- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

circFL-seq, A Full-length circRNA Sequencing Method
Published: Vol 12, Iss 8, Apr 20, 2022 DOI: 10.21769/BioProtoc.4384 Views: 4515
Reviewed by: Gal HaimovichQin TangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2021

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Due to overlapping sequences with linear cognates, identifying internal sequences of circular RNA (circRNA) remains a challenge. Recently, we have developed a full-length circRNA sequencing method (circFL-seq) and computational pipeline, to profile ordinary and fusion circRNA at the isoform level. Compared to short-read RNA-seq, rolling circular reverse transcription and nanopore long-read sequencing of circFL-seq make circRNA reads more than tenfold enriched, and show advantages for identification of both short (<100 nt) and long (>2,000 nt) circRNA transcripts. circFL-seq allows identification of differential alternative splicing suggested wide application prospects for functional studies of internal sequences in circRNAs. In addition, the experimental protocol and computational pipeline of circFL-seq shows better sensitivity and precision for identification of back-splicing junctions than current long-read sequencing methods. Together, the accurate identification and quantification of full-length circRNAs makes circFL-seq a potential tool for large-scale screening of functional circRNAs.
Keywords: circRNABackground
Circular RNAs (circRNAs) are a class of covalently closed RNA molecules formed via back-splicing (BS) or lariat precursors (Kristensen et al., 2019; Chen, 2020). By detecting the back-splicing junctions (BSJs) of circRNAs with deep sequencing, short-read RNA-seq discriminates circRNAs from their linear cognates. Although the full-length sequences of circRNAs less than 500 nt can be inferred via bioinformatics approaches, a full understanding of circRNA isoforms is impossible using short reads alone (Zheng et al., 2019; Wu et al., 2019). Single-molecule long-read sequencing has shown methodological advances in identifying circRNAs at the isoform level. Pacific Biosciences (PacBio) sequencing has been applied to PCR products for target full-length circRNA sequences, in a low-throughput and high-cost way (You et al., 2015). Very recently, several Oxford Nanopore Technology (ONT)-based methods (i.e., circNick-LRS, isoCirc, CIRI-long, circFL-seq), have also been employed in genome-wide full-length circRNA reconstruction (Rahimi et al., 2021; Xin et al., 2021; Zhang et al., 2021; Liu et al., 2021).
Our developed high-throughput circRNA sequencing method circFL-seq utilized rolling circular reverse transcription (RCRT) and nanopore long-read sequencing, to identify circRNA at the isoform level. Our study has validated circFL-seq for full-length circRNA detection and quantification, by comparison to annotated circRNAs, RNA-seq, isoCirc, CIRI-long, and RT-qPCR results. Here, by providing the protocol, circFL-seq allowed the study of sequence features, alternative splicing, and differential expression at the isoform level, for both ordinary and fusion circRNAs.
Materials and Reagents
Gloves
Pipette tips
1.5 mL Eppendorf tube
200 μL PCR tube
70% ethanol
RNase away
DNase/RNase-free water
100% ethanol
Qubit assay tubes (Thermo Fisher Scientific, catalog number: Q32856)
Thermostable RNase H (NEB, catalog number: M0523)
DNase I (NEB, catalog number: M0303S)
ATP (10 mM) (NEB, catalog number: P0756S)
Poly(A) Polymerase (NEB, catalog number: M0276S)
RNA Clean Beads (Vazyme, catalog number: N411-01/02)
RNase R (Lucigen, catalog number: RNR0725)
Equalbit RNA BR Assay Kit (Vazyme, catalog number: EQ212-01)
dATP (NEB, catalog number: N0440S)
ddATP (Jena Bioscience, catalog number: NU-1015S)
HiScript III 1st Strand cDNA Synthesis Kit (Vazyme, catalog number: R312-01/02) or SuperScript IV First-Strand Synthesis System (Invitrogen, catalog number: 18091050)
DNA Clean Beads (Vazyme, catalog number: N412-01/02)
Terminal deoxynucleotidyl transferase (TDT) (Invitrogen, catalog number: EP0161)
I-5 High-Fidelity Master Mix (MCLAB, catalog number: I5HM-200)
Equalbit 1× dsDNA HS Assay Kit (Vazyme, catalog number: EQ121-01)
First-strand synthesis primer P1-N6: GTCGACGGCGCGCCGGATCCATA(N)6 (6 randomized mixed bases)
Second-strand synthesis primer P2-T24: ATATCTCGAGGGCGCGCCGGATCC(T)24 (24 consecutive Ts as reverse-complement to polyA)
PCR primers: P1 (GTCGACGGCGCGCCGGATCCATA), P2 (ATATCTCGAGGGCGCGCCGGATCC)
NEBNext FFPE DNA Repair Mix (NEB, catalog number: M6630S)
NEBNext Ultra II End repair/dA-tailing Module (NEB, catalog number: E7546S)
NEB Blunt/TA Ligase Master Mix (NEB, catalog number: M0367S)
NEBNext Quick Ligation Module (NEB, catalog number: E6056S)
Nanopore Ligation Sequencing Kit (ONT, catalog number: SQK-LSK109)
Nanopore Flow Cell (ONT, catalog number: MinION/PromethION R9.4)
rRNA probes mix (see Recipes)
Probe hybridization buffer (see Recipes)
70% and 80% ethanol (see Recipes)
1 U/μL RNase R (see Recipes)
Equipment
Pipettes
Mini-centrifuge with head for 8-strip PCR tubes (Eppendorf, catalog number: 5452000093)
Vortexer (Thermo Fisher, catalog number: 88882012)
Thermocycler (Bio-Rad, catalog number: T100)
Qubit 4 Fluorometer (Thermo Fisher, catalog number: Q33238)
Qsep1 BioAnalyzer (BiOptic, catalog number: C100001)
12-Tube Magnetic Separation Rack (NEB, catalog number: S1509S)
200 μL Tube Magnetic Separation Rack (Vazyme, catalog number: CM101)
Software
Guppy (ONT, https://nanoporetech.com/community)
porechop (Github, https://github.com/rrwick/Porechop)
circfull (Github, https://github.com/yangence/circfull)
Procedure
Note: RNA is easily degradable. Please clean all surfaces with 70% ethanol and RNase away, change gloves often, and sterilize all materials used.

Figure 1. Experimental operation of circFL-seq consisted of circRNA enrichment, library construction, and nanopore sequencing. RCRT, rolling circular reverse transcription.
circRNA enrichment
A1. rRNA depletionMix total RNA, rRNA probes, and probe hybridization buffer, for a final reaction volume of 15 μL:
Components Volume per sample Total RNA 2.0 μg rRNA probes 1.0 μL Probe hybridization buffer 2.0 μL Nuclease-free water add to 15.0 μL Place samples in a thermocycler with a heated lid set to approximately 105°C. Heat the mixture to 95°C for 2 min, slowly cool to 22°C (-0.1°C/s), and incubate at 22°C for an additional 5 min.
Add thermostable RNase H and 10× RNase H buffer, for a final reaction volume of 20 μL:
Components Volume per sample Last step reaction 15.0 μL Thermostable RNase H 2.0 μL 10× RNase H buffer 2.0 μL Nuclease-free water 1.0 μL Place samples in a thermocycler with lid at 50°C, and incubate at 50°C for 30 min.
Prepare DNase I and 10× DNase I buffer, for a final volume of 30 μL:
Components Volume per sample Last step reaction 20.0 μL DNase I 2.5 μL 10× DNase I Buffer 3.0 μL Nuclease-free water 4.5 μL Place samples in a thermocycler with lid at 50°C, incubate at 37°C for 30 min, and place on ice.
Mix the reaction above with ATP (10 mM), 10× Poly(A) Polymerase Reaction Buffer, and Poly(A) Polymerase, for poly(A) tailing:
Components Volume per sample Last step reaction 30.0 μL ATP 10mM 4.0 μL 10× Poly(A) Polymerase Reaction Buffer 4.0 μL Poly(A) Polymerase 1.0 μL Nuclease-free water 1.0 μL Place samples in a thermocycler with lid at 50°C, incubate at 37°C for 30 min, and place on ice.
Isolate RNA in 8 μL of nuclease-free water by 2.5× RNA Clean Beads, according to the manufacturer’s instructions. The total amount of RNA is approximately 200 ng.
Erase linear RNA with RNase R (need 1 U per sample) and RNase R buffer:
Components Volume per sample Last step reaction 8.0 μL 1 U/μL RNase 1.0 μL 10× RNase R buffer 1.0 μL Place samples in a thermocycler with lid at 105°C, incubate at 37°C for 30 min, and 70°C for 10 min.
Full-length circRNA cDNA preparation
B1. Reverse transcription.
After enrichment of circRNAs, reverse transcribe RNA into first cDNA strands in a 20-μL reaction by P1-N6 with HiScript III reverse transcriptase:
Components Volume per sample Last step reaction 10.0 μL Nuclease-free water 5.0 μL 10× HiScript III Buffer 2.0 μL HiScript III mix 2.0 μL P1-N6 1.0 μL Place samples in a thermocycler with lid at 105°C, and incubate at 25°C for 5 min, at 50°C for 50 min, at 70°C for 2 min, and at 85°C for 5 s.
Isolate cDNA by 0.75× DNA Clean Beads, according to the manufacturer’s instructions.
Add poly(A) tails at the 3’ ends in a 20-μL reaction by TDT with final dATP and ddATP concentrations of 2.5 mM and 25 μM, respectively:
Components Volume per sample Last step reaction 14.0 μL 100 mM dATP 0.5 μL 1 mM ddATP 0.5 μL 5× TDT Buffer 4.0 μL TDT 1.0 μL Place samples in a thermocycler with lid at 50°C, incubate at 37°C for 20 min, and place on ice.
Isolate cDNA by 0.75× DNA Clean Beads, according to the manufacturer’s instructions.
Synthesize second-strand cDNA with P2-T24 and I-5 High-Fidelity DNA polymerase:
Components Volume per sample Last step reaction 12.0 μL I-5 High-Fidelity Master Mix 13.0 μL 10 μM P2-T24 1.0 μL Note: The volume of PCR mix will slightly decrease after thoroughly mixing. The total volume of the reaction is 25 μL.
Place samples in a thermocycler with lid at 105°C, and incubate at 98°C for 2 min, 50°C for 2 min, and 72°C for 5 min. Place on ice.
Split cDNAs equally into four 50-μL PCR reactions with primers P1 and P2:
Components Volume per sample Last step reaction 25.0 μL 10 μM P1 8.0 μL 10 μM P2 8.0 μL I-5 High-Fidelity Master Mix 92.5 μL Nuclease-free water 76.5 μL Note: The volume of PCR mix will slightly decrease after thoroughly mixing. The total volume of the reaction is 200 μL.
Place samples in a thermocycler with lid at 105°C, and amplify by 20 cycles of 98°C for 10 s, 67°C for 15 s, and 72°C for 75 s.
Isolate cDNA by 0.5× DNA Clean Beads, according to the manufacturer’s instructions. Resuspend cDNA with 100 μL of nuclease-free water.
Isolate cDNA by 0.5× DNA Clean Beads, again according to the manufacturer’s instructions.
Note: Over the second beads selection from water, it is better to select longer fragments (length around 1,000 bp), which can further enrich cDNA library of circRNAs.
Acquire DNA library concentration with Qubit, according to the manufacturer’s instructions.
If the total amount of DNA library is less than 1 μg, take 10–50 ng of purified cDNAs for further amplification by 6–8 cycles, in a set of four 50-μl PCRs with P1 and P2. Isolate cDNA by 0.5× DNA Clean Beads.
Acquire DNA library concentration with Qubit, according to the manufacturer’s instructions. For Procedure C (Nanopore library construction and sequencing), 0.5–1 μg cDNA library is needed.
Run BioAnalyzer to check the quality of the library. Samples should be run on a BioAnalyzer, to check for the length distribution with a peak around 1,000 bp. Most of the fragments should distribute from 600–2,000 bp (Figure 2).

Figure 2. Length distribution of circFL-seq cDNA library.For troubleshooting of low-quality libraries, see Table 1.
Table 1. List of low-quality libraries.
Observation
Possible cause
Comments and actions
Low amount of library
RNA contamination or degradation
Before library construction, make sure RNA quality is good, based on assessment using electrophoresis and spectrophotometry.
Low recovery after beads clean-up
- 1. Clean beads settle quickly, so ensure they are well resuspended before adding them to the sample.
2. DNA will be eluted from the beads when using ethanol <70%. Make sure to use the correct percentage.Multiple peaks
rRNAs or linear RNA were not well depleted in step A
Design single-strand cDNA probes for residual RNA, as described in Morlan et al. (2012).
Nanopore library construction and sequencing
The libraries can be handed to a sequencing facility. DNA library for barcoding and ligation sequencing was prepared following protocols EXP-NBD104 and SQK-LSK109.
Repair and dA-tail 0.5–1 μg of circFL-seq cDNA, by NEBNext FFPE DNA Repair Mix and NEBNext Ultra II End repair/dA-tailing Module, followed by purification with 1× DNA Clean Beads.
For multiplexing, repaired and end-prepped DNA was barcoded with Native Barcode, by NEB Blunt/TA Ligase Master Mix, followed by purification with 1× DNA Clean Beads.
Pool barcoded samples together in equimolar amounts. Single samples without barcodes or pooled barcoded samples (700 ng) were ligated to an ONT Adapter, with NEBNext Quick Ligation Module, followed by purification with 0.4× DNA Clean Beads, and Short Fragment Buffer.
Mix the DNA library with sequencing buffer, load beads onto a PromethION or MinION Flow Cell, and run on a PromethION or MinION sequencer. For the following data analysis, 5 million reads (about 5 Gbases) per sample are necessary.
Data analysis
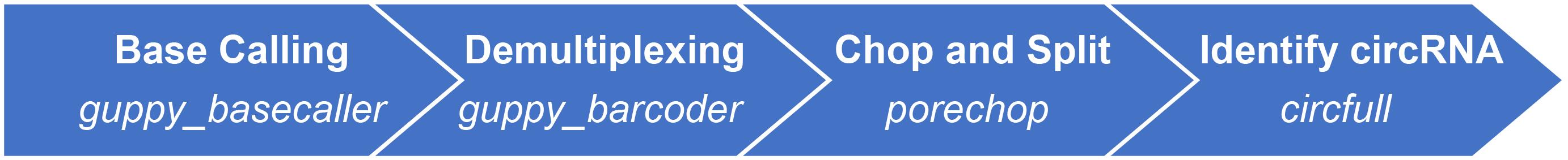
Figure 3. Computational pipeline of circFL-seq data.
Transform raw sequenced data in FAST5 format files to FASTQ format files (kept reads with qscore ≥7.0) by guppy_basecaller, and demultiplex by guppy_barcoder of guppy.
Use porechop to trim barcode and circFL-seq primers (P1, and P2) and split chimeric reads, to obtain clean reads for each sample.
Use RG mode of circfull (https://github.com/yangence/circfull) to detect circRNAs with clean reads, DNSC mode to correct circFL-seq reads by consensus sequence (CS), cRG mode to detect circRNAs with CS, sRG mode to detect the strand origin of reads and adjust circRNA results with strand information, and mRG mode to filter out low-quality full-length circRNAs after integrating circRNA results. See examples in https://github.com/yangence/circfull/tree/main/example.
Recipes
rRNA probes mix
160 DNA probes of complementary sequences (https://cdn.elifesciences.org/articles/69457/elife-69457-supp7-v2.xlsx, ordered from Tsingke Biotechnology with PAGE purification) of 18S and 28S rRNA, at a final concentration of 1 μM, as well as 5S and 5.8S rRNA, 12S and 16S mtrRNA, ETS, and ITS, at a final concentration of 0.1 μM, were pooled.
Probe hybridization buffer
750 mM Tris-HCl and 750 mM NaCl, pH adjusted to 8.0.
70% and 80% ethanol
Adjust 100% ethanol to 70% and 80% ethanol with nuclease-free water.
1 U/μL RNase R
Adjust 20 U/μL RNase R to 1 U/μL RNase R with nuclease-free water.
Acknowledgments
The work was supported by grants from Beijing Municipal Science and Technology Commission of China (7212065, Z181100001518005), and Chinese Institute for Brain Research, Beijing (2020-NKX-XM-01). This protocol was adapted from our recent work (Liu et al., 2021).
Competing interests
The authors declare no competing interests.
References
- Chen, L. L. (2020). The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat Rev Mol Cell Biol 21(8): 475-490.
- Kristensen, L. S., Andersen, M. S., Stagsted, L. V. W., Ebbesen, K. K., Hansen, T. B. and Kjems, J. (2019). The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 20(11): 675-691.
- Liu, Z., Tao, C., Li, S., Du, M., Bai, Y., Hu, X., Li, Y., Chen, J. and Yang, E. (2021). circFL-seq reveals full-length circular RNAs with rolling circular reverse transcription and nanopore sequencing. eLife 10: e69457.
- Morlan, J. D., Qu, K. and Sinicropi, D. V. (2012). Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PloS one 7(8): e42882.
- Rahimi, K., Veno, M. T., Dupont, D. M. and Kjems, J. (2021). Nanopore sequencing of brain-derived full-length circRNAs reveals circRNA-specific exon usage, intron retention and microexons. Nat Commun 12(1): 4825.
- Wu, J., Li, Y., Wang, C., Cui, Y., Xu, T., Wang, C., Wang, X., Sha, J., Jiang, B., Wang, K., Hu, Z., Guo, X. and Song, X. (2019). CircAST: Full-length Assembly and Quantification of Alternatively Spliced Isoforms in Circular RNAs. Genomics Proteomics Bioinformatics 17(5): 522-534.
- Xin, R., Gao, Y., Gao, Y., Wang, R., Kadash-Edmondson, K. E., Liu, B., Wang, Y., Lin, L. and Xing, Y. (2021). isoCirc catalogs full-length circular RNA isoforms in human transcriptomes. Nat Commun 12(1): 266.
- You, X., Vlatkovic, I., Babic, A., Will, T., Epstein, I., Tushev, G., Akbalik, G., Wang, M., Glock, C., Quedenau, C., et al. (2015). Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci 18(4): 603-610.
- Zhang, J., Hou, L., Zuo, Z., Ji, P., Zhang, X., Xue, Y. and Zhao, F. (2021). Comprehensive profiling of circular RNAs with nanopore sequencing and CIRI-long. Nat Biotechnol 39(7): 836-845.
- Zheng, Y., Ji, P., Chen, S., Hou, L. and Zhao, F. (2019). Reconstruction of full-length circular RNAs enables isoform-level quantification. Genome Med 11(1): 2.
Article Information
Copyright
![]() Liu and Yang. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Liu and Yang. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Liu, Z. and Yang, E. (2022). circFL-seq, A Full-length circRNA Sequencing Method. Bio-protocol 12(8): e4384. DOI: 10.21769/BioProtoc.4384.
- Liu, Z., Tao, C., Li, S., Du, M., Bai, Y., Hu, X., Li, Y., Chen, J. and Yang, E. (2021). circFL-seq reveals full-length circular RNAs with rolling circular reverse transcription and nanopore sequencing. eLife 10: e69457.
Category
Cell Biology > Cell-based analysis > Gene expression
Systems Biology > Transcriptomics > RNA-seq
Molecular Biology > RNA > RNA splicing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.