- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Tissue-Specific, Genome-wide Mapping of R-loops in Drosophila Using MapR
Published: Vol 12, Iss 18, Sep 20, 2022 DOI: 10.21769/BioProtoc.4516 Views: 2190
Reviewed by: Giusy TornilloMario RuizNingfei An
Original research article
The authors used this protocol in:

Feb 2022
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
R-loops, or RNA:DNA hybrids, are structures that arise co-transcriptionally when a nascent RNA hybridizes back with the template ssDNA, leading to a displaced ssDNA. Because accumulation of R-loops can lead to genomic instability and loss of cellular homeostasis, it is important to determine the genome-wide distribution of R-loops in different physiological conditions. Current R-loop mapping strategies are based on R-loop enrichment—mediated by the S9.6 antibody, such as DRIP-seq, or by the exonuclease RNase H1, such as MapR—or the latest R-loop CUT&Tag, based on an artificial R-loop sensor derived from an RNase H1 sub-domain. Because some of these techniques often require high input material or expensive reagents, we sought to apply MapR, which does not require expensive reagents and has been shown to be compatible with low input samples. Importantly, we demonstrate that incorporation of improved CUT&RUN steps into the MapR protocol yields R-loop-enriched DNA when using low input Drosophila nuclei.
Graphical abstract:

Workflow for mapping tissue-specific, genome-wide R-loops in Drosophila. Purify GST-tagged and catalytically inactive RNase H1 tethered MapR enzymes, GST-ΔRH-MNase, and GST-MNase, from transformed E. coli. Perform tissue-specific nuclei immuno-enrichment from UAS-EGFP.KASH-Msp300 Drosophila using magnetic bead–bound green fluorescent protein (GFP) antibody. Incubate isolated nuclei with MapR enzymes and activate MNase DNA cleavage with low salt/high calcium buffers. Purify released, R-loopenriched DNA fragments and generate sequencing-ready libraries. Align MapR data to reference genome and compare R-loop enrichment peaks in genome browser.
Background
R-loops are RNA:DNA hybrids that mostly form during transcription as a result of a nascent RNA strand annealing to a template DNA strand while misplacing a single-stranded DNA. R-loops play an important role in biological processes such as class switch recombination and mitochondrial replication. However, persistent or un-scheduled R-loop formation is a major source of spontaneous DNA damage that can lead to genome instability, one of the key hallmarks of aging (García-Muse and Aguilera, 2019). R-loop resolution is particularly important for neuronal function, as mutations in the R-loop processing factors are associated with neurodegenerative disorders such as Aicardi–Goutières syndrome, ataxia with oculomotor apraxia, and amyotrophic lateral sclerosis (Becherel et al., 2015; Lim et al., 2015; Salvi and Mekhail, 2015). Strategies to map R-loops are based on using either the S9.6 antibody, the RNase H1 enzyme, or more recently, the RNase H1–derived N-terminal hybrid-binding domain (HBD) fused to a Tn5 (Wang et al., 2021). However, the majority of these methods can require high amounts of starting material, or extensive and expensive protocols.
Here, we modified a recently published method, called MapR, that uses the catalytically inactive ribonuclease H1 (RNase H1) enzyme to enrich for R-loops. MapR, which is based on CUT&RUN, utilizes an RNase H1 tethered to a micrococcal nuclease (MNase) for cleavage and release of R-loop-enriched DNA (Yan and Sarma, 2020). Because using the traditional MapR protocol with low input Drosophila samples led to over-digested DNA (Jauregui-Lozano et al., 2022), we adapted the MapR protocol with steps from an improved published CUT&RUN protocol, which incorporates low salt, high calcium buffers to decrease MNase activation time, as well as background binding (Meers et al., 2019). The main advantage of this approach is the ability to map R-loops where starting material is very limited, such as specific tissues or cells from the whole animal. Using this method, we show that Drosophila melanogaster photoreceptor (PR) neurons accumulate R-loops during aging, which correlates with decreased expression of genes with neuronal function and decreased visual function (Jauregui-Lozano et al., 2022). Thus, studies that combine cell-specific approaches provide us with a unique and powerful tool to map R-loops genome-wide and study R-loop-associated toxicity in the context of neuronal aging.
Materials and Reagents
5 or 15 mL conical tube
40 µm cell strainer (Corning, catalog number: 431750)
Slide-A-LyzerTM G2 dialysis cassettes, 10K MWCO, 3 mL (Thermo Fisher, catalog number: 87730)
PierceTM glutathione magnetic agarose beads (Thermo Fisher, catalog number: 78601). Keep at 4 °C
One Shot BL21 (DE3) chemically competent E. coli (Thermo Fisher, catalog number: C600003). Keep unused E. coli pellets at -80 °C
L-Glutathione reduced (Millipore Sigma, catalog number: G4251-300MG)
HyperPAGE II pre-stained protein marker (Bioline, catalog number: BIO-33066). Keep aliquots at -20 °C
DynabeadsTM protein G for immunoprecipitation (Invitrogen, catalog number: 10003D). Keep at 4 °C
Anti-GFP antibody (Sigma-Aldrich, catalog number: 11814460001). Keep at -20 °C upon resuspension
UAS-EGFP.KASH-Msp300 flies (Bloomington Drosophila Stock Center, BDSC#92580)
Digitonin 5% (Thermo Fisher, catalog number: BN2006)
UltraPureTM dithiothreitol (DTT) 0.1 M solution (Thermo Scientific, catalog number 707265ML)
UltraPureTM ethylenediaminetetraacetic acid, disodium salt, dihydrate (EDTA) (Thermo Fisher, catalog number: 15576028)
UltraPureTM 1 M Tris-HCl, pH 8.0 (Invitrogen, catalog number 15568025)
EGTA, molecular biology grade (Millipore, catalog number: 324626)
cOmpleteTM, mini, EDTA-free protease inhibitor cocktail tablets (Roche, catalog number: 04693159001)
Corning® 100 mL HEPES, liquid 1 M solution (238.3 mg/mL) (Corning, catalog number: 25-060-CI)
IGEPAL® CA-630 (Supelco, catalog number: 56741-50ML-F)
TWEEN® 20 (Sigma-Aldrich, catalog number: P1379)
Coomassie Brilliant Blue R-250 dye (Thermo Scientific, catalog number: 20278)
Acetic acid glacial, ReagentPlus® (Sigma-Aldrich, catalog number A6283)
Methanol, ACS reagent (Sigma-Aldrich, catalog number 179337)
Spermidine (Sigma-Aldrich, catalog number: S022-1G)
RNase A (Thermo Fisher, catalog number: 12091021)
Linear acrylamide (Thermo Fisher, catalog number: AM9520)
RNase H (New England BioLabs, catalog number M0297S). Keep at -20 °C
ChIP DNA Clean & Concentrator (Zymo, catalog number D5205)
QubitTM 1× dsDNA high sensitivity (HS) (Thermo Fisher, catalog number: Q33230)
Note: We recommend using high sensitivity assays since obtained DNA can be low-input and difficult to quantify using traditional reagents.
Ovation® ultralow V2 DNA-Seq library preparation kit (Tecan, catalog number: 0344NB-08)
GST-wash/equilibration buffer (see Recipes)
GST-elution buffer (see Recipes)
Coomassie fixing solution (see Recipes)
Coomassie staining solution (see Recipes)
Coomassie destaining solution (see Recipes)
Homogenization/wash buffer (see Recipes)
Dilution buffer (see Recipes)
Bead washing buffer (see Recipes)
PBST (1×, pH 7.4) (see Recipes)
Dig-wash buffer (see Recipes)
Low salt rinse buffer (see Recipes)
Activation buffer (see Recipes)
EGTA-STOP buffer (1×) (see Recipes)
Equipment
Branson sound amplifier
Agilent TapeStation 4200
WHEATON® Dounce tissue grinder, 1 mL
37 °C incubator with shaking platform (such as Thomas Scientific SCO2W benchtop water jacketed CO2 incubator, catalog number: 1229P58)
Cold (4 °C) centrifuge (such as EppendorfTM, model: Centrifuge 5424 R)
Cold (4 °C) incubator (such as FisherbrandTM mini low temperature refrigerated incubator, catalog number: 15-015-2632)
Tube revolver rotator (such as Thermo ScientificTM tube revolver rotator, catalog number: 88881001)
Magnetic rack (such as Millipore PureProteomeTM magnetic stand, catalog number: LSKMAGS08)
Gel electrophoresis chamber (such as Bio-Rad Mini-PROTEAN® tetra vertical electrophoresis cell, catalog number: 1658025FC)
Software
Genomic alignment of sequencing reads: Bowtie2 (Langmead and Salzberg, 2012)
BAM file processing: Samtools (Li et al., 2009)
Bigwig generation/processing: Deeptools (Ramírez et al., 2014)
Integrative genome browser (Thorvaldsdóttir et al., 2013)
Procedure
E. coli transformation and protein purification
For this protocol, we performed the E. coli transformation and protein overexpression induction as described in the original MapR method paper (Yan and Sarma, 2020). We also described these steps in Jauregui-Lozano et al. (2022).
Transform BL21 DE3 cells using 10 ng of plasmid and following manufacturer’s instructions from New England Biolabs.
Plate transformed cells in LB media plates overnight at 37 °C.
Pick a single colony and grow in liquid LB media supplemented with carbenicillin (1 μg/mL), at 37 °C and 225 rpm overnight.
The next day, add 4.5 mL of liquid culture into 495 mL of liquid LB media supplemented with carbenicillin and incubate for 2–4 h at 37 °C and 225 rpm. After 2 h of incubation, check optical density of an aliquot of liquid culture until it reaches 0.6 as measured with a spectrophotometer.
Induce protein expression by adding IPTG to a final concentration of 0.5 mM.
Incubate IPTG-containing liquid culture for 3 h at 37 °C and 225 rpm.
Prepare fresh GST-wash/equilibration buffer and GST-elution buffer.
Centrifuge bacterial lysate for 10 min at 8,000 × g in a cold centrifuge.
Remove supernatant and resuspend bacterial pellet in 5 mL of cold PBS buffer with cOmpleteTM protease inhibitor.
Sonicate using Branson sound amplifier with amplitude 20% and 5 cycles of 15 s on–45 s off. Make sure that the conical tube containing the bacterial lysate is on ice during sonication.
Centrifuge for 20 min at 4 °C and 13,000 × g.
Transfer soluble lysate to a new 5 or 15 mL conical tube.
Equilibrate 1 mL of PierceTM glutathione magnetic agarose beads GST by adding 1 mL of wash/equilibration buffer.
Vortex for 10 s, place on magnet, remove supernatant, and add 1 mL of wash/equilibration buffer.
Add DTT and EDTA to soluble lysate (final 1 mM for each).
Remove supernatant from beads and transfer the beads to soluble lysate.
Incubate for 1–2 h at 4 °C with constant rotation.
Using magnet, remove supernatant and add 1 mL of wash buffer. Save 20–50 μL of supernatant as “flowthrough” sample, to later use when running the SDS-page.
Incubate at 4 °C with constant rotation.
Remove wash buffer using magnet and repeat wash step two more times.
Remove wash buffer and add 250 μL of GST-elution buffer.
Incubate for 10 min at 4 °C with constant rotation.
Collect eluate #1 [e#1] and repeat elution step two more times.
To each 250 μL eluate, add 41 μL of 7× cOmpleteTM protease inhibitor previously resuspended in GST-elution buffer.
Run SDS-page with flowthrough and eluates to assess purification efficiency (Figure 1).
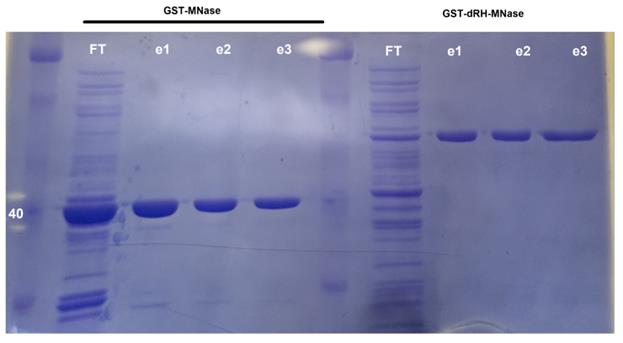
Figure 1. Coomassie-stained SDS page evaluating purification efficiency. Efficiency of GST protein purification was assessed using SDS-page followed by staining with Coomassie Blue. For flowthrough (FT), 20 μg of protein were loaded; for the elution fractions, 2 μg of protein were loaded. GST-ΔRH-MNase will have a size of ~60 kDa, while GST-MNase will have a size of ~40 kDa.After running SDS-page, transfer gel into Coomassie fixing solution for 30 min, and place in a rocker.
Transfer fixed gel into Coomassie staining solution for 1 h and place in a rocker.
Transfer stained gel into Coomassie destaining solution for 20 min and place in a rocker.
Replace Coomassie destaining solution, and place in a rocker.
Repeat steps 28 and 29 two more times (for a total of 1 h 20 min destaining). GST-MNase will have a molecular weight close to 40 kDa, and GST-ΔRH-MNase close to 60 kDa.
Dialyze protein, add glycerol for storing at -20 °C, and quantify protein according to original MapR method (Yan and Sarma, 2020).
Sample preparation
If purifying tissue-specific nuclei from Drosophila, perform nuclei immuno-enrichment as follows: for this protocol, we performed tissue-specific nuclei immunoprecipitation from UAS-EGFP.KASH-Msp300 flies (available at the Bloomington Drosophila Stock Center, see Methods). These flies, when crossed with a Gal4 fly line, will express a GFP anchored to the outer nuclear membrane in a specific tissue. Then, magnetic beads and GFP antibody can be used to purify these nuclei. We have also described these steps (Jauregui-Lozano et al., 2021), and this approach is compatible with different chromatin profiling techniques.
Prepare fresh bead washing buffer, homogenization/wash buffer, and dilution buffer and keep on ice.
Incubate 40 µL of DynabeadsTM in 1 mL of bead washing buffer for 10 min at room temperature (RT) with constant rotation.
Transfer tube to magnetic rack. Invert rack several times to ensure all beads are bound to the magnet. Remove supernatant with pipettor.
Resuspend beads in 1 mL bead washing buffer with 4 µg of anti-GFP antibody.
Incubate beads for 30 min at RT with constant rotation to couple with GFP antibody.
Using magnet, remove supernatant.
Wash beads with 1 mL bead washing buffer for 5 min at RT with constant rotation.
Using magnet, remove supernatant.
Resuspend beads in 0.1% NP-40 homogenization/wash buffer. Mix three parts dilution buffer with one part homogenization/wash buffer to a final concentration of 0.1% NP-40.
Load Dounce homogenizer with 1 mL of homogenization/wash buffer. Keep homogenizer on ice.
Transfer Drosophila samples to homogenizer.
400 fly heads (flash frozen in liquid nitrogen and stored at -80 °C) can be used with 1 mL of homogenization/wash buffer to isolate nuclei from neuronal tissue in the adult head.
Fresh samples such as larval tissue or whole embryos can also be used.
Grind samples with 5 “loose” pestle strokes.
Incubate samples on ice for 5 min.
Grind samples with an additional 5 “loose” pestle strokes followed by 10 “tight” pestle strokes.
Filter fly homogenate using a 40 µm cell strainer.
Add 3 mL of dilution buffer to filtered 1 mL homogenate to a final concentration of 0.1% NP-40.
Evenly split 4 mL of homogenate into four 1.5 mL tubes (1 mL per tube).
Add 50 µL of bead/antibody solution to each tube.
Incubate fly homogenate with bead/antibody solution for 30 min at RT with constant rotation to couple nuclei with the anti-GFP beads.
Using magnet, remove supernatant.
Combine bead-bound nuclei from all four tubes in 1 mL of 0.1% NP-40 homogenization/wash buffer.
Incubate for 5 min at 4 °C with constant rotation.
Using magnet, remove supernatant.
Resuspend bead-bound nuclei in 1 mL of 0.1% NP-40 homogenization/wash buffer and transfer to a fresh 1.5 mL tube.
Incubate for 5 min at 4 °C with constant rotation.
Using magnet, remove supernatant.
Wash bead-bound nuclei in 1 mL of 0.1% NP-40 homogenization/wash buffer for 5 min at 4 °C with constant rotation.
Note: As previously described (Jauregui-Lozano et al., 2021), this method will yield bead-bound nuclei, which is suitable with MapR. Otherwise, purify nuclei using Concanavalin A-coated beads, as described in the original MapR method (Yan and Sarma, 2020).
Modified MapR
Wash bead-bound nuclei with 1 mL of dig-wash buffer.
For every wash step, incubate bead-bound nuclei at 4 °C for 5 min with constant rotation.
Using magnet, remove dig-wash buffer, and repeat wash step two more times.
Resuspend bead-bound nuclei in 150 μL of dig-wash buffer.
Add GST-ΔRH-MNase or GST-MNase to a final concentration of 1 μM.
To calculate protein molarity based on protein concentration, use the formula:
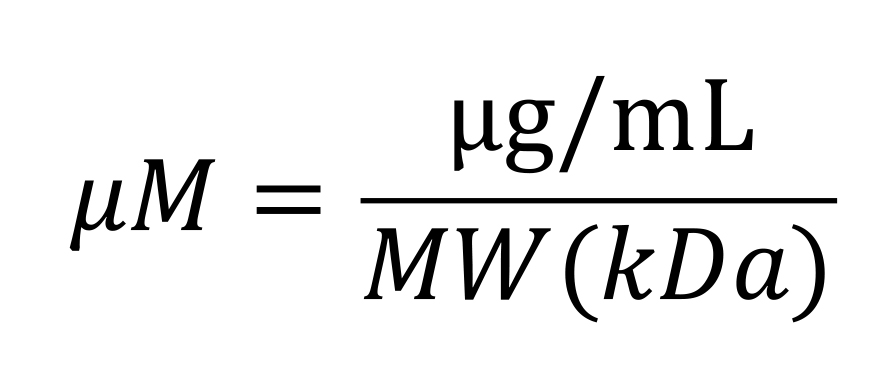
where,
μg/mL is the concentration of protein obtained with traditional protein quantification reagents, such as Bradford Assay or QubitTM protein assay kits (Thermo Fisher).
MW (kDa) is the molecular weight of the protein. GST-MNase has a MW of 44 kDa, and GST-dRH-MNase has a MW of 61.5 kDa.
The Bioline website has an online calculator that performs this calculation: https://www.bioline.com/media/calculator/01_04.html.
Incubate at 4 °C with constant rotation for 1 h.
Wash bead-bound nuclei three times with 500 μL of dig-wash buffer.
Wash bead-bound nuclei one time with low salt rinse buffer.
Resuspend bead-bound nuclei in 200 μL of ice-cold calcium-containing activation buffer, and place on wet ice for 60 s.
Using magnet, remove supernatant and immediately add 150 μL of EGTA-STOP buffer.
Incubate for 30 min at 37 °C.
Using magnet, transfer supernatant to a new tube and extract DNA from the supernatant using ChIP DNA clean & concentrator. The supernatant contains the released R-loop-enriched DNA; make sure you purify DNA from supernatant.
Quantify DNA using Nanodrop or QubitTM DNA reagent.
Use 100 pg–10 ng of purified DNA to make sequencing-ready MapR libraries using Ovation® Ultralow V2 DNA-Seq library preparation kit.
Data analysis
Notes:
In this example, we use the D. melanogaster reference genome “BDGP Release 6 + ISO1 MT/dm6”, or dm6, available at https://hgdownload.soe.ucsc.edu/goldenPath/dm6/bigZips/.
Code that is run in command line starts with $.
Alignment to reference genome
Index reference genome using Bowtie2
$ bowtie2-build --threads [# of threads/cores] [reference genome fasta file] [file name used to name the index files]
Find below a screenshot of what the first 20 lines will look like after running the bowtie2-build command using 64 threads and using a reference genome fasta file named “dm6.fa”. dm6 will be used as file name for index files.
Align raw/clean sequencing reads to reference genome (code is piped to directly generate BAM file rather than SAM).
$ bowtie2 -p [# of threads/cores] --sensitive -x [index specified in step 1] -1 CleanReads_1.fq -2 CleanReads_2.fq | samtools view -@ [# of threads/cores] -bSo Output.bam
Sort BAM file by genomic coordinates (this step is required to generate bigwig files using deepTools)
$ samtools sort -@ [# of threads/cores] -o Output_sorted.bam Output.bam
Bigwig generation for data visualization
-Using deepTools
$ bamCoverage -b BAMfile.bam -o output_file.bw -bs [bin size] -p [# of threads/cores] --normalizeUsing CPM
Genome browser inspection
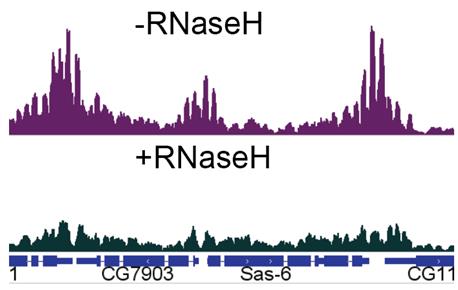
Figure 2. Genome browser inspection of MapR data with or without pre-treatment with RNAse H1. Data was visualized in a genome browser (such as Integrative Genomics Browser, or IGV) using counts-per-million (CPM) normalized bigwig files. In order to compare enrichment, both tracks were scaled to the same values.
Notes
Traditional MapR contains a control for R-loop enrichment, where a fraction of bead-bound nuclei is incubated with GST-MNase. In this experimental condition, the researcher can control for random MNase binding to chromatin or background. Unexpectedly, we were not able to get the MNase control to work. We did not obtain purifiable DNA upon performing the experiment. For quantification of purified DNA, we used the High Sensitivity QubitTM, which allows quantification of small amounts of DNA (picograms). Thus, we performed an additional control, where we treated a fraction of bead-bound nuclei with catalytically active RNAse H1 for three hours, prior to performing MapR.
Analyzed bigwig files used for data visualization were downloaded from Gene Expression Omnibus (GEO), accession code GSE174488. They correspond to MapR from D. melanogaster photoreceptor neurons, as originally described in Jauregui-Lozano et al. (2022).
Recipes
GST-wash/equilibration buffer
Reagent Final concentration Amount Tris (1 M pH 7.5) 125 mM 1,250 µL NaCl (5 M) 150 mM 300 µL DTT (100 mM) 1 mM 100 µL EDTA (500 mM) 1 mM 20 µL H2O
Total
n/a mL
10 mL
GST-elution buffer
Reagent Final concentration Amount Glutathione (reduced) (1 M) 50 mM 500 µL GST-wash/equilibration buffer
Total
n/a 9.5 mL
10 mL
Coomassie fixing solution
Reagent Final concentration Amount Methanol 50% 50 mL Acetic acid, glacial
H2O
Total
10% 10 mL
40 mL
100 mL
Coomassie staining solution
Reagent Final concentration Amount Coomassie Brilliant Blue 0.1 0.1 g Methanol
Acetic acid, glacial
H2O
Total
50%
10%
50 mL
10 mL
40 mL
100 mL
Alternatively, you can make a 10% Coomassie Blue stock solution (1 g Coomassie reagent + 10 mL H2O) and add 1 mL of 10% stock solution into Coomassie staining solution.
Coomassie destaining solution
Reagent Final concentration Amount Methanol 40% 40 mL Acetic acid, glacial
H2O
Total
10% 10 mL
50 mL
100 mL
Homogenization/wash buffer
Reagent Final concentration Amount HEPES (1M, pH 7.5) 40 mM 400 µL KCl (1M) 120 mM 1.2 mL NP-40 (10%) 0.4% 400 µL H2O n/a 8 mL Total 10 mL Dilution buffer
Reagent Final concentration Amount HEPES (1 M, pH 7.5) 40 mM 400 µL KCl (1 M) 120 mM 1.2 mL H2O n/a 8.4 mL Total 10 mL Bead washing buffer
Reagent Final concentration Amount MgCl2 2.5 mM 25 µL PBST (1×) n/a 9.975 mL Total 10 mL PBST (1×, pH 7.4)
Reagent Final concentration Amount PBS (10×) 1× 5 mL Tween 0.1% 50 µL H2O n/a 44.950 mL Total 50 mL Dig-wash buffer
Reagent Final concentration Amount HEPES (1 M, pH 7.5) 20 mM 1 mL Spermidine (6.4 M, pH 7.0) 0.5 mM 3.9 µL NaCl (5 M) 150 mM 1.5 mL H2O
Total
n/a 47.5 mL
50 mL
Low salt rinse buffer
Reagent Final concentration Amount HEPES (1 M, pH 7.5) 20 mM 200 µL Spermidine (6.4 M) 0.5 mM 0.8 µL Digitonin (5%) 0.05% 100 µL H2O n/a 9.62 mL Total n/a 10 mL Activation buffer
Reagent Final concentration Amount HEPES (1 M, pH 7.5) 3.5 mM 14 µL CaCl (100 mM) 10 mM 400 µL Digitonin (5%) 0.05% 40 µL H2O n/a 3.46 mL Total n/a 4 mL EGTA-STOP buffer (1×)
Reagent Final concentration Amount NaCl (5M) 170 mM 34 µL EGTA (100 mM) 20 mM 200 µL Digitonin (5%) 0.05% 10 µL RNase A (10 µg/µL) 0.05% 5 µL Linear acrylamide (5 µg/µL) 0.025% 5 µL H2O n/a 746 µL Total n/a 1 mL
Acknowledgments
This research was supported by the National Institute of Health – National Eye Institute grant R21EY031024. This protocol describes a methodology used in the research paper (DOI: 10.1111/acel.13554) by Jauregui-Lozano et al. (2022).
Competing interests
The authors declare no competing financial interests.
References
- Becherel, O. J., Sun, J., Yeo, A. J., Nayler, S., Fogel, B. L., Gao, F., Coppola, G., Criscuolo, C., De Michele, G., Wolvetang, E. et al. (2015). A new model to study neurodegeneration in ataxia oculomotor apraxia type 2. Hum Mol Genet 24(20): 5759-5774.
- García-Muse, T. and Aguilera, A. (2019). R Loops: From Physiological to Pathological Roles. Cell 179(3): 604-618.
- Jauregui-Lozano, J., Bakhle, K. and Weake, V. M. (2021). In vivo tissue-specific chromatin profiling in Drosophila melanogaster using GFP-tagged nuclei. Genetics 218(3).
- Jauregui-Lozano, J., Escobedo, S., Easton, A., Lanman, N. A., Weake, V. M. and Hall, H. (2022). Proper control of R-loop homeostasis is required for maintenance of gene expression and neuronal function during aging. Aging Cell 21(2): e13554.
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357-359.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Lim, Y. W., Sanz, L. A., Xu, X., Hartono, S. R. and Chedin, F. (2015). Genome-wide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi-Goutieres syndrome. Elife 4: e08007.
- Meers, M. P., Bryson, T. D., Henikoff, J. G. and Henikoff, S. (2019). Improved CUT&RUN chromatin profiling tools. Elife 8: e46314.
- Ramírez, F., Dundar, F., Diehl, S., Gruning, B. A. and Manke, T. (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42(Web Server issue): W187-191.
- Salvi, J. S. and Mekhail, K. (2015). R-loops highlight the nucleus in ALS. Nucleus 6(1): 23-29.
- Thorvaldsdóttir, H., Robinson, J. T. and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14(2): 178-192.
- Wang, K., Wang, H., Li, C., Yin, Z., Xiao, R., Li, Q., Xiang, Y., Wang, W., Huang, J., Chen, L., et al. (2021). Genomic profiling of native R loops with a DNA-RNA hybrid recognition sensor. Sci Adv 7(8): eabe3516.
- Yan, Q. and Sarma, K. (2020). MapR: A Method for Identifying Native R-Loops Genome Wide. Curr Protoc Mol Biol 130(1): e113.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Jauregui-Lozano, J., Cottingham, K. and Hall, H. (2022). Tissue-Specific, Genome-wide Mapping of R-loops in Drosophila Using MapR. Bio-protocol 12(18): e4516. DOI: 10.21769/BioProtoc.4516.
Category
Bioinformatics and Computational Biology
Molecular Biology > DNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.