- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

An Improved EMSA-based Method to Prioritize Candidate cis-REs for Further Functional Validation
Published: Vol 12, Iss 8, Apr 20, 2022 DOI: 10.21769/BioProtoc.4397 Views: 2920
Reviewed by: Alessandro DidonnaJoyce ChiuAbhilash Padavannil
Original research article
The authors used this protocol in:

Jan 2022
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Cells are the complex product of gene expression programs that involve the coordinated transcription of thousands of genes controlled by cis-regulatory elements (cis-REs). Therefore, identification of cis-REs is the key to decipher the mechanisms underlying the regulation of gene expression. Here, we describe a simple and time-effective protocol of fine-mapping cis-REs by using an electrophoresis mobility shift assay (EMSA)-based, in vitro, high-throughput (HTP) technique called regulatory element-sequencing (Reel-seq). Reel-seq can be applied to identify cis-REs at a high resolution and sensitivity over large genome regions, in a systematic and continuous manner. It can be used to prioritize candidate cis-REs as a complement to the existing approaches, such as massive parallel reporter assay (MPRA), chromatin immunoprecipitation DNA-sequencing (ChIP-seq), and the assay for transposase-accessible chromatin sequencing (ATAC-seq).
Graphical abstract:
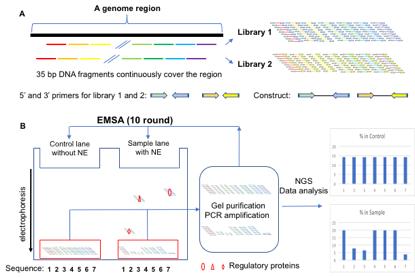
Generation of the Reel-seq Library 1 and 2 (A) and identification of cis-REs by an electrophoresis mobility shift assay (EMSA)-based Reel-seq screen (B). NE: nuclear extract; NGS: next generation sequencing.
Background
Genes make proteins through two steps: transcription and translation. However, specific expression of genes in particular cells is largely regulated by transcription, which is, in general, controlled by millions of promoters and enhancers. Both promoters and enhancers contain short DNA sequences that serve as cis-regulatory elements (cis-Res), which regulate gene transcription by recruiting regulatory proteins (Mikhaylichenko et al., 2018; Schoenfelder and Fraser, 2019). In addition, gene transcription can also be regulated on the epigenetic level, by opening the chromatin structure to alter DNA accessibility, thereby making the underlying cis-REs accessible to regulatory proteins (Crawford et al., 2006; Angeloni and Bogdanovic, 2019; Zhang et al., 2021). Therefore, identification of cis-REs is the key for understanding of biology.
To date, a number of high-throughput (HTP) approaches have been developed to identify cis-REs. Some identify cis-REs directly, by determining the functional impact of each DNA fragment using HTP reporter assays, such as massive parallel reporter assay (MPRA) (Ulirsch et al., 2016; Tewhey et al., 2016), multiplexed editing regulatory assay (MERA) (Rajagopal et al., 2016), clustered regularly interspaced short palindromic repeats interference, flow cytometry and RNA fluorescence in situ hybridization (CRISPRi-FlowFISH) (Fulco et al., 2019), and cis-regulatory element scan by tiling-deletion and sequencing (CREST-seq) (Diao et al., 2017). Some use antibodies specifically against transcriptional factors or epigenetic markers, to identify cis-REs based on protein-DNA interactions, such as chromatin immunoprecipitation DNA-sequencing (ChIP-seq) (Park et al., 2009), and chromatin immunocleavage sequencing (ChIC-seq) (Schmid et al., 2004). Others use the open chromatin regions that are indicative of active regulatory sites, to identify cis-Res, such as DNase-seq (Song and Crawford, 2010), and the assay for transposase-accessible chromatin sequencing (ATAC-seq) (Buenrostro et al., 2015). While all these approaches have their advantages and specific applications in identifying cis-REs, the technical complexity of these methods limits their routine application. In addition, some of these approaches do not have the high resolution to define each single cis-RE, whereas others do not have enough sensitivity and fidelity to reveal cis-REs. Recently, we developed a simple and time-effective technique that not only complements the pre-existing approaches, but can also be used to prioritize candidate cis-REs (Wu et al., 2022). We refer to this technique as regulatory element-sequencing (Reel-seq). Reel-seq is an in vitro HTP screen technique, designed to identify cis-REs based on protein-DNA interaction that is detected by an electrophoresis mobility shift assay (EMSA). For screening, a Reel-seq construct will be engineered by placing a 35-bp DNA fragment between two primers for PCR amplification, as well as for next-generation sequencing (see Graphic abstract A). A Reel-seq library (e.g., Library 1) will be generated by hundreds of thousands of these constructs synthesized by massive parallel oligonucleotide synthesis, so that an entire DNA region will be reconstructed. To cover the break points between all 35-bp fragments, another Reel-seq library (e.g., Library 2) will be generated in the same fashion, except that each 35-bp fragment in this library will bridge the break point of the two contiguous 35-bp fragments in Library 1. For screening, each Reel-seq library will be first mixed with and without nuclear extract (NE, source of regulatory proteins) isolated from relevant types of cells or tissues, and then analyzed on a TBE native gel. After electrophoresis, unshifted bands from both the buffer-treated control and the NE-treated samples will be isolated, and amplified by PCR. Amplified DNA will then be used for another round of gel shifting. In total, ten rounds of EMSA will be performed so that, if a DNA fragment in the library is a cis-RE, it will be shifted in each round of the gel shift assay. After ten screening rounds, a cis-RE can be identified by its decreased percentage in the unshifted DNA library (see Graphic abstract B). Since Reel-seq is an EMSA-based assay, it is highly specific and sensitive in identifying cis-REs. Moreover, since Reel-seq identifies cis-REs at a high resolution, within a defined 35-bp fragment, each identified cis-RE can be used as a “bait” to pull down the regulatory proteins, by using flanking restriction enhanced DNA pulldown-mass spectrometry (FREP-MS) (Li et al., 2018; Wu et al., 2022), another technique recently developed in our lab. Here, we present the detailed protocol of Reel-seq, for the convenient application of this method to identify cis-REs.
Materials and Reagents
150 × 21 mm Dish, NunclonTM Delta (Thermo Fisher, catalog number: 168381)
AccuPrimeTM Taq DNA Polymerase System (Thermo Fisher, catalog number: 12339016. Upon receipt, store at -20°C)
Gel Loading Dye, Blue (6×) [New England Biolabs, catalog number: B7021S. Upon receipt, store at room temperature (RT)]
5% Mini-PROTEAN® TBE Gel, 10 well (Bio-Rad, catalog number: 4565014. Upon receipt, store at 4°C)
PierceTM 10× TBE Buffer (Thermo Fisher, catalog number: 28355. Upon receipt, store at RT)
TE buffer (Thermo Fisher, catalog number: 12090015. Upon receipt, store at RT)
10,000× GelStarTM Nucleic Acid Gel Stain (Lonza, catalog number: 50535. Upon receipt, store at -20°C)
Low molecular weight DNA ladder (New England Biolabs, catalog number: N3233S. Upon receipt, store at -20°C)
NE-PERTM Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher, catalog number: 78833. Upon receipt, store at 4°C)
Roche cOmplete, Mini Protease Inhibitor Cocktail (25 tablets) (Emerald Scientific, catalog number: 11836153001. Upon receipt, store at 4°C)
LightShiftTM Chemiluminescent EMSA Kit (Thermo Fisher, catalog number: 20184. Upon receipt, store kit components at -20°C, except for the Chemiluminescent reagents, which are stored at 4°C as described by the manufacturer)
Herculase II Fusion DNA Polymerases (Agilent, catalog number: 600677. Upon receipt, store at -20°C)
UltraPureTM Agarose (Thermo Fisher, catalog number: 16500100. Upon receipt, store at RT)
Safe-Red (APPLIED BIOLOGICAL MATERIALS INC, catalog number: G108-R. Upon receipt, store at -20°C)
All primers are purchased from Integrated DNA Technologies (IDT) as a standard formula
GibcoTM DPBS, no calcium, no magnesium (Gibco, catalog number: 14190235. Upon receipt, store at RT)
Primary human arterial ECs (Lonza, catalog number: CC-2535)
EGMTM-2 Endothelial Cell Growth Medium-2 BulletKitTM (Lonza, catalog number: CC-3162)
0. 5× TBE buffer (see Recipes)
1× GelStar (see Recipes)
1× Low Molecular Weight DNA Ladder (see Recipes)
10% TE buffer (see Recipes)
1.5% agarose gel (see Recipes)
Equipment
T100 Thermal Cycler (Bio-Rad, catalog number: 1861096)
PowerPacTM Basic Power Supply (Bio-Rad, catalog number: 1645050)
Mini-PROTEAN® Tetra Vertical Electrophoresis Cell (Bio-Rad, catalog number: 1658003FC)
Eppendorf ThermoMixerTM C (Fisher Scientific, catalog number: 13527550)
Thermo Scientific Savant DNA 120 SpeedVac Concentrator (Thermo Scientific, catalog number: BZ10133848)
BLooKTM LED Transilluminator (GeneDireX, catalog number: BK001)
Implen C40 NanoPhotometer UV/Vis Spectrophotometer Mobile System, 110/220 V (Cole-Parmer, catalog number: UX-83070-62)
Cell scraper (VWR, catalog number: 15621-005)
VWR Ergonomic High-Performance Pipettor set (VWR, catalog number: 89079-970)
Thermo Forma Steri Cycle 370 CO2 Incubator (Marshall Scientific, catalog number: TH-370)
Allegra® X-5 Clinical Benchtop IVD Centrifuge, Beckman Coulter® (VWR, catalog number: 89429-566)
Procedure
A simplified flow chart of Reel-seq is present in Figure 1.

Figure 1. A simplified flow chart of Reel-seq
Construction and generation of DNA library 1 and 2
Download the genome DNA sequence of interest from the NCBI website (https://www.ncbi.nlm.nih.gov).
Scroll down from All Databases to Gene.
Input the name of the gene you are interested in, for example, CDKN2A.
Select the gene in Search results.
Select FASTA under the Genomic regions, transcripts, and products.
Define the region of your sequence, by inputting the nucleotide number on the chromosome your gene is located under Change region shown on the right site of the screen. For example, from 2230356 to 22172015, for the 58 kb CDKN2A region on chromosome 9.
Click Update View, and copy the sequence into word file.
Obtain 35-bp fragments continuously starting from the first base of the genomic sequence for Library 1, as shown in the graphic abstract.
Obtain 35-bp fragments continuously starting from base 18 for Library 2, so that these fragments will cover the break points between each of two 35-bp fragments in Library 1. For a 58 kb CDKN2A/B region, you will have 1668 35-bp fragments for both Libraries 1 and 2.
List the 1668 sequences in an Excel file for each library.
Construct Libraries 1 and 2, by adding the common flanking sequences to each 35-bp fragment in the Excel file, as shown in Table 1.
Table 1. Construction of Reel-seq library
PE_SP Library 1 sequence G3 ACACGCACGATCCGACGGTAGTGT 1–35 bp genomic DNA sequence GGATGACGACGATAAGCTCG new_SP Library 2 sequence 926RR GGTGTGATGCTCGGATCCAGGAAC 18–52 bp genomic DNA sequence GGTAGGACAGTAGTCTCGTG Order Libraries 1 and 2 from commercial vendors.
Note: You can order these two libraries together as two sub-libraries, by adding the common sequence AGGACCGGATCAACT on the 5’ end and CATTGCGTGAACCGA at the 3’ end. These common sequences will be served as adaptors for massive parallel oligonucleotide synthesis.
Separation of library DNA for libraries 1 and 2
Upon receipt of the library DNA, resuspend the synthetic DNA Library using DNase-free water to a final concentration of 100 ng/mL, and keep it as a stock at -20°C.
Dilute the synthetic DNA library with 1 μL stock using DNase-free water, to a final concentration of 5, 10, 20, 50, 100, and 200 pg/μL.
PCR amplify the library DNA, by using primers PE_SP (L1_FP): 5’ACACGCACGATCCGACGGTAGTGT3’ and G3 (L1_RP): 5’CGAGCTTATCGTCGTCATCC3’ for Library 1, and new_SP(L2_FP): 5’GGTGTGATGCTCGGATCCAGGAAC3’ and 926RR (L2_RP): 5’CACGAGACTACTGTCCTACC3’ for Library 2.
Note: These primers were generated in our lab for different purposes and were selected among many primers tested in our pilot Reel-seq screen. These primers are all good for PCR, but some are not good for repeated amplicon amplification, and others are not good for next-generation sequencing, or library preparation. Therefore, please make sure you don’t change these primers for Reel-seq screen.
Perform the PCR reaction with the T100 Thermal Cycler, as formulated in Table 2, with the PCR program listed in Table 3.
Table 2. PCR amplification of library DNA
Component Volume (μL) Library DNA in different concentrations 5 Forward primer (10 μM) 1 Reverse primer (10 μM) 1 10× AccuPrime PCR buffer 2.5 AccuPrime Taq DNA polymerase 0.25 H2O 15.25 Total 25 Table 3. PCR amplification program
Step Temperature Time Initial denaturation 94°C 2 min 30 cycles 94°C 30 s 60°C 30 s 68°C 40 s Final extension 68°C 5 min Hold 10°C Assemble a 5% Mini-PROTEAN® TBE Gel in a Mini-PROTEAN® Tetra Vertical Electrophoresis Cell.
Resolve all 25 μL of PCR products by adding 5 μL 6× gel Loading Dye on the 5% Mini-PROTEAN® TBE Gel.
Use 10 μL of 1× low molecular weight DNA ladder as a marker.
Run the gel electrophoresis using 0.5× TBE buffer at 100 V for ~60 min.
Disassemble the gel cassette in such a way that the gel is still attached to one side of the glass plate.
Stain the gel, by covering the surface of the gel with 5 mL of 1× GelStar at RT for 20 min.
Visualize the PCR products with BLooKTM LED Transilluminator, and cut the band containing the 79-bp library DNA from the best amplified band (without upper band) (Figure 2).
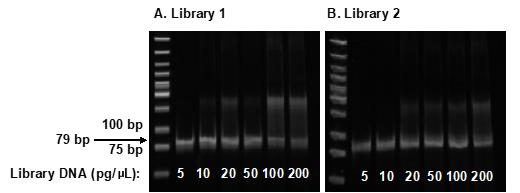
Figure 2. Amplification of Libraries 1 (A) and 2 (B). A 5% Acrylamide gel showing a 79-bp band containing library DNA was amplified by PCR with different concentrations of library DNA, as indicated.Place each sliced gel band (~3 × 5 mm in size) into 150 μL of 10% TE buffer in a microtube, and shake at RT overnight.
Collect ~120 μL of liquid containing the library DNA. At this point, you should have one tube for each library with purified library DNA.
Put the tube with the cap open in the Thermo Scientific Savant DNA 120 SpeedVac Concentrator, which is a vacuum centrifuge.
Concentrate the library DNA, by turning on the centrifuge first, and then the vacuum.
Normalize the DNA samples to 10 ng/μL, as measured with the Implen C40 NanoPhotometer UV/Vis Spectrophotometer Mobile System.
Preparation of library DNA for Reel-seq screen
PCR amplify the library DNA for Libraries 1 and 2 obtained from Procedure B.
Prepare the PCR reaction as formulated in Table 2, by using 5 μL of purified library DNA (~10 ng/μL), and run the PCR reaction according to the program listed in Table 3, except that only 15 cycles are required.
Amplify each library DNA in triplicate, so that you will have enough DNA for the Reel-seq screen.
Run 5 μL of PCR product from each reaction on a 5% Mini-PROTEAN® TBE Gel, as described in Procedure B (steps 6–10), to check the quality of PCR amplification. You should observe one major band around 79-bp, with all the reactions having relatively the same amount of DNA.
The amplified library DNA for Libraries 1 and 2 is now ready for the Reel-seq screen.
Isolation of nuclear extract (NE)
Culture ~1.5 × 108 primary human arterial ECs with ERG-2 medium in 150-mm plates in a cell culture incubator with 5% CO2.
Wash the cells three times using DPBS.
Scrape the cells in 5 mL of DPBS with 1× Roche cOmplete mini protease inhibitor cocktail.
Collect the cells by centrifuging at 450 g for 5 min, using the Clinical Benchtop IVD Centrifuge.
Isolate the NE using NE-PERTM Nuclear and Cytoplasmic Extraction Reagents, according to the manufacturer’s instructions, except that only half volume of Nuclear Extraction Reagent (NER) is used to extract NE. This will yield NE with a concentration of ~10 μg/μL.
Use Roche cOmplete mini protease inhibitor cocktail to prevent protein degradation, according to the manufacturer’s instructions.
Divide the NE in ~20 μL aliquots, and store it at -80°C for at least one year.
Note: Cell culture conditions and media will be dependent on the cell line you use to isolate NE.
Titration of NE for EMSA
Use LightShiftTM Chemiluminescent EMSA Kit to perform the titration, by mixing all the reagents as formulated in Table 4.
Table 4. Titrate nuclear extract concentration reaction
Component Volume (μL) Library DNA from C 5 10× Binding Buffer 2 Poly dI-dC (1 μg/μL) 1 100 mM MgCl2 1 1% NP-40 1 50% Glycerol 1 Nuclear Extract 0, 0.5, 1, 2, 3 H2O Add to 20 Total 20 Use 5 μL of library DNA from Procedure C to titrate the NE, by adding 0, 0.5, 1, 2, and 3 μL of NE isolated in Procedure D.
Incubate the mixture at RT for 2 h or at 4°C overnight.
Perform EMSA using a 5% Mini-PROTEAN® TBE Gel, by adding 5 μL of 5× loading dye provided in the kit to each 20 μL of reaction mixture.
Run all 25 μL of PCR product from each reaction on a 5% Mini-PROTEAN® TBE Gel, as described in Procedure B (steps 6–10), and visualize the PCR products with BLooKTM LED Transilluminator.
Use densitometry to determine the reaction that shows ~50% library DNA shifted in the sample compared to the control (no NE added), and use the amount of NE (for example, 2 μL) for the following Reel-seq screen (Figure 3).
Note: If a 79-bp fragment is not observed clearly, you should apply more cycles, such as 20 cycles for the PCR in step 2 of Procedure C.
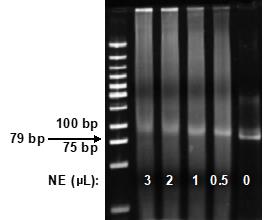
Figure 3. Titration of NE. Acrylamide gel showing that different amounts of NE were used to titrate the amount of NE that results in ~50% library DNA shifting in the sample compared to the control.Reel-seq screen
Prepare EMSA reaction using LightShiftTM Chemiluminescent EMSA Kit, according to Table 5, in triplicate. At this point, you should have three controls and three samples for each library.
Table 5. EMSA reaction
Component Control (μL) Sample (μL) Library 1/2 DNA (Procedure C) 5 5 10× Binding Buffer 2 2 Poly dI-dC (1 μg/μL) 1 1 100 mM MgCl2 1 1 1% NP-40 1 1 50% Glycerol 1 1 Nuclear Extract (Procedure D) 2 (H2O) 2 H2O 7 7 Total 20 20 Shake the reaction mixtures (300 rpm) at RT for 2 h or at 4°C overnight.
Perform EMSA using a 5% Mini-PROTEAN® TBE Gel, by adding 5 μL of 5× loading dye provided in the kit to each 20 μL of reaction mixture.
Purify the 79-bp unshifted library DNA band from the three controls and the three samples (Figure 4A), as described in Procedure B (steps 6–14).
PCR amplify the library DNA for Libraries 1 and 2 obtained above.
Prepare the PCR amplification reaction as formulated in Table 2, and run the PCR with the program listed in Table 3, except that only 15 cycles are required.
Run 5 μL of PCR product from each reaction on a 5% Mini-PROTEAN® TBE Gel, as described in Procedure B (steps 6–10), to check the quality of PCR amplification with the BLooKTM LED Transilluminator (Figure 4B).
Repeat Procedure F ten times.
Notes:
You should observe one major band around 79-bp with all the reactions (three controls and three samples for each library), and having relatively the same amount of DNA (Figure 4B). The amplified library DNAs are now ready for the next round of Reel-seq screen.
Also, in general, applying positive controls for Reel-seq screen is a good idea. When we performed this Reel-seq, we did not have any positive controls. This is because no cis-REs had ever been identified in the 58-kb CDKN2A/B region we screened. However, one can always use cis-REs from other regions of the human genome as positive controls, if one can make sure that these cis-REs function in the cells you use for your Reel-seq screen.
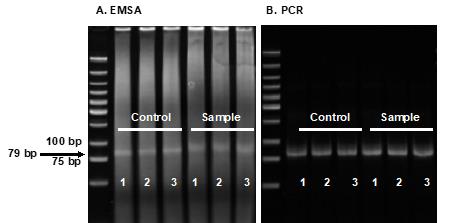
Figure 4. Reel-seq screen. Acrylamide gel showing the results of EMSA (A) and PCR (B) in round 1 of Reel-seq screen, with three buffer-treated controls and the three NE-treated samples, for one of the two libraries.Preparation of a next-generation sequencing library using two PCR reactions
Select the Reel-seq PCR products from rounds 1, 4, 7, and 10, to prepare the next-generation sequencing library. As each round has three controls and three samples, a total of 24 sub-libraries will be generated for each library.
Using 1 μL of the Reel-seq PCR product for each control and sample, perform the first PCR reaction according to Table 6.
Note: We used Herculas II Fusion DNA Polymerase, as previously described (Larman et al., 2013).
Run the PCR according to the program listed in Table 7.
Table 6. PCR amplification before high throughput sequencing
Component Volume/Reaction (μL) PCR product from cycle 1, 4, 7, and 10 1 F (L1-PE/L1-New-seq, 10 μM) 1 R (R1-G3/R1-926RR, 10 μM) 1 5× Herculase II Reaction Buffer 5 Herculase II Fusion DNA Polymerase 0.25 dNTP 0.25 H2O Add to 25 Total 25 Table 7. PCR amplification program before high throughput sequencing
Temperature
Time
95°C
2 min
95°C
20 s
Repeat 29 cycles
58°C
30 s
72°C
40 s
10°C
5 min
Use L1/PE_SP and R1/G3 primers for Library 1, and L1/new-SP and R1/926RR primers for Library 2. The sequences of all these primers are listed in Table 8.
Note: We selected PCR products from rounds 1, 4, 7, and 10 for sequencing, based on the capacity of next-generation sequencing, the number of DNA fragments in the Reel-seq library, a 30–50% mutation rate introduced by PCR in the Reel-seq screen, and the cost of sequencing. The data generated from these four rounds is good enough for statistical analysis.
Table 8. Primers for next-generation sequencing library preparation
L1/PE_SP
(L1_FP)
5’ACACTCTTTCCCTACACGACACACGCACGATCCGACGGTAGTGT3’ R1/G3
(L1_RP)
5’GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCCGAGCTTATCGTCGTCATCC3’ L1/new_SP
(L2_FP)
5’ACACTCTTTCCCTACACGACGGTGTGATGCTCGGATCCAGGAAC3’ R1/926RR
(L2-RP)
5’GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCCACGAGACTACTGTCCTACC3’ L2 5’AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGAC3’ R2 (Barcode) 5’CAAGCAGAAGACGGCATACGAGATXXXXXXXGTGACTGGAGTTCAGACGTGT3’ Check the quality of the first PCR reaction, by running 5 μL of PCR product on a 2% agarose gel with 2 μL of Gel Loading Dye. A relatively even band at 132 bp should be observed across all the reactions (Figure 5A and B).

Figure 5. Sequencing library preparation. Agarose gel showing the PCR products from the first PCR (A and B) and second PCR (C and D). C: Control; S: Sample.Perform the second PCR reaction according to Table 9, using 2 μL of the first PCR product for each control and sample, and run the PCR using the program listed in Table 10.
Table 9. Second PCR amplification reaction before high-throughput sequencing
Component Volume (μL) First PCR product 2 L2 1 R2 (Barcode) 1 5× Herculase II Reaction Buffer 10 Herculase II Fusion/ DNA Polymerase 0.5 dNTP 0.5 H2O 35 Total 50 Table 10. Second PCR amplification program before high throughput sequencing
emperature Time Note 95°C 2 min 95°C 20 s Repeat 9 cycles 58°C 30 s 72°C 40 s 72°C 5 min Use L2 and R2 (Barcode) primers for both libraries, as listed in Table 8. In total, 48 R2 primers with 48 different barcodes are needed for amplifying 48 sub-libraries.
Note: L2 and R2 primers contain adaptor sequences p5 and p7 (underlined in Table 8). Adaptors allow the sequencing library to bind and generate clusters on the flow cell for illumina sequencing. The design and the sequence of these primers with barcodes were described in Kozarena and Tuner (2011).
Check the quality of the second PCR reaction, by running 5 μL of PCR product on 2% agarose gel with 2 μL of Gel Loading Dye. A relatively even band at 188 bp should be observed across all the reactions (Figure 5C and D).
Mix the second PCR products according to Table 11. You will have 186 μL of secondary PCR product mixture for each library.
Table 11. Mix PCR product for sequencing library purification
Round Control (μL) Sample (μL) 1 1 1 4 3 5 7 5 10 10 7 30 Note: We chose the volumes mentioned in the Table 11 based on our experience. PCR can generate mutations, and more mutations are generated by PCR with library DNA treated with NE. Therefore, we used these numbers to obtain relatively even sequencing reads (free of mutation) for each library.
Purify the mixed PCR product, by using a 5% Mini-PROTEAN® TBE Gel for each library, as described in Procedure B (steps 6–14). Each 186 μL of library DNA should be evenly distributed into 9 wells, with one well left for the marker.
Cut the band at 188-bp, place each sliced gel into 150 μL of 10% TE buffer, and isolate the sequencing library by shaking at RT overnight.
Concentrate the DNA Library to ~50 ng/μL with the SpeedVac Concentrator.
Send both sequencing libraries to the core facility for next-generation sequencing. Use the PE_SP primer for Library 1, and the new_SP primer for Library 2.
Sequence both Libraries 1 and 2 together, using NextSeq 500 with 75-bp single read high output, at 2.5 pM with 1% PhiX spiked in.
Notes:
As a part of sequencing, you should request for data analysis to list the reads for each 35-bp fragment in all 24 sub-libraries, for both Libraries 1 and 2. Next generation sequencing was performed at the Genomics Research Core at University of Pittsburgh (https://www.genetics.pitt.edu/next-generation-sequencing).
The actual platform you use for next generation sequencing depends on the size of your library. A small library can be sequenced by MySeq; therefore, talk to a sequencing expert to determine the actual sequencing platform you want to use.
Data analysis
Use Excel to perform all data analysis.
Calculate the percentage of each 35-bp DNA fragment in each of the 24 sub-libraries.
Use the three percentages from the three buffer-treated controls and the three NE-treated samples to calculate the P-value of the Student’s t-test for each fragment in rounds 1, 4, 7, and 10.
Calculate the average of the percentage for each DNA fragment from the three buffer-treated controls and the three NE-treated samples in rounds 1, 4, 7, and 10.
Calculate the ratio of the percentage average from the three NE-treated samples versus the three buffer-treated controls in rounds 1, 4, 7, and 10.
Apply these four ratios to calculate the slope for each 35-bp DNA fragment across rounds 1, 4, 7, and 10.
Apply slope < 0 as the first filter, and P < 0.05 in rounds 1, 4, 7, and 10 as the second filter.
Identify the fragments with slope < 0 and P < 0.05 in rounds 1, 4, 7, and 10 as the candidate cis-REs.
Note: We have successfully used Reel-seq to identify cis-REs over a 200 kb region. However, at this point, we don’t yet know the limitations on how big a region can be screened by Reel-seq. Also, we have estimated the fidelity to be >90% for identifying cis-REs using Reel-seq.
Recipes
0. 5× TBE buffer (1 L)
Add 50 mL of 10× TBE to 950 mL of distilled water (dH2O).
1× GelStar (10 mL)
Add 1 μL of 10,000× GelStarTM Nucleic Acid Gel Stain to 10 mL of dH2O.
1× Low Molecular Weight DNA Ladder
Add 100 μL of Low Molecule Weight DNA ladder to 900 μL of dH2O and 200 μL of 6× DNA Gel Loading Dye.
10% TE buffer
Add 1 mL of TE buffer to 9 mL of dH2O.
2% agarose gel
Dissolve 2 μg agarose in 100 mL of 0.5× TBE.
Acknowledgments
The Reel-seq protocol presented herein is modified from its original publication in Nucleic Acids Research by Wu et al. (2021). We thank NIH NIA and UPMC for their grant support. This work was supported partly by grants from NIH NIA R01AG056279 (GL), R01AG065229 (GL) and UPMC (GL).
Competing interests
The authors declare no competing financial interests.
References
- Angeloni, A. and Bogdanovic, O. (2019). Enhancer DNA methylation: implications for gene regulation. Essays Biochem 63(6): 707-715.
- Buenrostro, J. D., Wu, B., Chang, H. Y. and Greenleaf, W. J. (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109: 21 29 21-21 29 29.
- Crawford, G. E., Davis, S., Scacheri, P. C., Renaud, G., Halawi, M. J., Erdos, M. R., Green, R., Meltzer, P. S., Wolfsberg, T. G. and Collins, F. S. (2006). DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat Methods 3(7): 503-509.
- Diao, Y., Fang, R., Li, B., Meng, Z., Yu, J., Qiu, Y., Lin, K. C., Huang, H., Liu, T., Marina, R. J., et al. (2017). A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat Methods 14(6): 629-635.
- Fulco, C. P., Nasser, J., Jones, T. R., Munson, G., Bergman, D. T., Subramanian, V., Grossman, S. R., Anyoha, R., Doughty, B. R., Patwardhan, T. A., et al. (2019). Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat Genet 51(12): 1664-1669.
- Kozarewa, I. and Turner, D. J. (2011). 96-plex molecular barcoding for the Illumina Genome Analyzer. Methods Mol Biol 733: 279-298.
- Larman, H. B., Laserson, U., Querol, L., Verhaeghen, K., Solimini, N. L., Xu, G. J., Klarenbeek, P. L., Church, G. M., Hafler, D. A., Plenge, R. M., et al. (2013). PhIP-Seq characterization of autoantibodies from patients with multiple sclerosis, type 1 diabetes and rheumatoid arthritis. J Autoimmun 43: 1-9.
- Li, G., Martinez-Bonet, M., Wu, D., Yang, Y., Cui, J., Nguyen, H. N., Cunin, P., Levescot, A., Bai, M., Westra, H. J., et al. (2018). High-throughput identification of noncoding functional SNPs via type IIS enzyme restriction. Nat Genet 50(8): 1180-1188.
- Mikhaylichenko, O., Bondarenko, V., Harnett, D., Schor, I. E., Males, M., Viales, R. R. and Furlong, E. E. M. (2018). The degree of enhancer or promoter activity is reflected by the levels and directionality of eRNA transcription. Genes Dev 32(1): 42-57.
- Park, P. J. (2009). ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet 10(10): 669-680.
- Rajagopal, N., Srinivasan, S., Kooshesh, K., Guo, Y., Edwards, M. D., Banerjee, B., Syed, T., Emons, B. J., Gifford, D. K. and Sherwood, R. I. (2016). High-throughput mapping of regulatory DNA. Nat Biotechnol 34(2): 167-174.
- Schmid, M., Durussel, T. and Laemmli, U. K. (2004). ChIC and ChEC; genomic mapping of chromatin proteins. Mol Cell 16(1): 147-157.
- Schoenfelder, S. and Fraser, P. (2019). Long-range enhancer-promoter contacts in gene expression control. Nat Rev Genet 20(8): 437-455.
- Song, L. and Crawford, G. E. (2010). DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc 2010(2): pdb prot5384.
- Tewhey, R., Kotliar, D., Park, D. S., Liu, B., Winnicki, S., Reilly, S. K., Andersen, K. G., Mikkelsen, T. S., Lander, E. S., Schaffner, S. F., et al. (2016). Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 165(6): 1519-1529.
- Ulirsch, J. C., Nandakumar, S. K., Wang, L., Giani, F. C., Zhang, X., Rogov, P., Melnikov, A., McDonel, P., Do, R., Mikkelsen, T. S., et al. (2016). Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits. Cell 165(6): 1530-1545.
- Wu, T., Jiang, D., Zou, M., Sun, W., Wu, D., Cui, J., Huntress, I., Peng, X. and Li, G. (2022). Coupling high-throughput mapping with proteomics analysis delineates cis-regulatory elements at high resolution. Nucleic Acids Res 50(1): e5.
- Zhang, Y., Sun, Z., Jia, J., Du, T., Zhang, N., Tang, Y., Fang, Y. and Fang, D. (2021). Overview of Histone Modification. Adv Exp Med Biol 1283: 1-16.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wu, T. and Li, G. (2022). An Improved EMSA-based Method to Prioritize Candidate cis-REs for Further Functional Validation. Bio-protocol 12(8): e4397. DOI: 10.21769/BioProtoc.4397.
Category
Molecular Biology > DNA > Gene expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.