- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Single Molecule Tracking Nanoscopy Extended to Two Colors with MTT2col for the Analysis of Cell-Cell Interactions in Leukemia
Published: Vol 12, Iss 8, Apr 20, 2022 DOI: 10.21769/BioProtoc.4390 Views: 3065
Reviewed by: Ralph Thomas BoettcherYihui ZhuAnonymous reviewer(s)
Original research article
The authors used this protocol in:
Sep 2021

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Single molecule tracking (SMT) is a powerful technique to study molecular dynamics, and is particularly adapted to monitor the motion and interactions of cell membrane components. Assessing interactions among two molecular populations is classically performed by several approaches, including dual-color videomicroscopy, which allows monitoring of interactions through colocalization events. Other techniques, such as fluorescence recovery after photobleaching (FRAP), Förster resonance energy transfer (FRET), and fluorescence correlation spectroscopy (FCS), are also utilized to measure molecular dynamics.
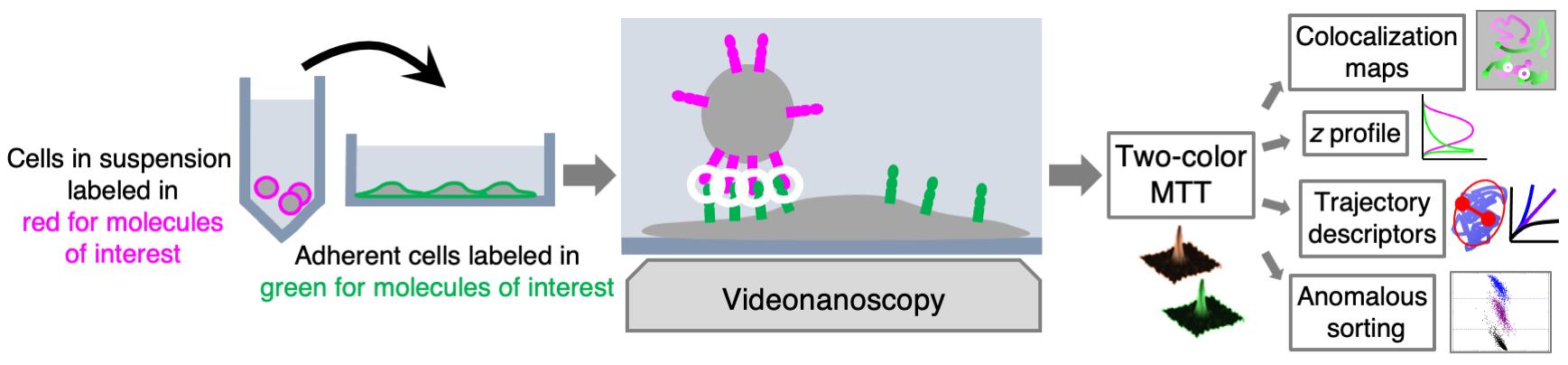
We developed MTT2col, a set of algorithmic tools extending multi-target tracing (MTT) to dual-color acquisition (https://github.com/arnauldserge1/MTT2col). In this protocol, we used MTT2col to monitor adhesion molecules at the contact between leukemic stem cells and stromal cells, a process involved in cancer resistance to chemotherapy and in relapse. Our dual-color single molecule protocol includes the following steps: (i) labeling molecules of interest with fluorescent probes, (ii) video-acquisition, (iii) analyses using our MTT2col in-house software, to obtain positions and trajectories, followed by (iv) detailed analyses of colocalization, distribution, and dynamic motion modes, according to the issues addressed. MTT2col is a robust and efficient SMT algorithm. Both MTT and MTT2col are open-source software that can be adapted and further developed for specific analyses.
Graphical abstract:
Background
Single molecule tracking (SMT) is a powerful approach to investigate molecular dynamics in live cells, at the plasma membrane, in the cytosol, or in the nucleus (Figure 1A). Noteworthy, determining the position of a single fluorescent molecule at sub-resolution accuracy can be achieved using a two-dimensional (2D) Gaussian fit, provided there is a sufficient signal-to-noise ratio. We typically achieve ~40 nm lateral accuracy for living cell images (Sergé et al., 2008). Labeling and monitoring two molecular species by dual-color SMT allows for the comparison of their dynamic state and kinetic interactions in situ. Single molecule labeling can be done classically with fluorescently coupled antibodies (either primary alone, or primary and secondary), Fab fragments, and nanobodies, or by fluorescent proteins, such as GFP and its variants (in other colors or photoactivatable for instance). Additionally, protein tags such as HaloTag, SnapTag, and ClipTag, with their bright and stable fluorescent ligands, may also be utilized (Figure 1B). Each approach has its advantages and limitations, notably in terms of stoichiometry, steric hindrance, brilliance, and photostability (Achimovich, Ai and Gahlmann, 2019; Jacquemet et al., 2020; Lemon and McDole, 2020; Petazzi, Aji and Chiantia, 2020). There are already several SMT algorithms available, each of which vary in performance due to critical issues, such as signal-to-noise ratio and probe density (Manzo and Garcia-Parajo, 2015).
Alternative microscopy approaches for monitoring molecular dynamics (classically for one, and eventually for two populations) include fluorescence recovery after photobleaching (FRAP), fluorescence correlation spectroscopy (FCS), and Förster resonance energy transfer (FRET), as well as the more recently introduced super-resolution SMT methods (Owen et al., 2009; Sergé, 2016). Super-resolution SMT methods comprise single-particle tracking by photo-activation localization microscopy (sptPALM) (Manley et al., 2008), points accumulation for imaging in nanoscale topography (PAINT) (Sharonov and Hochstrasser, 2006), and minimal photon fluxes (MINFLUX) nanoscopy (Balzarotti et al., 2017).
Of note, concerning temporal limits, FRAP and FCS are more sensitive to fast motions (i.e., above ~0.1 µm2/s, providing a higher contribution to the measurement), while SPT is more sensitive to slow motions (leading to more efficient reconnections). Furthermore, concerning spatial limits, SMT can reach nanometer accuracy, FRET is efficient only for distances around 10 nm, and both can document entire cell(s) within the microscope field of view, while FRAP and FCS provide local measurements, inherently applied over distances larger than the diffraction limit. Other non-dynamic approaches to assess molecular interactions at the cell or tissue level, in vivo or in vitro, include immunostaining or coimmunoprecipitation. A key advantage of single molecule techniques, as opposed to ensemble-average measurements, is that they provide access to the distribution of descriptors, hence to the dynamic of each molecule.
All these techniques may be applied to different space and time domains, with various advantages and limitations (De Los Santos et al., 2015; Li and Vaughan, 2018). For instance, SMT approaches such as MTT2col cover a broad range of space and time, as can be addressed both at the global and local level. Globally, SMT is spatially limited to the field of view of the imaging system: microscope and camera. Images have a width of 512 pixels, with 160-nm pixels and 100× objective, leading to 82-µm width. Temporarily, the limit is only the biology (and the storage capacity, often not limiting). We consider that cells should be kept no more than one hour in the microscope. However, some experimental conditions may need shorter or longer time periods: for instance, endo/exocytosis and protein synthesis occur on minute and hour time-scales, respectively. For each video, we arbitrarily set the length to typically 100–500 frames at 100-ms exposure, hence 10–50 s. This can be readily adjusted to the kinetics of the biological system being measured. Locally, space and time resolution limits are linked, essentially by the equation of diffusion MSD = 4Dt. The acquisition time is usually set at its lowest value, as imposed by the fastest video rate of the camera, typically in the 10-ms range. For a diffusion coefficient ranging from 10-3 to 1 µm2/s, this leads to 50–500 nm.
MTT2col may be tuned using several parameters, including probability of false alarm (PFA) detection for different signal-to-noise ratios, spatiotemporal search windows, probability parameters for the reconnection statistical laws, target size, and cutoffs for discarding short and slow trajectories. Each parameter may be adjusted by visual inspection of the results obtained with the default values. For instance:
- For too many/not enough detections, PFA should be increased/decreased to become more/less stringent.
- Similarly, if trajectories are connecting too many/not enough targets, spatiotemporal search windows and probability parameters for reconnection should be adjusted accordingly. In particular, the spatiotemporal search window should match the spatiotemporal resolution limits [see supplementary Figure 6B in Gorshkova et al. (2021)]. Cutoffs relate to statistical laws describing the past statistics and recombination probabilities. Thus, they should be of general scope [see Sergé et al. (2008) for a description of these parameters]. Given the robustness of MTT, the default values are often satisfactory.
- For a single molecule, the target size is equal to the diffraction limit. Hence, the physical value is almost constant over the visible spectrum, i.e., 200–300 nm. It was converted into pixels with a pixel size of 160 nm in the original version. If the pixel size substantially varies, then the value of the target size should be modified accordingly.
- Cutoffs for discarding short and slow trajectories are defined according to the distribution of trajectory length and diffusion coefficient. Irrelevant values (too short and/or too slow) are determined by comparing experimental results and negative controls for non-specific staining for length and immobile dyes for diffusion, see supplementary Figure 6B of Gorshkova et al. (2021). Nevertheless, raw data are saved as dat files and these cutoffs can be reconsidered a posteriori.
However, the algorithm is ultimately robust regarding these values, which are often kept at their original level or easily adjusted.
Finally, the main parameter to optimize is the input diffusion coefficient, which has to correspond to the measured molecules and can be readily identified by testing a range of values [as described in the paragraph Material and Methods/SMT determination of the input diffusion coefficient for MTT, associated to the supplementary Figure 6A of Gorshkova et al. (2021)]. MTT2col is an open-source software, hence the code may be freely adapted to given conditions and/or completed by supplementary analyses tailored for further measurements.
Dual-color SMT can be applied to a broad range of interacting molecules, such as ligands and receptors, or associated proteins like scaffolds, cell adhesion molecule partners, enzymes and cognate substrates, as well as DNA sequences and associated binding regulatory elements. Tracking can also be performed at other scales, for other target types, namely cells, animals (i.e., fishes or birds), or vehicles (i.e., cars or planes). The core of the MTT algorithm has been extensively downloaded to be used by the scientific community, leading to numerous publications. Notably, MTT has been adapted for use in various contexts, i.e., cell tracking, allowing monitoring in space and time of the fluorescence level associated with the intracellular calcium concentration of a population of T cells (Salles et al., 2013).
Importantly, efficient tracking can be achieved provided there is a sufficient signal-to-noise ratio. MTT has good performances down to about 20 dB [as illustrated in Figure 4 from Sergé et al. (2008)]. Of note, noise may be reduced by cultivating cells in a medium without phenol red. Choosing fluorochromes with emission spectra in the red or even infrared can also be helpful to discriminate from autofluorescence (mostly in the green and yellow part of the visible spectrum), hence reducing signal-to-noise ratio. Furthermore, the signal intensity of a single molecule is expected to remain constant until photoblinking or photobleaching, except if photoblinking occurs on timescales shorter than the acquisition time, leading to a lower intensity. Observing stepwise variations instead of gradual decays provides a signature of single molecule measurements. As a supplementary analysis (not implemented in MTT2col), successive stepwise variations in intensity are expected to be due to variations in stoichiometry, for monomers, dimers, trimers, and so on (Schmidt, 1996). To perform these measurements, the intensity of the laser illuminating the sample must be carefully set, by measuring the laser power at the exit of the objective.
The junctional adhesion molecule (JAM) C has been previously reported to be highly expressed in a subset of leukemic stem cells (De Grandis et al., 2017). Thus, JAM-C provides a signature for leukemic initiating cells, potentially leading to relapse after therapy, hence constituting a potential therapeutic target. Thus, we analyzed the dynamics of JAM-B and JAM-C at contacts between leukemic and stromal cells. The MTT2col protocol was initially developed for tracking JAM-B on MS5 stromal cells, and JAM-C on KG1 leukemia cells (Gorshkova et al., 2021). However, it can be readily applied to any other molecules of interest, for instance implicated in cell-cell contacts.
Materials and Reagents
Micropipette tips (Thermo Fisher Scientific, catalog numbers: 11752584 [10 µL]; 10731194 [20 µL]; 16641953 [100 µL]; 11782584 [200 µL]; 11749855 [1,000 µL])
1.5 mL tubes (Eppendorf, Dutscher, catalog number: 033305)
µ-Slide 8 Well Glass Bottom (Ibidi cells in focus, catalog number: 80827)
Trypan blue (Thermo Fisher Scientific, InvitrogenTM, catalog number: T10282)
Rabbit serum (Thermo Fisher Scientific, GibcoTM, catalog number: 16120099)
Cell lines
MS5 (DSMZ ACC 441)
KG1 (ATCC CCL-246)
IMDM (Thermo Fisher Scientific, GibcoTM, catalog number: 12440053)
HBSS, with calcium and magnesium, no phenol red (Thermo Fisher Scientific, GibcoTM, catalog number: 14025092)
L-glutamine (Thermo Fisher Scientific, GibcoTM 25030149, catalog number: 15430614)
Penicillin and streptomycin (Thermo Fisher Scientific, GibcoTM, catalog numbers: 15140122 and 15140148)
β-mercaptoethanol (PanReac AppliChem, catalog number: A1108)
HEPES (Thermo Fisher Scientific, GibcoTM, catalog number: 15630080)
Fetal bovine serum (Thermo Fisher Scientific, GibcoTM, catalog number: 10270106)
Sodium pyruvate (Thermo Fisher Scientific, GibcoTM, catalog number: 11360070)
Antibodies:
Anti-JAM-B: in-house rabbit polyclonal antibody, clone 829
Anti-JAM-C, blocking: mouse IgG (R&D, catalog number: MAB1189, clone 208206)
Anti-JAM-C, non-blocking: in-house rat IgG, clone 19H36
Alexa Fluor 488 conjugated anti-rabbit-IgG (Thermo Fisher Scientific, catalog number: A-11008)
Alexa Fluor 594 conjugated anti-mouse or anti-rat IgG (Thermo Fisher Scientific, catalog number: A-11001 or A-11006)
Quantum-dot 655 conjugated anti-mouse IgG (Thermo Fisher Scientific, catalog number: Q-11021MP)
Quantum-dot 655 conjugated anti-rat IgG (Thermo Fisher Scientific, catalog number: Q-11621MP)
Culture medium for MS5 cells (see Recipes)
Culture medium for KG1 cells (see Recipes)
Equipment
Micropipettes 20, 100 and 1000 (Eppendorf, Thermo Fisher Scientific, catalog number: 05-403-152, model: Research)
Centrifuge (Eppendorf, Thermo Fisher Scientific, catalog number: 5805 000.010)
CO2 incubator Forma Scientific 3548 (Labexchange, Forma Scientific, catalog number: B00032422)
4°C refrigerator
-80°C freezer
Hemocytometer (Ozyme, C-Chip, catalog number: DHC-M01)
Axio Observer Z1 inverted microscope (Zeiss), with thermostated incubator at 37°C
Yokogawa spinning disk device
491- and 561-nm lasers (ILas2 or Cobolt Calypso 1358, 100 mW)
100× α Plan Neofluar NA 1.45 oil-immersion objective (Zeiss) or equivalent, with high NA
Filter cubes n°15 and 44 (Zeiss), with 120-W metal halide lamp (X-cite) for visual inspection
405/488/568/647 nm Yokogawa quadriband dichroic beamsplitter (Semrock, catalog number: Di01-T405/488/568/647-13x15x0.5)
BS 573 dichroic beamsplitter (Semrock, catalog number: FF573-Di01-25x36), between the two cameras
525/50 nm (green channel) and 641/75 nm (red channel) emission filters (Semrock, catalog numbers: FF03-525/50-25 and FF02-641/75-25)
Two Evolve 512 EMCCD cameras (Photometrics) or equivalent, such as the Prime 95B sCMOS camera (Photometrics)
Software
Metamorph software (Version 7.4.8, Molecular Devices)
MTT2col set of MATLAB functions and scripts, using the Image Processing Toolbox with Statistics and Machine Learning Toolbox (any version) (The Mathworks)
Procedure
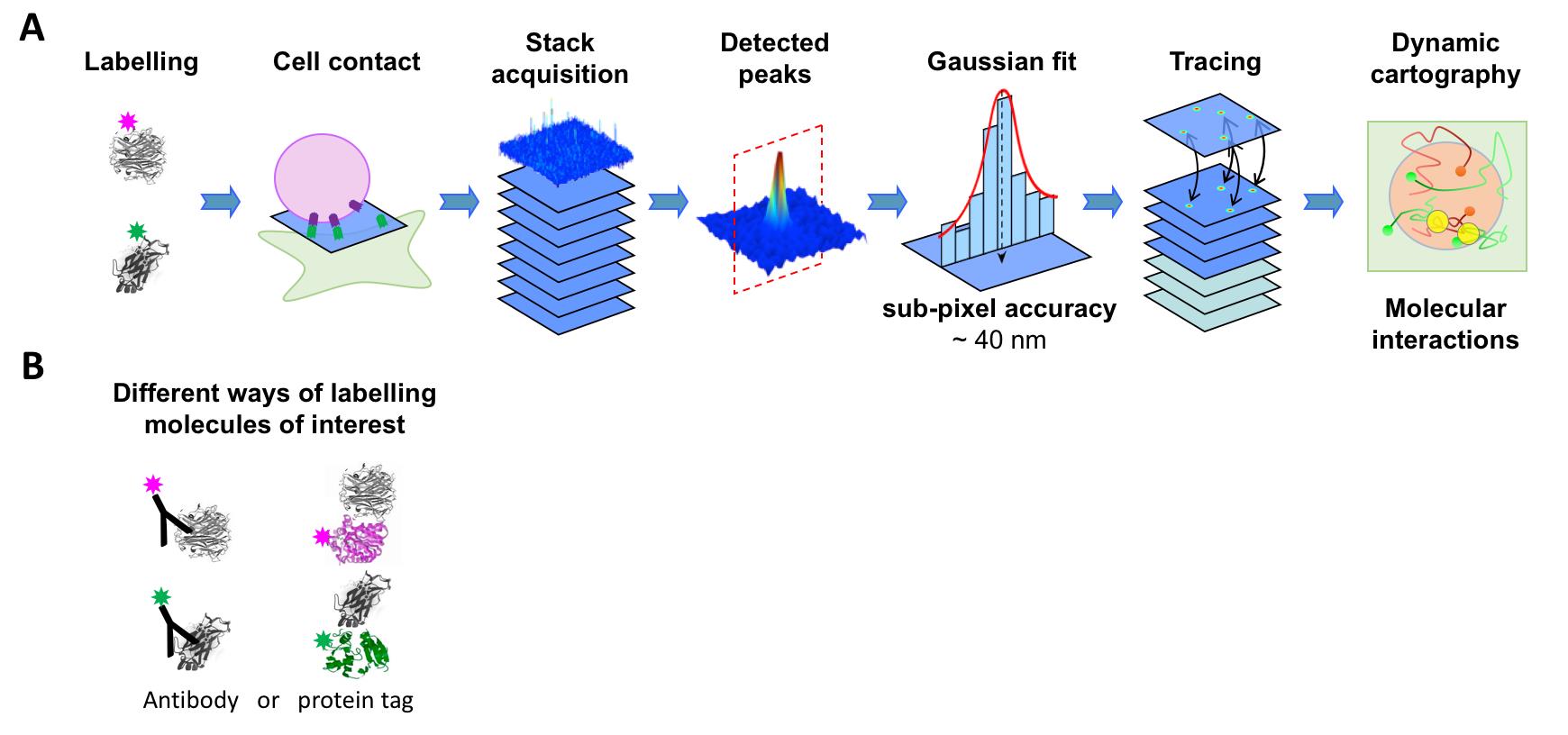
Figure 1. Overview of the experiment. (A) Steps of the experimental procedure: preparation, acquisition, and data analysis. (B) Of note, labeling molecules of interest can be performed by a broad range of strategies.
Cell preparation 24 h or 48 h before videonanoscopy
Pass MS5 cells (adherent): spread in a 1-cm2 area Ibidi glass-bottom well at 30,000 cells in a 300-µL culture volume overnight. This cell density allows reaching about 50% confluence (about 50,000 cells) the day of the experiment, according to DSMZ guidelines. Of note, glass-bottom wells may need specific treatment, such as extracellular matrix coating, for proper adhesion of some cell lines.
Pass KG1 cells (in suspension) at the appropriate culture density, between 100,000 and 1,000,000 cells/mL, according to ATCC guidelines.
Incubate the cultures in a 37°C 5% CO2 air humidified incubator.
If using other types of chamber, they must have a glass bottom to allow fluorescence imaging, and cell numbers should be adjusted to the area.
Immunofluorescence staining of MS5 cells (Figure 1)
Dilute the primary and secondary antibodies in 100 µL of fresh cell culture medium, according to the determined optimal concentration. The optimal antibody concentration gives the best staining with minimum background. It is determined by using a series of dilutions in a titration experiment: 10 nM for anti-JAM-B 829 and for Alexa Fluor 488 conjugated anti-rabbit antibodies (or equivalent, such as quantum-dot conjugated antibodies) in IMDM supplemented with serum, which is necessary to saturate and limit non-specific binding.
Aspirate the culture medium from the well of the live cells to be stained and add 100 µL of the medium containing the diluted antibody directly to the well.
Incubate the cells at room temperature for 10 min.
Aspirate the supernatant and gently wash the cells twice with HBSS with 1% HEPES (or fresh culture medium—to limit autofluorescence, use culture medium without phenol red).
Immunofluorescence staining of KG1 cells
Dilute the primary antibody, blocking or not, and secondary antibody in 100 µL of fresh cell culture medium, according to the determined optimal concentration: 10 nM for anti-JAM-C (blocking: mouse RnD 208206, non-blocking: homemade rat 19H36), and for Alexa Fluor 594 conjugated anti-mouse (or equivalent, such as quantum-dot conjugated antibodies) in supplemented IMDM.
Determine the total number of cells and percent of cell viability using a hemocytometer: mix 0.4% trypan blue with the cell suspension, in a 1:1 proportion (or in other proportions, using the appropriate dilution factor for your calculations). Thoroughly mix the two solutions and pipette 20 µL of this mix into one hemocytometer chamber. Place the hemocytometer under the microscope. Count the unstained (live) and stained (dead) cells (according to the datasheet of the hemocytometer used). Multiply the total number of live cells by two (or by the appropriate dilution factor, if using a different dilution for trypan blue).
Pipette the appropriate volume to have either 50,000 cells (low density: as many as MS5 cells to image one KG1 cell per field of view, allowing cell by cell measurement) or 500,000 cells (high density: 10 times more than MS5 cells, to image several KG1 cells per field of view, enabling more statistics per video).
Transfer the cells to a 1.5-mL Eppendorf tube.
Briefly centrifuge at 88 × g for 3 min.
Resuspend in 1 mL of medium and saturate with 10% rabbit serum, in a 5% CO2 incubator at 37°C for 60 min.
Briefly centrifuge at 88 × g for 3 min.
Resuspend in 100 µL of medium containing the diluted antibodies (see step 1).
Incubate the cells at room temperature for 10 min.
Briefly centrifuge at 88 × g for 3 min, aspirate the supernatant, and gently wash the cells twice with 1 mL of HBSS with 1% HEPES (or fresh culture medium without phenol red). Resuspend the cell pellet in 200 µL of HBSS with 1% HEPES.
Remove the supernatant from the MS5 cells, and add the 200 µL of KG1 cell suspension.
Examine the cells using a spinning disc fluorescence microscope with a thermostated incubator set to 37°C, and appropriate filters (see Equipment, items 7 to 15). Cells should be imaged immediately after staining, to limit endocytosis (Figure 2).
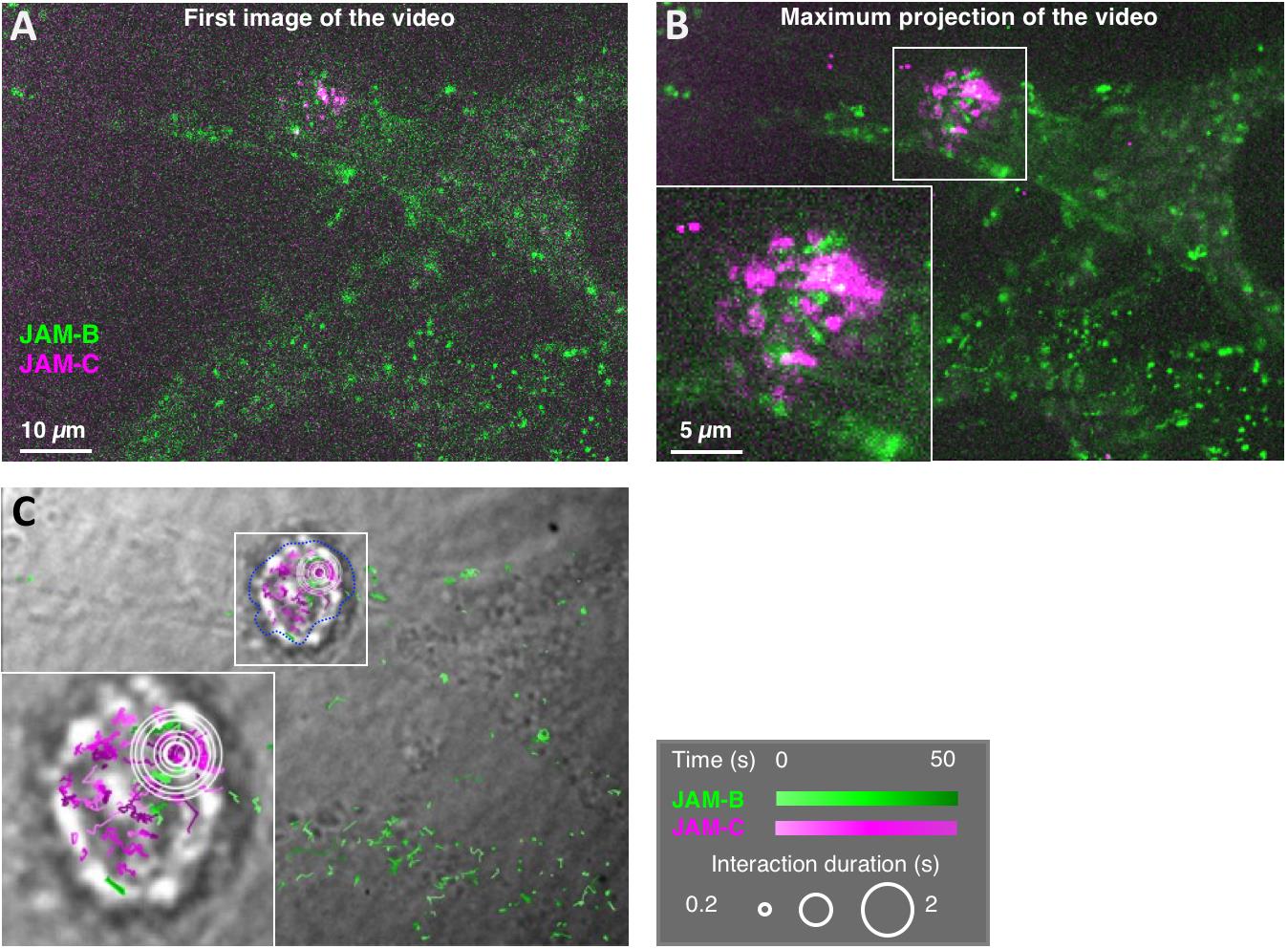
Figure 2. Representative videonanoscopy acquisition and MTT2col trajectory maps. (A) First image of a videonanoscopy acquisition, with the two colors combined into an RGB image. JAM-B staining on MS5 cells is in the green channel, and JAM-C staining on the KG1 cell is in the magenta channel. (B) Maximum projection of the video. (C) JAM-B and JAM-C single molecule trajectories are represented by green and magenta gradients, respectively, according to time, and superimposed on the transmission image of the cells (right). Inserts show magnifications of the framed areas around a leukemic KG1 cell. Spatiotemporal colocalizations are denoted by white circles, with a size proportional to duration. Several concentric circles correspond to successive colocalization events at nearby locations.
Representative videos can be seen from the supplementary data of the related article (Gorshkova et al., 2021).
Performing acquisitions on a confocal microscope
Create a folder, usually named with the date and a brief description of the experiment.
Select a field of view with KG1 cells on top of spread MS5 cells.
First, acquire a brightfield image that allows for further checking of cellular aspects and spatial limitations (as used, in combination with all trajectories, in Figure 2C).
Save this image using the same name as the video (time-stack), in a subfolder named “trans”. With this convention, the algorithm can automatically find the matching brightfield image for each stack.
Next, acquire one (or several) video (time-stack), typically at a 36-ms or 100-ms rate. Save the video file in the folder created for the experiment, as either tif or stk file format. For each video, 300 frames were usually acquired (see Figure 2A for an example). Video rate and length may be adapted for a given experiment. For instance, an experiment may require faster video rate or longer trajectories.
Last, optionally, acquire a z-stack ranging over the entire height of both MS5 and KG1 cells (typically more than 10 µm). Save the z-stack in a subfolder named ‘z’.
Repeat steps 2–5 as many times as needed. Cells should be kept less than an hour in HBSS with 1% HEPES.
Data analysis
Running MTT2col (Figure 1) (Gorshkova et al., 2021)
Start Matlab (or Octave, alternatively, at least for MTT).
Within the Matlab environment, navigate to the folder containing the data to be analyzed (either on a local drive or on the network). Evaluating a given dataset with Matlab or Octave essentially requires selecting the path to the directory containing the video files.
Then, type the command MTT_2_colors in Matlab, which first displays a graphic interface listing all the parameters used, then runs the fully automated analysis (Figure 3).
Of note, when analyzing a mix of one- and two-color datasets, the supplementary Matlab function MTT_1_or_2_colors can determine if the data correspond to one color (square images, such as 512 x 512 pixels), two colors (double square, meaning that the width is double the height, such as 1024 x 512 pixels) or any other possibility. The function automatically runs the analysis for one or two colors if it can be determined without ambiguity. Otherwise the file is skipped.
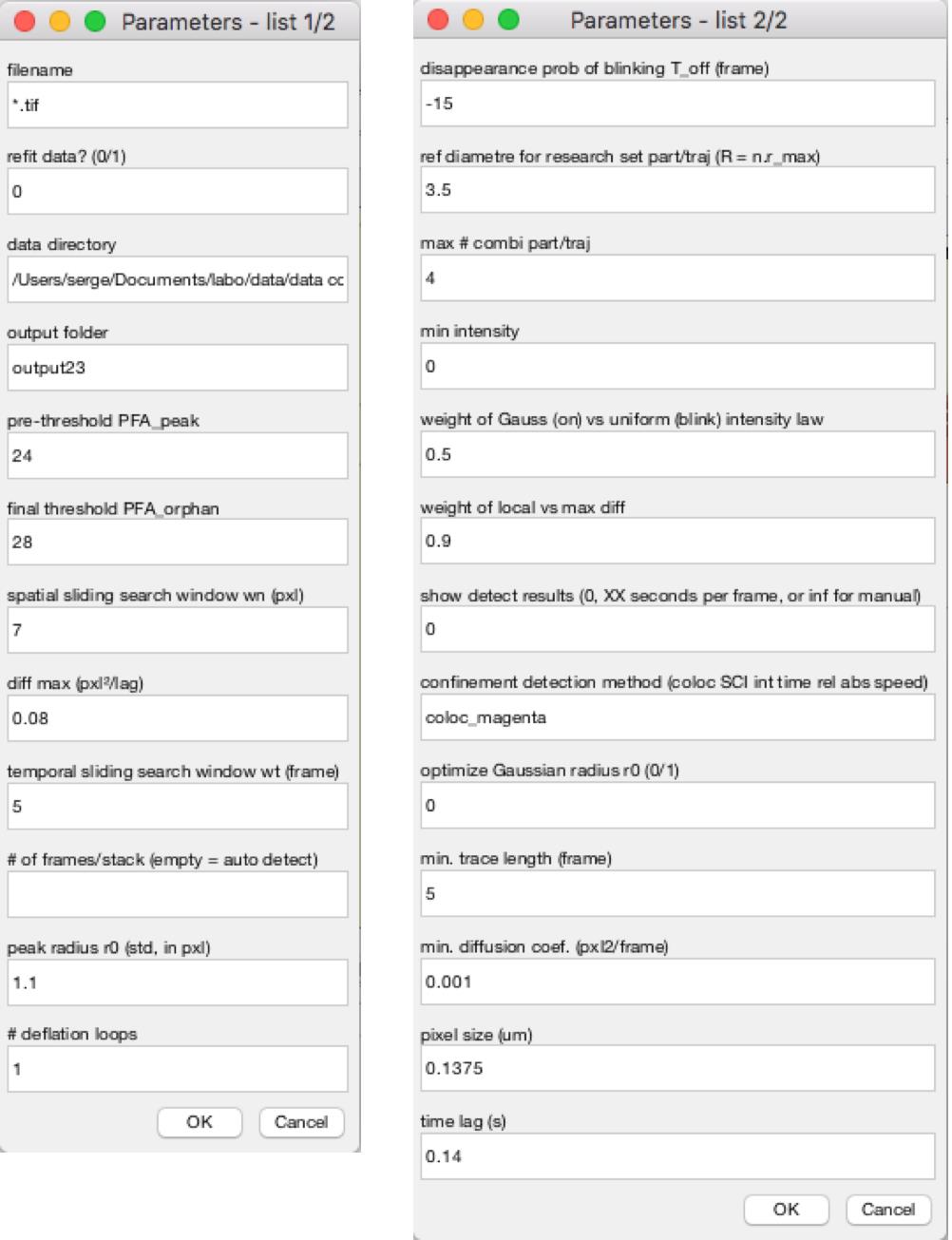
Figure 3. MTT2col graphical user interface. The graphical user interface (GUI) lists all input parameters for molecule detection and estimation, trajectory reconnection, and further analyses, with their default values (as defined in the Matlab functions MTT23_dialog_box MTTparams_def and MTT_param).
Overview of the MTT2col algorithm (Sergé et al., 2008; Rouger et al., 2012)
Inputs for each field of view
The program expects to run the analysis using the following files and subfolders in the local path:
Dual-color time-stacks (i.e. left: green / right: magenta)
Dual-color ‘DIC’ image (transmitted light, in a subfolder named 'dic', optional but used for registration)
Dual-color z-stacks (in a subfolder named 'z', optional)
Analysis
The main function of the MTT2col algorithm is called MTT_2_colors. Below is a list of the main Matlab functions and sub-functions in MTT2col. Key steps are in bold, variables are in purple, and comments are in green. This fully automated procedure allows to build trajectories from dual-color video acquisitions (Figure 2), and to extract several descriptors (Figure 4), as explained in the main text. See the original articles (Sergé et al., 2008; Rouger et al., 2012; Gorshkova et al., 2021) for an extensive description of the algorithmic processes and input/output parameters.
MTT_2_colors % MTT dedicated to 2 colors
MTT23i % with method = 'coloc' by default
MTT23_dialog_box, MTTparams_def, MTT_param % User interface listing input parameters
detect_reconnex_23 % trajectories (Sergé et al., 2008; Rouger et al., 2012)
sort_traces % select long & fast enough trajectories
ct4_by_file, ct5_by_file % histograms of peak & trajectory descriptors such as number, length, diffusion coefficient
fit_directed_by_file % sort trajectories for D & γ
cartobyf % maps for all files
reg_2_colors % compute registration from DIC images, save mean_tform.mat
traj_xy_coloc % map with colocalizations, registering magenta trajectories
coloc3 % compute distance from green to magenta SM
merge_colors % magenta/green stacks & max. projection
detect_cell_contact % statistics at cell contact only
cd(zdir) % analyze z-stack data
MTT23i % run MTT on z-stack
gradient_z2 % z profile of z-stack
merge_colors % for z-stack
Outputs
MTT2col generates several files for each video (time-stack or z-stack), placed in different folders:
'output23': this folder contains the raw data, as a matrix of trajectory parameters. Each column corresponds to a trajectory, and lines correspond to parameters: frame number, x and y position, signal, radius, offset, blink, and noise. Each set of 8 lines corresponds to a given frame. Thus, the matrix has as many columns as trajectories, and 8 times more lines than frames in the video. The dat file contains all trajectories, and the mat file is filtered for trajectories with length and diffusion coefficient above the thresholds defined in the list of input parameters. See code from detect_reconnex_23 for more details (Sergé et al., 2008).
'ct4_by_file', 'ct5_by_file': these folders contain histograms of peak & trajectory descriptors (ct4) and trajectory shape (ct5), as png images for each file and for all files pulled together, as well as a text file containing a table of the mean value for each parameter and file.
'carto': this folder contains a map, i.e., an image of the trajectories superimposed over the image of the cells, as saved in the 'dic' folder (or the first frame of the video otherwise). The color code corresponds to the method selected in the input parameters: colocalization (for 2 colors), linearity (SCI), intensity, time, relative or absolute confinement level, or speed.
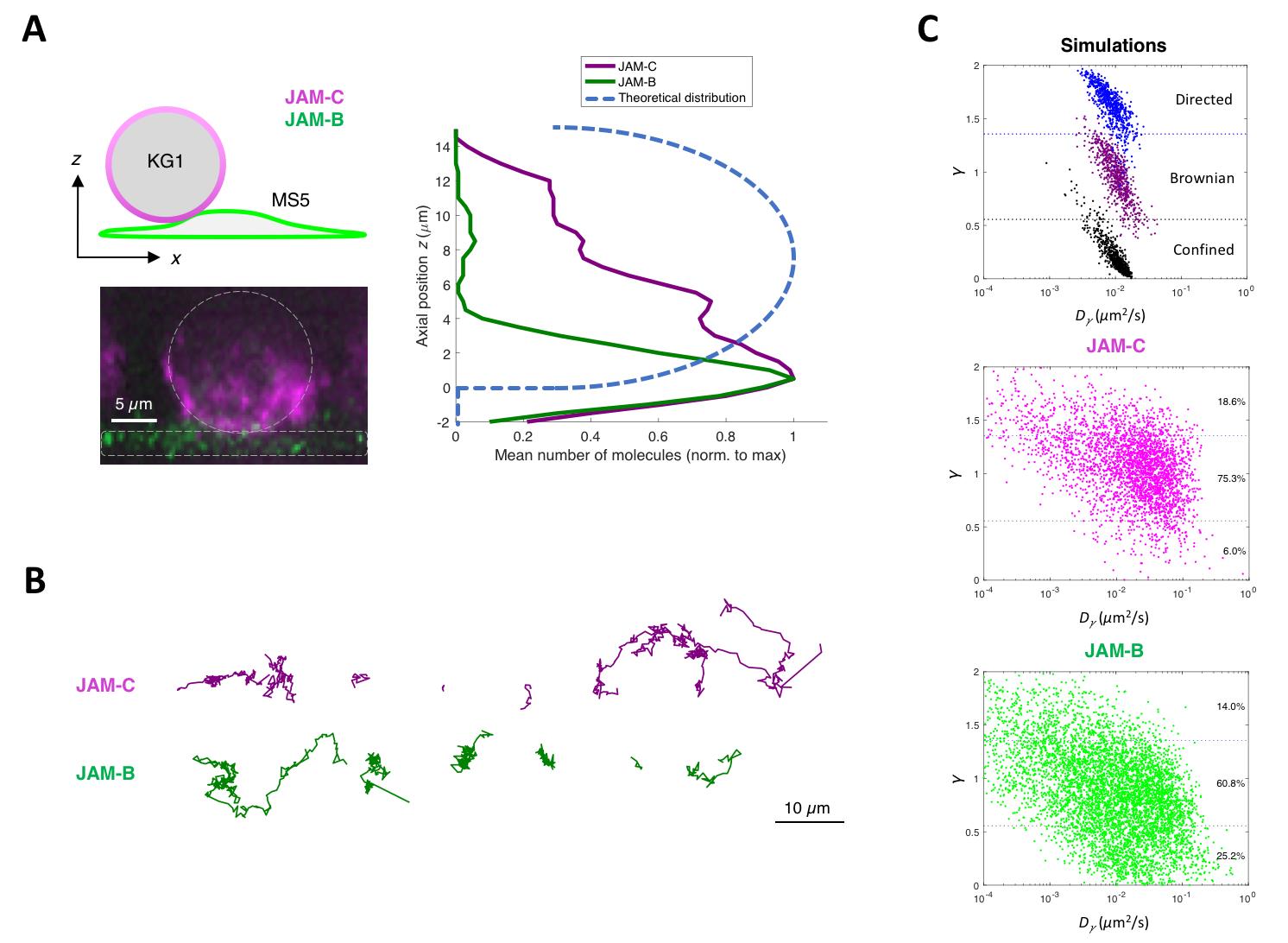
Figure 4. Representative results from MTT2col. (A) Diagram of localization (top left), lateral view of a z-stack with each cell delimited by a white dotted line (bottom left), and z-profile of JAM axial positions, with the expected theoretical distribution for a uniform repartition on a sphere (right). (B) Representative trajectories, illustrating the variety of either Brownian, linear, or confined motions. (C) Dot plot of the anomalous exponent γ vs. the anomalous diffusion coefficient Dγ, which was computed from the mean squared displacement of each trajectory for simulations (top), JAM-C (middle), and JAM-B (bottom).
Data and code availability
The source code for both MTT and MTT2col is available for download as open-source software on GitHub at https://github.com/arnauldserge1/MTT2col.
The data that support the findings of this study are accessible for academic and non-profit research, upon reasonable request to the corresponding author, A.S. (arnauld.serge@univ-amu.fr).
Notes
The signal-to-noise ratio of the fluorescence images may be enhanced by:
Increasing laser power, but avoiding photobleaching and photodamage.
Using bright (as well as stable) dyes: high cross section and quantum yield.
Increasing the acquisition time, but this is limited by the dynamics of the molecules: displacement (increasing with time, according to r2 = 4Dt for Brownian motion) should remain smaller than the pixel size to generate diffraction limited signals.
Increase the gain, while avoiding saturation, when using an EMCCD camera.
The pixel size in the images is determined by the pixel size of the camera, divided by the magnifications of the objective and the tube lens (optovar) and multiplied by the binning. We use a camera with a pixel size of 16 µm, a 100× lens, no optovar, and no binning, resulting in a pixel size of 160 nm in the images. Ideally, according to the Shannon-Nyquist theorem, the pixel size should be equal to (or less than) half the diffraction limit, i.e., 0.3λ/NA (λ: fluorescence emission wavelength, NA: numerical aperture of the lens).
Too large a size leads to under-sampling of the single molecule signal (i.e., the point-spread function of the microscope, with a size set by diffraction).
Too small a size dilutes the signal over neighboring pixels, leading to a weaker signal (lower number of collected photons per pixel).
Scientific cameras may generate 8, 12, 14, 16, or 32-bit images (as well as 24, 36, or 48-bit for color cameras, which is not appropriate here). MTT is intended for 16-bit images by default. It might be used for other formats, but with careful checking of the results.
A typical experiment generates about tens of videos, with about hundreds of molecules per frame, and hundreds of frames per video. There is thus a large variability in the total size of data to analyze. Similarly, computer processing capability can change over at least an order of magnitude. Of note, computing time scales linearly with the number of videos, as well as the number and size of frames, but exponentially with the density of targets (labeled molecules), which leads to the increasing computation of connectivity possibilities, when molecules are getting close to each other. Running MTT2col analysis on a standard computer (e.g., 6–8 cores, 3–3.5 GHz processor, and 16–32 Gb RAM) for a few tens of video files typically takes a few hours (e.g., 0.5–6 h). Therefore, an experiment performed during one day can be analyzed overnight, with results available the next day.
Statistical tests can be automatically performed using an appropriate Matlab script.
Recipes
Culture medium for MS5 cells
Iscove's Modified Dulbecco's Medium (IMDM)
10% Fetal Calf Serum
1% HEPES, essential amino acids
1% sodium pyruvate
25 µM β-mercaptoethanol
1% L-glutamine
Culture medium for KG1 cells
Iscove's Modified Dulbecco's Medium (IMDM)
10% Fetal Calf Serum
1% L-glutamine
Acknowledgments
We thank Jessica Chevallier (TAGC, CIML, LAI, Aix-Marseille Univ) for critical reading of the manuscript. We thank Martine Biarnes-Pelicot (LAI, Aix-Marseille Univ) for providing technical help for cell culture. We thank Dr. Dalia El Arawi and Dr. Laurent Limozin (LAI, Aix-Marseille Univ) for providing technical help for the microscopy setup. We thank Rémi Torro and Dr. Pierre-Henri Puech (LAI, Aix-Marseille Univ) for providing technical help for Github. We also thank the microscopy, cytometry, and statistics research facilities at CRCM, as well as Thi Tien Nguyen for the cell culture facility at LAI, for excellent technical support.
This work was supported by the Fondation ARC pour la Recherche sur le Cancer (PJA 20131200238 to A.S.), the Institute Marseille Imaging, and institutional grants from INSERM and Aix-Marseille Univ. L.M. was supported by grants from the GdR Imabio (Bourse Accueil Master Interdisciplinaire), and from the Ligue contre le Cancer (IP/SC/SK - 17250). This protocol was originally developed in Gorshkova et al. J Cell Sci. 2021.
Competing interests
We declare no competing interest.
References
- Achimovich, A. M., Ai, H. and Gahlmann, A. (2019). Enabling technologies in super-resolution fluorescence microscopy: reporters, labeling, and methods of measurement. Curr Opin Struct Biol 58: 224-232.
- Balzarotti, F., Eilers, Y., Gwosch, K. C., Gynna, A. H., Westphal, V., Stefani, F. D., Elf, J. and Hell, S. W. (2017). Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 355(6325): 606-612.
- De Grandis, M., Bardin, F., Fauriat, C., Zemmour, C., El-Kaoutari, A., Serge, A., Granjeaud, S., Pouyet, L., Montersino, C., Chretien, A. S., et al. (2017). JAM-C Identifies Src Family Kinase-Activated Leukemia-Initiating Cells and Predicts Poor Prognosis in Acute Myeloid Leukemia. Cancer Res 77(23): 6627-6640.
- Gorshkova, O., Cappai, J., Maillot, L. and Serge, A. (2021). Analyzing normal and disrupted leukemic stem cell adhesion to bone marrow stromal cells by single-molecule tracking nanoscopy. J Cell Sci 134(18): jcs258736.
- Jacquemet, G., Carisey, A. F., Hamidi, H., Henriques, R. and Leterrier, C. (2020). The cell biologist's guide to super-resolution microscopy. J Cell Sci 133(11): jcs240713.
- Lemon, W. C. and McDole, K. (2020). Live-cell imaging in the era of too many microscopes. Curr Opin Cell Biol 66: 34-42.
- Li, H. and Vaughan, J. C. (2018). Switchable Fluorophores for Single-Molecule Localization Microscopy. Chem Rev 118(18): 9412-9454.
- De Los Santos, C., Chang, C. W., Mycek, M. A. and Cardullo, R. A. (2015). FRAP, FLIM, and FRET: Detection and analysis of cellular dynamics on a molecular scale using fluorescence microscopy. Mol Reprod Dev 82(7-8): 587-604.
- Manley, S., Gillette, J. M., Patterson, G. H., Shroff, H., Hess, H. F., Betzig, E. and Lippincott-Schwartz, J. (2008). High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat Methods 5(2): 155-157.
- Manzo, C. and Garcia-Parajo, M. F. (2015). A review of progress in single particle tracking: from methods to biophysical insights. Rep Prog Phys 78(12): 124601.
- Owen, D. M., Williamson, D., Rentero, C. and Gaus, K. (2009). Quantitative Microscopy: Protein Dynamics and Membrane Organisation. Traffic 10: 962-971.
- Petazzi, R. A., Aji, A. K. and Chiantia, S. (2020). Fluorescence microscopy methods for the study of protein oligomerization. Prog Mol Biol Transl Sci 169: 1-41.
- Rouger, V., Bertaux, N., Trombik, T., Mailfert, S., Billaudeau, C., Marguet, D. and Serge, A. (2012). Mapping molecular diffusion in the plasma membrane by Multiple-Target Tracing (MTT). J Vis Exp (63): e3599.
- Salles, A., Billaudeau, C., Serge, A., Bernard, A. M., Phelipot, M. C., Bertaux, N., Fallet, M., Grenot, P., Marguet, D., He, H. T., et al. (2013). Barcoding T cell calcium response diversity with methods for automated and accurate analysis of cell signals (MAAACS). PLoS Comput Biol 9(9): e1003245.
- Schmidt, T., Schütz, G. J., Gruber, H. J. and Schindler, H. (1996). Local Stoichiometries Determined by Counting Individual Molecules. Analytical Chemistry 68(24): 4397-4401.
- Sergé, A., Bertaux, N., Rigneault, H. and Marguet, D. (2008). Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat Methods 5(8): 687-694.
- Sergé, A. (2016). The Molecular Architecture of Cell Adhesion: Dynamic Remodeling Revealed by Videonanoscopy. Front Cell Dev Biol 4: 36.
- Sharonov, A. and Hochstrasser, R. M. (2006). Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc Natl Acad Sci U S A 103(50): 18911-18916.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Maillot, L., Irla, M. and Sergé, A. (2022). Single Molecule Tracking Nanoscopy Extended to Two Colors with MTT2col for the Analysis of Cell-Cell Interactions in Leukemia. Bio-protocol 12(8): e4390. DOI: 10.21769/BioProtoc.4390.
Category
Biophysics > Single-molecule technique
Biophysics > Microscopy
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.