gel and PCR Clean-up mini kit.
Extract the DNA with phenol/chloroform:
Add an equal volume of phenol, vortex well, and spin down at 17,000 × g for 1 min. Keep the upper phase.
Add an equal volume of chloroform, vortex well, and spin down at 17,000 × g for 1 min. Keep the upper phase.
Precipitate the DNA with 2.5 volumes of 100% cold ethanol and 1/16 volume of 3 M sodium acetate.
Wash the pellet with 500 μL of 80% cold ethanol.
In vitro transcribe the mRNA for 3 h at 37 °C using a MEGAscript kit.
Stop the reaction with 2 U/μL DNase turbo and incubate for 15 min at 37 °C.
Extract the mRNA with phenol/chloroform and precipitate it with 100% cold ethanol (similar to steps 5 and 6).
Wash the pellet with 500 μL of 80% cold ethanol.
Add 50% formamide to the sample and heat for 3 min at 90 °C.
Separate the mRNA by 6% polyacrylamide midi-size gel and elute with the buffer ELU for 16 h at 4 °C.
Extract the mRNA with phenol/chloroform and precipitate it with 100% cold ethanol (similar to steps 5 and 6).
Wash the pellet with 200 μL of 80% cold ethanol.
Resuspend in 10–30 μL mQ water.
Store the aliquots at -20 °C.
30S and mRNA quality analysis
The absence of high-molecular-weight non-ribosomal proteins and the presence of all ribosomal proteins are detected on a 15% polyacrylamide gel (Figure 1B-V). The positions of all 70S ribosomal proteins on the SDS-PAGE are identified in Khusainov et al. (2016) (Figure 1A-II). The ribosomal RNA integrity is checked by 0.8% agarose gel in denaturing conditions (Figure 1B-VI).
Protein polyacrylamide gel
Mix all buffer components for the 15% resolving gel and leave to polymerase for 15–20 min.
Repeat the same procedure for 4% stacking gel.
Add 1× blue buffer to the 30S samples (0.15 OD per lane) and heat for 3 min at 95 °C.
Load 5 μL of protein ladder and 5 μL of 30S samples in the blue buffer.
Run the gel in a 1× running buffer at 120 V for 30 min and then at 150 V for 1 h.
Stain the gel in InstantBlue® coomassie for 20 min and destain in water for 1 h.
Extracting the ribosomal RNA
Add an equal volume of phenol to the 0.15 OD 30S, mix well by vortexing, and spin down at 17,000 × g for 1 min.
Add an equal volume of chloroform to the upper phase, mix well by vortexing, and spin down at 17,000 × g for 1 min.
Repeat the two last steps.
Precipitate the ribosomal RNA with 1/10 volume 3 M sodium acetate pH 5.3 and 2.5 volumes of 100% cold ethanol.
Keep the sample overnight at -20 °C or for 1 h at -80 °C and spin down at 17,000 × g for 15 min at 4 °C.
Wash the pellet with 70% cold ethanol and dry it for 5 min in a speed vac.
Resuspend the obtained sample in 10 μL of mQ water.
RNA agarose gel
Melt 0.8% agarose in 1× TBE buffer in the microwave and then cool down to 60 °C.
Add 100 mM potassium thiocyanate and ethidium bromide to the mixture.
Leave the gel for 40 min to polymerase.
Load 0.015 OD of the sample on the gel together with a high-range RNA ladder.
Run the gel in a 1× TBE buffer at 120 V for 45 min.
RNA polyacrylamide gel
The mRNA purity and integrity are checked by 6% polyacrylamide gel stained in ethidium bromide (Figure 2C). The sequence of spa mRNA is shown in Figure 2D.
Mix all components for 6% polyacrylamide gel and leave to polymerize for 15–20 min.
Load 4 μL of low-range RNA ladder in 1× RNA loading dye close to the 100 ng of mRNA in 50% formamide preheated at 90 °C for 2 min.
Run the gel at 200 V for 30 min.
Stain the gel in ethidium bromide for 10 min.
Toeprinting assay
Mix 1 μM Toe primer in PNK buffer, 10 U/μL T4 PNK, and 5 μL of [γ-32P]-ATP, incubate at 37 °C for 1 h, and then pass through the micro bio-spin column pre-equilibrated in mQ water.
Incubate 0.5 μM of spa mRNA with 169,916 cpm/μL Toe primer in a toeprinting buffer at 90 °C for 1 min, put on ice for 1 min, and then add MgCl2 to a 10 mM final concentration. Leave for refolding/annealing at 20 °C for 15 min. The final volume of the reaction mixture is 20 μL.
Mix 33,983 cpm/μL of mRNA-Toe primer with 0.025 μM, 0.05 μM, and 0.2 μM 30S small ribosomal subunits. Incubate at 37 °C for 10 min and with 0.1 μM itRNA for an additional 5 min (final MgCl2 concentration should be 8 mM). The final volume of the reaction mixture is 13 μL.
Incubate obtained samples with 0.3 U/μL of AMV reverse transcriptase and 0.3 mM dNTPs in AMV buffer for 30 min at 37 °C, extract with phenol/chloroform, and resuspend in urea blue buffer.
For sequencing lanes, incubate 0.33 μM mRNA with 135,932 cpm/μL Toe primer, 1.7 μM nucleotides, and 0.27 U/μL AMV reverse transcriptase in AMV buffer at 37 °C for 30 min.
Add 237 mM KOH, 26 mM tris-HCl pH 8, 0.26 % SDS, and 3.9 mM EDTA to the samples for the sequencing lanes and incubate at 90 °C for 3 min and at 37 °C for 1 h. Precipitate RNA in 0.3 M sodium acetate and 100% cold ethanol and resuspend in urea blue buffer.
Migrate both samples on a 10%, 40 cm, 0.5 mm denaturing polyacrylamide gel.
Obtain the data by autoradiography (Figure 2A and 2B).
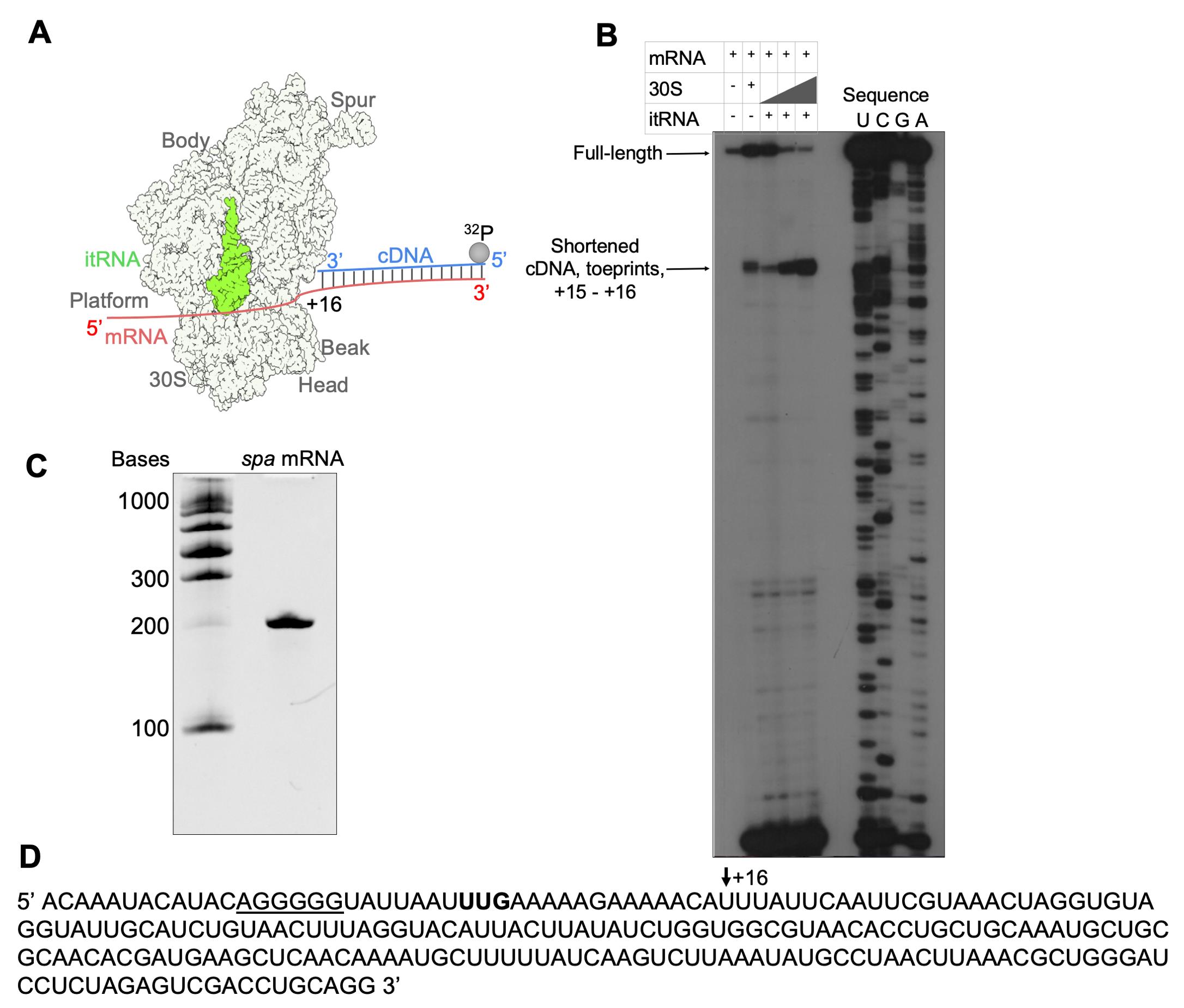
Figure 2. Toeprinting assay of the 30S–mRNA–itRNA complex. (A) Scheme of the toeprinting assay. (B) Toeprint analysis of the 30S–mRNA–itRNA complex formation with increasing 30S concentration. (C) The intactness of mRNA was checked by polyacrylamide gel in denaturing conditions. (D) Sequence of spa mRNA with the start codon in bold and Shine–Dalgarno sequence underlined; the +16 nucleotide is shown by the black arrow.
30S–mRNA complex preparation for cryo-EM
Combine 450 nM of spa mRNA with 90 nM of 30S in a storage buffer, apply immediately on the Quantifoil R2/2300-mesh holey carbon cryo-EM grids, and plunge-freeze in liquid ethane using a Vitrobot Mark IV device (4 °C and 100% humidity) under the following conditions: 4 μL of complex on the grid, 3 s blotting time, blot force of 5, and waiting time of 30 s.
Clip the grids into the cartridges if you intend to use an Autoloader equipped microscope.
Keep in liquid nitrogen until data acquisition.
Data acquisition and analysis
Collect the data on a 200 kV Talos Arctica instrument with a Falcon III direct electron detector under the focus range of -0.5 to -2.7 μm with a 0.2 step.
Note: For our purposes, the 120,000× magnification was used, yielding a pixel size of 1.24 Å.
Transfer the obtained data to the workstation or cluster for data processing.
Use the RELION 3.0 software for data processing (Zivanov et al., 2018).
Perform drift and gain correction and dose weighting with MotionCor2 (Zheng et al., 2017).
Use a dose-weighted average image of the whole stack to determine the contrast transfer function with the software Gctf (Zhang et al., 2016).
Pick the particles using a Laplacian of Gaussian function (min diameter 180 Å, max diameter 290 Å).
Extract the particles with a box size of 270 pixels and divide into several classes for 3D classification.
Merge classes (if necessary), extract the original size, and continue with 3D refinement.
The obtained 30S–mRNA structure has been refined to an overall 3.6 Å resolution with individual focused refinement of the head and the body leading to the resolution of 3.6 and 3.4 Å, respectively (Figure 3B and 3C).
Estimate the local resolution in RELION 3.0 (Zivanov et al., 2018).
Analyze structures in Chimera (Pettersen et al., 2004) or ChimeraX (Goddard et al., 2018) (Figure 3A).
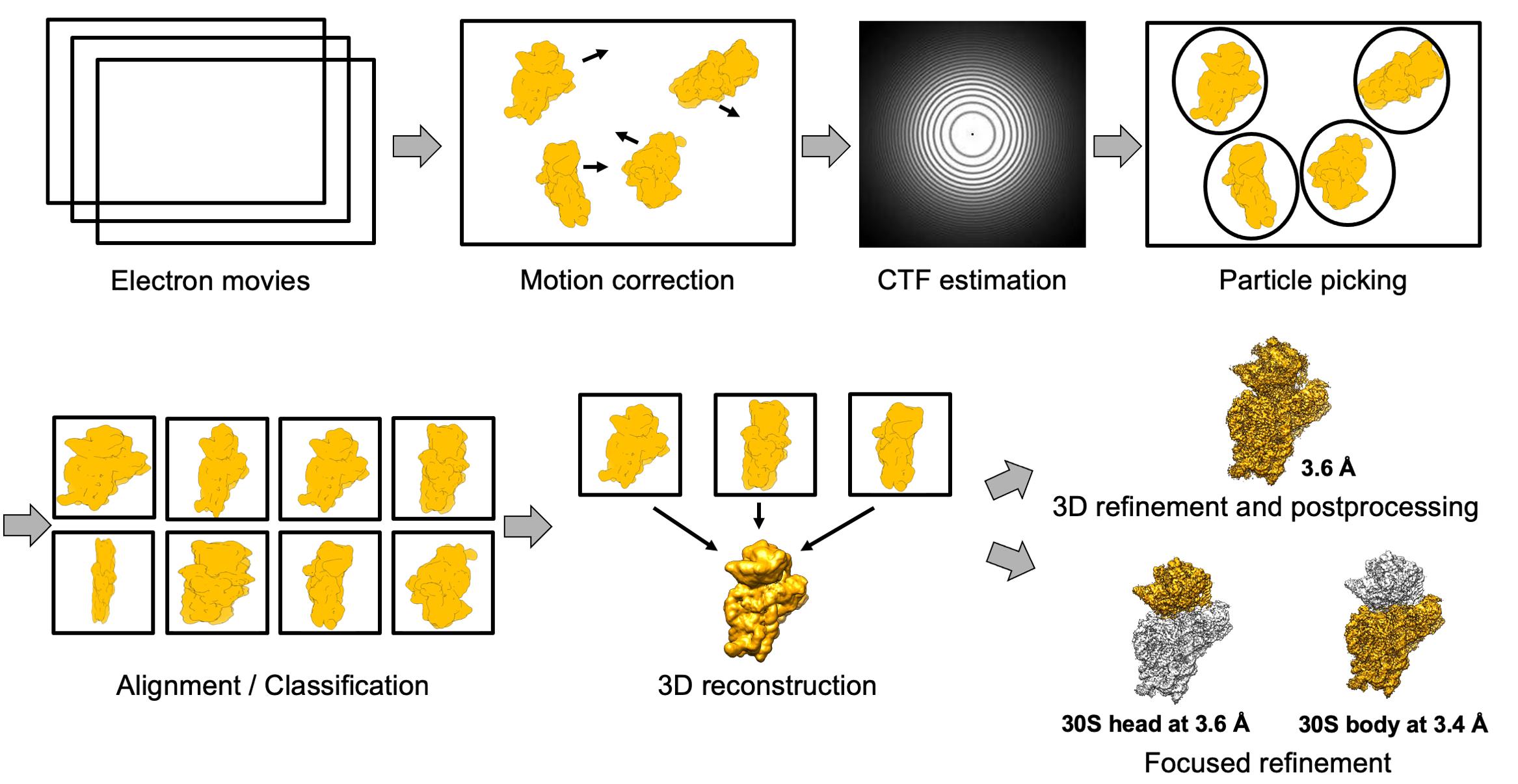
Figure 3. Scheme of the cryo-EM data processing. Electron movies were obtained from sample collection on the Talos microscope. After motion correction and contrast transfer function (CTF) estimation, the particles were picked and extracted with a minimum diameter of 180 Å and maximum diameter of 290 Å. The next steps were 2D and 3D classification, followed by 3D refinement and post-processing. The final 30S–mRNA structure was obtained at 3.6 Å with focus refinement on the head and body at 3.4 and 3.6 Å, respectively.
Recipes
70S ribosome purification
Buffer A
100 mM NH4Cl
21 mM Mg acetate
20 mM HEPES-KOH, pH 7.5
1 mM dithiothreitol (DTT)- Buffer B
1.05 M sucrose
0.5 M KCl
10.5 mM Mg acetate
0.5 mM EDTA
20 mM HEPES-KOH, pH 7.5
1 mM DTT - 5× buffer E
500 mM KCl
50 mM Mg acetate
2.5 mM EDTA
50 mM HEPES-KOH, pH 7.5
5 mM DTT - Dissociation buffer
1 mM Mg acetate
30 mM NH4Cl
10 mM HEPES-KOH, pH 7.5
1 mM DTT
30S subunit purification
30S storage buffer
10 mM Mg acetate
30 mM NH4Cl
10 mM HEPES-KOH, pH 7.5
1 mM DTT
2.5 mM spermidine
mRNA purification
Buffer ELU
16% (v/v) phenol pH 4.5–5.0
50 mM ammonium acetate
1 mM EDTA, pH 8.0
30S and mRNA quality analysis
4% stacking gel
4% (v/v) ROTIPHORESE® NF-acrylamide/bis-solution 30 (37.5:1)
125 mM tris-HCl, pH 6.8
0.1% (w/v) sodium dodecyl sulfate
0.1% (v/v) ammonium persulfate
1/500 V TEMED
15% resolving gel
15% (v/v) ROTIPHORESE® NF-acrylamide/bis-solution 30 (37.5:1)
375 mM tris-HCl, pH 8.8
0.1% (w/v) sodium dodecyl sulfate
0.1% (v/v) ammonium persulfate
1/1000 V TEMED
4× blue buffer
0.25 M tris-HCl, pH 6.8
9.2% (w/v) sodium dodecyl sulfate
40% (v/v) glycerol
0.1% (w/v) bromophenol blue
100 mM DTT
10× running buffer
30 g tris base
144 g glycine
10 g sodium dodecyl sulfate in 1,000 mL of mQ water, pH 8.3
6% polyacrylamide gel
6% (v/v) ROTIPHORESE® NF-acrylamide/bis-solution 40 (19:1)
1× TBE
8 M urea
10× TBE
108 g tris base
55 g boric acid
40 mL 0.5 M EDTA, pH 8.0 in 1,000 mL of mQ water, pH 8.3
1× TBE is a 1:10 dilution of the 10× TBE (from step 6)
Toeprinting assay
10× toeprinting buffer
100 mM tris-HCl, pH 7.5
300 mM KCl
5 mM DTT
10% polyacrylamide gel
10% (v/v) acrylamide/bis-solution 40 (19:1)
1× TBE
8 M urea
Urea blue buffer
0.025% bromophenol blue
xylene cyanol in 7.6 M urea
Acknowledgments
This work, of the Interdisciplinary Thematic Institute IMCBio+, as part of the ITI 2021-2028 program of the University of Strasbourg, CNRS, and Inserm, was supported by the CNRS, by the Agence Nationale de la Recherche (ANR, grant ANR-21-CE12-0030-01 (SM), IdEx Unistra (ANR-10-IDEX-0002), SFRI-STRAT’US project (ANR 20-SFRI-0012) and EUR IMCBio (ANR-17-EURE-0023) under the framework of the French Investments for the Future Program. We thank Dr. Marat Yusupov, Dr. Yaser Hashem, Dr. Pascale Romby, and Dr. Angelita Simonetti for useful discussions and critical advice, and Dr. Yaser Hashem and Dr. Armel Bézault for help in Data acquisition on the Talos Arctica microscope.
This protocol was adapted from Belinite et al. (2021).
Competing interests
The authors declare no competing interests.
References
- Agirrezabala, X., Lei, J., Brunelle, J. L., Ortiz-Meoz, R. F., Green, R. and Frank, J. (2008). Visualization of the hybrid state of tRNA binding promoted by spontaneous ratcheting of the ribosome. Mol Cell 32(2): 190-197.
- Akanuma, G. (2021). Diverse relationships between metal ions and the ribosome. Biosci Biotechnol Biochem 85(7): 1582-1593.
- Bartetzko, A. and Nierhaus, K. H. (1988). Mg2+/NH4+/polyamine system for polyuridine-dependent polyphenylalanine synthesis with near in vivo characteristics. Methods Enzymol 164: 650-658.
- Belinite, M., Simonetti, A., Marzi, S., Khusainov, I., Romby, P., Yusupov, M. and Hashem, Y. (2018). Initiation translation complex of Staphylococcus aureus ribosome. FEBS OpenBio 8: 433.
- Belinite, M., Khusainov, I., Soufari, H., Marzi, S., Romby, P., Yusupov, M. and Hashem, Y. (2021). Stabilization of Ribosomal RNA of the Small Subunit by Spermidine in Staphylococcus aureus. Front Mol Biosci 8.
- Benito, Y., Kolb, F. A., Romby, P., Lina, G., Etienne, J. and Vandenesch, F. (2000). Probing the structure of RNAIII, the Staphylococcus aureus agr regulatory RNA, and identification of the RNA domain involved in repression of protein A expression. RNA 6(5): 668-679.
- Cohen, S. S. (1971). Introduction to the Polyamines. Englewood cliffs, New Jersey: Prentice-Hall.
- Cohen, S. S. and Lichtenstein, J. (1960). Polyamines and ribosome structure. J Biol Chem 235: 2112-2116.
- Cressey, D. and Callaway, E. (2017). Cryo-electron microscopy wins chemistry Nobel. Nature 550(7675): 167.
- Echandi, G. and Algranati, I. D. (1975). Defective 30S ribosomal particles in a polyamine auxotroph of Escherichia coli. Biochem Biophys Res Commun 67(3): 1185-1191.
- Fahnestock, S. R. (1977). Reconstitution of active 50 S ribosomal subunits from Bacillus licheniformis and Bacillus subtilis. Arch Biochem Biophys 182(2): 497-505.
- Fechter, P., Chevalier, C., Yusupova, G., Yusupov, M., Romby, P. and Marzi, S. (2009). Ribosomal initiation complexes probed by toeprinting and effect of trans-acting translational regulators in bacteria. Methods Mol Biol 540: 247-263.
- Frank, J., Gao, H., Sengupta, J., Gao, N. and Taylor, D. J. (2007). The process of mRNA-tRNA translocation. Proc Natl Acad Sci U S A 104(50): 19671-19678.
- Goddard, T. D., Huang, C. C., Meng, E. C., Pettersen, E. F., Couch, G. S., Morris, J. H. and Ferrin, T. E. (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27(1): 14-25.
- Gordon, J. and Lipmann, F. (1967). Role of divalent ions in poly U-directed phenylalanine polymerization. Journal of Molecular Biology 23(1): 23-33.
- Hamana, K. and Satake, S. (1995). ABSENCE OF CELLULAR POLYAMINES IN GRAM-POSITIVE ANAEROBIC COCCI AND LACTIC ACID BACTERIA. J Gen Appl Microbiol 41(2): 159-163.
- Hardy, S. J. and Turnock, G. (1971). Stabilization of 70S ribosomes by spermidine. Nat New Biol 229(1): 17-19.
- Herbst, E. J., Weaver, R. H. and Keister, D. L. (1958). The gram reaction and cell composition: Diamines and polyamines. Arch Biochem Biophys 75(1): 171-177.
- Hershko, A., Amoz, S. and Mager, J. (1961). Effect of polyamines and divalent metals on in vitro incorporation of amino acids into ribonucleoprotein particles. Biochem Biophys Res Commun 5: 46-51.
- Hetrick, B., Khade, P. K., Lee, K., Stephen, J., Thomas, A. and Joseph, S. (2010). Polyamines accelerate codon recognition by transfer RNAs on the ribosome. Biochemistry 49(33): 7179-7189.
- Huntzinger, E., Boisset, S., Saveanu, C., Benito, Y., Geissmann, T., Namane, A., Lina, G., Etienne, J., Ehresmann, B., Ehresmann, C., et al. (2005). Staphylococcus aureus RNAIII and the endoribonuclease III coordinately regulate spa gene expression. The EMBO Journal 24(4): 824-835.
- Hussain, T., Llácer, J. L., Wimberly, B. T., Kieft, J. S. and Ramakrishnan, V. (2016). Large-Scale Movements of IF3 and tRNA during Bacterial Translation Initiation. Cell 167(1): 133-144.e113.
- Igarashi, K., Hikami, K., Sugawara, K. and Hirose, S. (1973). Effect of polyamines on polypeptide synthesis in rat liver cell-free system. Biochim Biophys Acta 299(2): 325-330.
- Igarashi, K. and Kashiwagi, K. (2018). Effects of polyamines on protein synthesis and growth of Escherichia coli. J Biol Chem 293(48): 18702-18709.
- Igarashi, K., Kishida, K. and Hirose, S. (1980). Stimulation by polyamines of enzymatic methylation of two adjacent adenines near the 3' and end of 16S ribosomal RNA of Escherichia coli. Biochem Biophys Res Commun 96(2): 678-684.
- Jahagirdar, D., Jha, V., Basu, K., Gomez-Blanco, J., Vargas, J. and Ortega, J. (2020). Alternative conformations and motions adopted by 30S ribosomal subunits visualized by cryo-electron microscopy. RNA 26(12): 2017-2030.
- Khusainov, I., Vicens, Q., Bochler, A., Grosse, F., Myasnikov, A., Ménétret, J. F., Chicher, J., Marzi, S., Romby, P., Yusupova, G., et al. (2016). Structure of the 70S ribosome from human pathogen Staphylococcus aureus. Nucleic Acids Res 44(21): 10491-10504.
- Kim, H. D., Puglisi, J. D. and Chu, S. (2007). Fluctuations of Transfer RNAs between Classical and Hybrid States. Biophys J 93(10): 3575-3582.
- Konecki, D., Kramer, G., Pinphanichakarn, P. and Hardesty, B. (1975). Polyamines are necessary for maximum in vitro synthesis of globin peptides and play a role in chain initiation. Arch Biochem Biophys 169(1): 192-198.
- Martin, R. G. and Ames, B. N. (1962). THE EFFECT OF POLYAMINES AND OF POLY U SIZE ON PHENYLALANINE INCORPORATION. Proc Natl Acad Sci U S A 48(12): 2171-2178.
- McMurry, L. M. and Aglranati, I. D. (1986). Effect of polyamines on translation fidelity in vivo. Eur J Biochem 155(2): 383-390.
- Nakane, T., Kotecha, A., Sente, A., McMullan, G., Masiulis, S., Brown, P. M. G. E., Grigoras, I. T., Malinauskaite, L., Malinauskas, T., Miehling, J., et al. (2020). Single-particle cryo-EM at atomic resolution. Nature 587(7832): 152-156.
- Nierhaus, K. H. (2014). Mg2+, K+, and the ribosome. J Bacteriol 196(22): 3817-3819.
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. and Ferrin, T. E. (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25(13): 1605-1612.
- Rheinberger, H.-J. and Nierhaus, K. H. (1987). The Ribosomal E Site at Low Mg2+: Coordinate Inactivation of Ribosomal Functions at Mg2+ Concentrations Below 10 mM and its Prevention by Polyamines. J Biomol Struct Dyn 5(2): 435-446.
- Rosenthal, S. M. and Dubin, D. T. (1962). Metabolism of polyamines by Staphylococcus. J Bacteriol 84(4): 859-863.
- Rozov, A., Khusainov, I., El Omari, K., Duman, R., Mykhaylyk, V., Yusupov, M., Westhof, E., Wagner, A. and Yusupova, G. (2019). Importance of potassium ions for ribosome structure and function revealed by long-wavelength X-ray diffraction. Nat Commun 10(1): 2519.
- Stahli, C. and Noll, H. (1977). Structural dynamics of bacterial ribosomes. Molecular and General Genetics MGG 153(2): 159-168.
- Stevens, L. (1969). The binding of spermine to the ribosomes and ribosomal ribonucleic acid from Bacillus stearothermophilus. Biochem J 113(1): 117-121.
- Takeda, Y. (1969). Polyamines and protein synthesis. I. The effect of polyamines on cell free polyphenylalanine synthesis in Escherichia coli. J Biochem 66(3): 345-349.
- Turnock, G. and Birch, B. (1973). Binding of putrescine and spermidine to ribosomes from Escherichia coli. Eur J Biochem 33(3): 467-474.
- Weiss, R. L., Kimes, B. W. and Morris, D. R. (1973). Cations and ribosome structure. 3. Effects on the 30S and 50S subunits of replacing bound Mg2+ by inorganic cations. Biochemistry 12(3): 450-456.
- Weiss, R. L. and Morris, D. R. (1970). The inality of polyamines to maintain ribosome structure and function. Biochim Biophys Acta 204(2): 502-511.
- Wohlgemuth, I., Pohl, C. and Rodnina, M. V. (2010). Optimization of speed and accuracy of decoding in translation. Embo j 29(21): 3701-3709.
- Zhang, K. (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193(1): 1-12.
- Zhang, W., Dunkle, J. A. and Cate, J. H. (2009). Structures of the ribosome in intermediate states of ratcheting. Science 325(5943): 1014-1017.
- Zheng, S. Q., Palovcak, E., Armache, J.-P., Verba, K. A., Cheng, Y. and Agard, D. A. (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Methods 14(4): 331-332.
- Zivanov, J., Nakane, T., Forsberg, B. O., Kimanius, D., Hagen, W. J., Lindahl, E. and Scheres, S. H. (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7: e42166.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Belinite, M., Khusainov, I. and Marzi, S. (2022). Staphylococcus aureus 30S Ribosomal Subunit Purification and Its Biochemical and Cryo-EM Analysis. Bio-protocol 12(20): e4532. DOI: 10.21769/BioProtoc.4532.
Category
Biochemistry > Protein > Isolation and purification
Biophysics > Microscopy > Cryogenic microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.