- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Plasmid and Sequencing Library Preparation for CRISPRi Barcoded Expression Reporter Sequencing (CiBER-seq) in Saccharomyces cerevisiae
Published: Vol 12, Iss 7, Apr 5, 2022 DOI: 10.21769/BioProtoc.4376 Views: 4365
Reviewed by: Dennis J NürnbergUte Angelika HoffmannAnonymous reviewer(s)

Original research article
The authors used this protocol in:

Dec 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Genetic networks regulate nearly all biological processes, including cellular differentiation, homeostasis, and immune responses. Determining the precise role of each gene within a regulatory network can explain its overall, integrated function, and pinpoint mechanisms underlying misregulation in disease states. Transcriptional reporter assays are a useful tool for dissecting these genetic networks, because they link a molecular process to a measurable readout, such as the expression of a fluorescent protein. Here, we introduce a new technique that uses expressed RNA barcodes as reporters, to measure transcriptional changes induced by CRISPRi-mediated genetic perturbation across a diverse, genome-wide library of guide RNAs. We describe an exemplary reporter based on the promoter that drives His4 expression in these guidelines, which can be used as a framework to interrogate other expression phenotypes. In this workflow, a library of plasmids is assembled, encoding a CRISPRi guide RNA (gRNA) along with one or more transcriptional reporters that drive expression of guide-specific nucleotide barcode sequences. For example, when interrogating regulation of the budding yeast HIS4 promoter normalized against a control housekeeping promoter that drives Pgk1 expression, this plasmid library contains a gRNA expression cassette, a HIS4 reporter driving expression of one gRNA-specific nucleotide barcode, and a PGK1 reporter driving expression of a second, gRNA-specific barcode. Long-read sequencing is used to determine which gRNA is associated with these nucleotide barcodes. The plasmid library is then transformed into yeast cells, where each cell receives one plasmid, and experiences a genetic perturbation driven by the guide on that plasmid. The expressed RNA barcodes are extracted in bulk and quantified using high-throughput sequencing, thereby measuring the effect of their corresponding gRNA on barcoded reporter expression. In the case of the HIS4 reporter described above, guides disrupting translation elongation will increase expression of the associated HIS4 barcode specifically, without changing expression of the PGK1 control barcode. It is further possible to quantify plasmid abundance by DNA sequencing, as an additional approach to normalize for differences in plasmid abundance within the population of cells. This protocol outlines the steps to prepare barcode reporter CRISPRi plasmid libraries, link guides to barcodes with long-read sequencing, and measure expression changes through barcode RNA and DNA sequencing. This method is ideal for probing transcriptional or post-transcriptional regulation, as it measures the effects of a genetic perturbation by directly quantifying reporter RNA abundance, rather than relying on indirect growth or fluorescence readouts.
Graphic abstract:

Background
By systematically measuring quantitative phenotypes across a library of gene perturbations, one can build a picture of the regulatory landscape of the cell. The advent of CRISPR technology simplifies the process of systematic genetic perturbation (Cong et al., 2013; Jinek et al., 2013), which can be combined with a molecular phenotypic reporter to understand regulation of that reporter across a diverse library of genetic backgrounds (Gilbert et al., 2013). Previous methods have measured transcriptional phenotypes, by coupling changes in transcription to either changes in cellular growth, or changes in fluorescent protein expression (Kampmann, 2020). However, because cellular growth and fluorescence are indirect readouts of underlying transcriptional regulation, genetic perturbations unrelated to the underlying transcriptional control can nevertheless affect reporter expression. The expression of barcoded RNAs provides a more direct means of quantifying transcriptional changes (Muller et al., 2020; Alford et al., 2021). Furthermore, RNA barcodes can be quantified from bulk extraction and sequencing, which avoids bottlenecks that occur as a result of cell sorting, and is thus more scalable than traditional fluorescence-activated cell sorting (FACS) approaches. In addition to encoding the unique identity of the gRNA, the expressed barcodes can also encode reporter-specific tags, allowing for multiplexed screens in a single experiment.
RNA barcodes can be used to interrogate additional biological questions beyond transcriptional regulation. For example, RNA stability and splicing phenotypes, which impact the abundance of specific RNA isoforms, can be interrogated by comparing a query reporter against a suitable control that normalizes for transcriptional effects. Nonsense-mediated mRNA decay, which acts on specific splice isoforms that contain premature stop codons, can be interrogated in this manner. By driving barcode expression with two reporters that differ only by the presence or absence of a premature stop codon, one can ignore transcriptional effects and identify factors that specifically affect mRNA isoform stability. With some adaptations to the technique, RNA barcodes can be used to study other RNA-centric processes, including RNA modifications, interactions, and localization. Interrogating these processes may require additional enrichment steps to recapitulate the phenotype of interest. Interrogating nuclear localization of certain mRNAs, for example, would combine a nuclear isolation protocol with the barcode expression profiling detailed in these protocols. A barcoded-RNA readout can also be linked to other versions of CRISPR-mediated genetic perturbation, including CRISPR activation (CRISPRa) for targeted gene expression, or nucleolytically active CRISPR for targeted gene mutations (Cong et al., 2013; Gilbert et al., 2013; Jinek et al., 2013). Finally, by adapting an orthogonal transcription factor to drive RNA barcode expression, it is possible to extend this barcode sequencing approach to interrogate questions in protein stability and translation regulation (Aranda-Díaz et al. 2017). This approach has previously been used to understand Gcn4 protein degradation by fusing the GCN4 coding sequence in frame with an orthogonal transcription factor and monitoring barcode expression of a reporter driven by said transcription factor (Muller et al., 2020).
Materials and Reagents
Note: All reagents can be stored at room temperature unless otherwise specified.
Plasmid library construction
Gene Pulser/MicroPulser Electroporation Cuvettes, 0.1 cm gap (Bio-Rad, catalog number: 1652089)
pNTI743 dual-barcoded gRNA parent vector (Addgene, catalog number: 164915), store at -20°C
pNTI757 P(PGK1)-citrine P(HIS4)-mCherry (Addgene, catalog number: 164917), store at -20°C
Custom CRISPRi gRNA oligo pool, (Custom Array Inc; ~60,000 oligo pool of 60 nucleotide oligos), store at -20°C, previously published inMcGlincy et al. (2021)
Primers for amplifying gRNA pool (IDT, 25 nmole DNA Oligo, Standard Desalting), store at -20°C
For this specific study, we used NM636: ggctgggaacgaaactctgggagctgcgattggca and NM637: gccttattttaacttgctatttctagctctaaaac
Zymo DNA clean and concentrator-5 kit (Zymo, catalog number: D4013)
Q5 polymerase (NEB, catalog number: M0491L), store at -20°C
Restriction enzymes:
AvrII (NEB, catalog number: R0174S), store at -20°C
AscI (NEB, catalog number: R0558S), store at -20°C
HindIII-HF (NEB, catalog number: R3104S), store at -20°C
PmeI (NEB, catalog number: R0560S), store at -20°C
BglII (NEB, catalog number: R0144S), store at -20°C
XhoI (NEB, catalog number: R0146S), store at -20°C
BciVi (NEB, catalog number: R0596S), store at -20°C
HiFi DNA assembly 2× master mix (NEB, catalog number: E2621L), store at -20°C
MegaX DH10B T1R ElectrocompTM Cells (Invitrogen, catalog number: C640003), store at -80°C
NEB® 10-beta Competent E. coli (High Efficiency) (NEB, catalog number: C3019H), store at -80°C
LB-carbenicillin agar plates and liquid media (50 µg/mL carbenicillin):
Luria Bertani medium capsules (MP Biomedicals, catalog number: 3002031)
Carbenicillin (Gold Bio, catalog number: C-103-5), store at -20°C
SuperPureTM Agar Bacteriological Grade Type A (US BioTech Sources, catalog number: A01PD-500)
10 cm polystyrene Petri dishes (Fisher Scientific, catalog number: 08-757-100D)
HiSpeed Midiprep kit (Qiagen, catalog number: 12643)
Monarch Plasmid Miniprep Kit (NEB, catalog number: T1010L)
T5 exonuclease (NEB, catalog number: M0663S), store at -20°C
Dual barcode oligos (IDT, 25 nmole DNA Oligo, Standard Desalting), store at -20°C
RM720:
CCACATGTGCATTGCCTCGGACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNNNNNNNNNNNNNNNNNNNNNNNCACGT GCTAACAGTGAGGCGCGRM721:
GCTCGATCCAGTCACTCTGGACACGACGCTCTTCCGATCTNNNNNNNNNNNNNNNNNNNNNNNNNNGGTAAGTGACTCGACTG GCGCGCCTCACTGTTAGCACGTG
Sanger sequencing primers (IDT, 25 nmole DNA Oligo, Standard Desalting), store at -20°C
RM377: taacatggtagttacatatactagtaatatggttcgg
KS533: gggaaacgcctggtatcttt
RM336: gttgacaatgattacaggttaaaagg
PacBio library construction for barcode-gRNA assignment
Barcoded gRNA plasmid library: Either generated from “Plasmid Library Construction” section of this protocol, or purchased from Addgene as a pooled plasmid library (Yeast Genome-wide Dual-Barcoded CRISPRi gRNA Library, Addgene #166966)
Restrictions Enzymes
HindIII-HF (NEB, catalog number: R3104S), store at -20°C
PmeI (NEB, catalog number: R0560S), store at -20°C
BglII (NEB, catalog number: R0144S), store at -20°C
XhoI (NEB, catalog number: R0146S), store at -20°C
1% agarose gel for DNA size selection and purification
Agarose LE (Gold Bio, catalog number: A-201-500)
Tris-Borate-EDTA (TBE) Buffer, 10× (National Diagnostics, catalog number: EC-860)
Zymoclean Gel DNA Recovery Kit (Zymo, catalog number: D4001)
SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, catalog number: 100-938-900), store at -20°C
AMPure XP beads (Beckman Coulter, catalog number: A63881), store at 4 °C
Ethanol (VWR-A #TX89125-170SFU)
Nuclease-free water (Fisher Scientific, catalog number: UPW012548)
Nuclease-free microcentrifuge tubes (Ambion, catalog number: AM12450)
High Sensitivity D1000 Sample Buffer (Agilent Technologies, catalog number: 5067-5603), store at 4°C
High Sensitivity D1000 ScreenTape (Agilent Technologies, catalog number: 5067-5584), store at 4°C
High Sensitivity D1000 Ladder (Agilent Technologies, catalog number: 5067-5587), store at 4°C
Genomic DNA Reagents (Agilent Technologies, catalog number: 5067-5366), store at 4°C
Genomic DNA ScreenTape (Agilent Technologies, catalog number: 5067-5365), store at 4°C
RNA barcode sequencing library construction
PhasemakerTM Tubes (optional) (ThermoFisher Scientific, catalog number: A33248)
UltraPureTM SDS Solution, 10% (ThermoFisher Scientific, catalog number: 15553027)
Sodium Acetate 3 M pH 5.5 (Ambion, catalog number: AM9740)
EDTA 0.5 M pH 8.0 (Ambion, catalog number: AM9260G)
Phenol-chloroform-isoamyl alcohol (PCA) pH 6.6 (Ambion, catalog number: AM9730), store at 4°C
Chloroform (Fisher Chemical, catalog number: C607SK-1)
Ethanol, 100% and 70%, diluted in nuclease-free H2O (VWR-A #TX89125-170SFU)
GlycoBlue (Ambion, catalog number: AM9515), store at -20°C
Nuclease-Free Water (Fisher Scientific, catalog number: UPW012548)
Nuclease-Free Microcentrifuge Tubes (Ambion, catalog number: AM12450)
Yeast pellets from library experiment, corresponding to at least 1,000× coverage of plasmid library (at least 240 million cells if using a plasmid library containing 240,000 barcoded gRNAs), flash frozen in liquid nitrogen and stored at -80°C.
*Suggested: Harvest, pellet, and freeze (at -80°C) extra yeast pellets as back-up.
Protoscript II reverse transcriptase kit (NEB, catalog number: M0368L), store at -20°C
NEB-next dual indexing primers (NEB, catalog number: E7600S), store at -20°C
Primer-RM511: GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTGGTCCAGTCTTGTTACCAGACAACC (IDT, 25 nmole DNA Oligo, Standard Desalting), store at -20°C
Primer-RM810: ACACTCTTTCCCTACACGACGCTCTTCCGATCT (IDT, 25 nmole DNA Oligo, Standard Desalting), store at -20°C
Primer-RM812: GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTcCGGCGCCTACAACGTCAACATC (IDT, 25 nmole DNA Oligo, Standard Desalting), store at -20°C
RNase A (Thermo Scientific, catalog number: EN0531), store at -20°C
RNase H (NEB, catalog number: M0297S), store at -20°C
Turbo DNase I (optional) (Invitrogen, catalog number: AM2238), store at -20°C
Zymo RNA clean and concentrator-25 kit (optional) (Zymo, catalog number: R1017)
Zymo DNA clean and concentrator-5 kit (Zymo, catalog number: D4013)
Q5 polymerase (NEB, catalog number: M0491L), store at -20°C
AMPure XP beads (#A63881), store at 4°C
High Sensitivity D1000 Sample Buffer (Agilent Technologies, catalog number: 5067-5603), store at 4°C
High Sensitivity D1000 ScreenTape (Agilent Technologies, catalog number: 5067-5584), store at 4°C
High Sensitivity D1000 Ladder (Agilent Technologies, catalog number: 5067-5587), store at 4°C
AE buffer (see Recipes)
DNA barcode sequencing library construction
Yeast cell pellets from library experiment, corresponding to at least 2,000× coverage of plasmid library (at least 480 million cells, if using a plasmid library containing 240,000 barcoded gRNAs), flash frozen in liquid nitrogen, and stored at -80°C.
*Suggested: Harvest, pellet, and freeze (at -80°C) extra yeast pellets as back-up.
Zymo yeast miniprep kit (Zymo, catalog number: D2004)
T7 HiScribe Kit (NEB, catalog number: E2040S), store at -20°C
Restriction Enzymes
MfeI-HF (if reporter uses citrine expression cassette) (NEB, catalog number: R3589S), store at -20°C
PvuII-HF (if reporter uses mCherry expression cassette) (NEB, catalog number: R3151S), store at -20°C
If using a custom expression cassette, choose a restriction enzyme that will linearize the plasmid library without cutting in between the T7 polymerase binding site and the annealing site for the custom designed primer referred to in material 10 of this section. See Figure 9 for a diagram of the relevant linearization and in vitro transcription steps.
Protoscript II reverse transcription kit (NEB, catalog number: M0368L), store at -20°C
NEB-next dual indexing primers (NEB, catalog number: E7600S), store at -20°C
Primer: 511DNA_IVT_RT: GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT TGGTCCAGTCTTGTTACCAGACAACC (if the reporter uses a citrine expression cassette) (IDT, 25 nmole DNA Oligo, Standard Desalting), store at -20°C
Primer: 546mcherIVT_RT: GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT TCAAGTTGGACATCACCTCCCAC (if the reporter uses an mCherry expression cassette) (IDT, 25 nmole DNA Oligo, Standard Desalting), store at -20°C
For custom barcoded ORF plasmid libraries, use a custom designed primer with 5' adapter GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT (adds the compatible priming site for the i7 NEB, catalog number: E7600S dual indexing primer), and a 3' end specific to a given ORF that will prime a reverse transcription reaction toward the random nucleotide barcode with roughly 150nt intervening sequence. (This ensures you'll have a final dual-indexed sequence that is ~300nt long, which is short enough to cluster well on the Illumina sequencer, but long enough to efficiently size select from primers and primer dimers)
RNase A (ThermoFisher, catalog number: EN0531), store at -20°C
RNase H (NEB, catalog number: M0297S), store at -20°C
Zymo DNA clean and concentrator-5 kit (Zymo, catalog number: D4013)
Zymo RNA clean and concentrator-25 kit (Zymo, catalog number: R1017)
Q5 polymerase (NEB, catalog number: M0491L), store at -20°C
AMPure XP beads (catalog number: A63881), store at 4°C
High Sensitivity D1000 Sample Buffer (Agilent Technologies, catalog number: 5067-5603), store at 4°C
High Sensitivity D1000 ScreenTape (Agilent Technologies, catalog number: 5067-5584), store at 4°C
High Sensitivity D1000 Ladder (Agilent Technologies, catalog number: 5067-5587), store at 4°C
Equipment
Plasmid library construction
Spectrophotometer (Bio-Rad, model: SmartSpecTM 3000)
Thermal cycler (Bio-Rad, model: T100TM)
ThermoMixer (Eppendorf, model: ThermoMixer C)
Incubator Shaker (New Brunswick Scientific, model: InnovaTM 4080)
4 L Pyrex Erlenmeyer flask (Corning, catalog number: F3620-4L)
14 mL polypropylene round-bottom tube (Corning, catalog number: 352059)
1.5 mL tube Centrifuge (Eppendorf, model: Centrifuge 5430)
15 and 50 mL conical tube centrifuge (Eppendorf,model: Centrifuge 5810R 15 amp version)
Stationary incubator (Thermo Scientific, model: HERATHERM Incubator)
Electroporator (Bio-Rad, model: Gene Pulser® II)
PacBio barcode-gRNA assignment
NanoDrop spectrophotometer (Thermo Scientific, model: NANODROP 2000 Spectrophotometer)
Tape Station (Agilent Technologies, model: 2200 TapeStation)
1.5 mL tube centrifuge (Eppendorf, model: Centrifuge 5430)
Gel electrophoresis rig (Thermo Scientific, model: OWL EasycastTM B1A)
DynaMagTM-2 Magnet, 1.5 mL tube magnetic rack (ThermoFisher Scientific, catalog number: 12321D)
RNA Barcode sequencing library prep
Centrifuge tube caplock clip (Benchmark Scientific, catalog number: C1005-T5-LOCK)
Thermal cycler (Bio-Rad, model: T100TM)
ThermoMixer (Eppendorf , model:ThermoMixer C)
TapeStation (Agilent Technologies, model: 2200 TapeStation)
NanoDrop (Thermo Scientific, model: NANODROP 2000 Spectrophotometer)
1.5 mL tube Centrifuge (Eppendorf, model: Centrifuge 5430)
Temperature-regulated 1.5 mL tube Centrifuge (Eppendorf, model: Centrifuge 5430R)
DNA Barcode sequencing library prep
Thermal cycler (Bio-Rad, model: T100TM)
ThermoMixer (Eppendorf, model: ThermoMixer C)
TapeStation (Agilent Technologies, model: 2200 TapeStation)
NanoDrop (Thermo Scientific, model: NANODROP 2000 Spectrophotometer)
1.5 mL tube centrifuge (Eppendorf, model: Centrifuge 5430)
Procedure
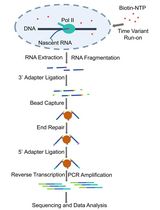
The overall workflow to interrogate genetic networks using barcode expression reporters is shown in Figure 1. The set of protocols provided details the steps to create a reporter expression plasmid library, generate a long-read sequencing library, and prepare RNA and DNA barcode sequencing libraries. These protocols do not detail the yeast library transformation, the experimental apparatus for yeast continuous culture, the computational steps that generate the barcode-to-gRNA assignments, or the computational steps that quantify RNA and DNA barcodes. Protocols and guidelines for these steps have been provided in earlier work as follows. The high-efficiency lithium acetate yeast transformation, suitable for introducing plasmid libraries, has been previously described (Gietz and Schiestl, 2007). A recommended system for yeast continuous culture has been described in an earlier publication (McGeachy et al., 2019). All computational pipelines are described previously and in the original publication that introduced CiBER-seq (Myint et al., 2019; Muller et al., 2020), and the source code available at https://github.com/ingolia-lab/CiBER_seq.

Figure 1. Overview of the four sub-protocols described within this protocol. Red octagons indicate convenient stopping points within the protocol. A yeast barcoded gRNA plasmid library is available on Addgene, pending internal processing (Addgene #166966). Colored boxes indicate steps of the workflow that are described within each of the four sections. Workflow steps that are not contained within colored boxes are not described in this protocol.
1. Plasmid library construction
A prepared barcoded-gRNA plasmid library (Muller et al., 2020) is available from Addgene by request, which contains ~60,000 gRNAs targeting nearly all ~6,000 yeast protein-coding genes and ~240,000 barcodes—an average of ~4 barcodes per gRNA. When using this library, advance straight to the divergent promoter insertion step (section C, step 27), skipping the gRNA library and barcode library insertion steps (sections A and B). Details about the specific gRNAs in this library have been previously published (McGlincy et al., 2021). When creating a custom barcoded gRNA library, start the plasmid assembly from the beginning of this protocol. For custom gRNA libraries driven by the yeast RPR1 promoter, the custom gRNA oligo pool can be designed using the same flanking sequences, as shown in Figure 2. The 20 nucleotide gRNA target sequence is flanked by 20 nucleotides on either side that match the sequence of the destination vector. The flanking sequences shown in Figure 2 are provided here in text format, for ease of use in custom designs.
Left flanking sequence: tctgggagctgcgattggca; Right flanking sequence: gttttagagctagaaatagc

Figure 2. PCR amplification of the CRISPR gRNA oligo pool. A custom gRNA oligo pool containing the displayed flanking sequences in red can be amplified using NM636 and NM637.
Assembly of gRNA library into Addgene parent vector pNTI743
Amplify 100 pg CRISPRi gRNA oligo pool using primers NM636 and NM637. Figure 1 shows the design of the CRISPRi Oligo Pool and the final amplicon after amplification by PCR.
Set up two PCR reactions using Q5 polymerase: one 7× scaled 350 µL PCR reaction, using the standard buffer conditions, and one 50 µL PCR reaction with the optional Q5 High GC Enhancer, as detailed by manufacturer protocols (NEB, catalog number: M0491L). The 7× scaled reaction can be split evenly across seven PCR tubes, to ensure uniform heating across samples within the thermocycler. The amplicons from these two PCR reactions will be mixed in downstream steps to mitigate the drop out of GC rich gRNAs represented in the final gRNA library. Use 100 pg of the CRISPRi Oligo Pool as the template per 50 µL of PCR reaction.
Thermocycler conditions:
Initial Denaturation 98°C 30 s 9 Cycles 98°C
58°C
72°C
10 s
15 s
10 s
Final Extension 72°C 2 min Hold 4–10°C Purify the PCR reactions using a DNA clean & concentrator kit, according to manufacturer's instructions (Zymo, catalog number: D4013). Keep the PCR products amplified using the standard buffer conditions separate from those made with High GC Enhancer.
Use a nanodrop spectrophotometer to quantify the purified PCR products. Mix the standard and High-GC PCR products in a 9:1 molar ratio. The inclusion of 10% PCR product amplified with the High-GC Enhancer mitigates the dropout of guides with high GC content within the final gRNA library pool, improving the distribution of gRNAs within the library.
Digest 5 µg of the parent vector (pNTI743 dual-barcoded gRNA parent vector available at Addgene) with AvrII, using the manufacturer’s specified buffering conditions. Digest for 4 h, to ensure complete digestion. Purify the digested vector using a Zymo DNA Clean & Concentrator kit.
Assemble 1 µg of digested backbone with 12-fold molar excess of amplified gRNA library pool from step 3, according to the HiFi DNA assembly (NEB, catalog number: E2621L) manufacturer’s instructions. Scale this assembly reaction to a 100 µL volume. Incubate at 50°C for 1 h.
Note: Do not use alternative Gibson master mixes, as high efficiency and fidelity assembly is important for this step.
Purify the assembly reaction according to the DNA Clean & Concentrator kit instructions, and elute in 12 µL of nuclease-free water.
Set up five electroporation reactions according to the MegaX electrocompetent cells (Invitrogen, catalog number: C640003) kit instructions, up to and including the outgrowth step. One vial contains 100 µL of electrocompetent cells and each electroporation requires 20 µL. Use 1 µL of column-purified assembly reaction for each 20-µL-electroporation. Include all specified electroporation conditions and outgrowth steps to maximize transformation efficiency. Expect at least ~30 million transformants per 20-µL-electroporation. Aim to cover a given library with at least 500× coverage, as this will ensure sufficient diversity of the library with fairly uniform coverage of gRNAs.
Take note of the total volume of the outgrowth (5 mL, if following the MegaX protocol for 100 µL of electrocompetent cells), and plate two separate dilutions equal to 1:1,000,000 and 1:10,000,000 of the total cells, represented in the outgrowth on LB-carbenicillin selective plates. The dilution plating will be used to calculate the total number of colony-forming units, and estimate library size and diversity. Incubate at 37°C overnight.
Inoculate the rest of the outgrowth into 500 mL of sterile LB-carbenicillin selective media in a 4-L-Erlenmeyer flask.
Incubate the 500 mL liquid culture with shaking (250 rpm) at 30°C. When the culture reaches an OD600 of 2, centrifuge at 4,000 × g for 10 min to pellet cells. Aspirate and discard the media, and freeze E. coli pellets at -20°C.
Note: Allowing liquid cultures to grow to saturation can skew representation of individual members within the library pool, decreasing library diversity in downstream experiments. At 30°C, it takes roughly 12 h to reach an OD600 close to 2. Begin monitoring the OD600 around 10 h after starting the 30°C incubation. The reduced E. coli growth rate at 30°C provides a larger time window for harvesting the cells at an OD600 close to 2. A shorter incubation at 37°C is also acceptable as long as the culture is harvested at an OD600 close to 2, but will require more frequent monitoring.
Count transformant colonies on the selective plate dilutions to determine library coverage. Library coverage = (number of counted colony-forming units) × (dilution used in step 8). If the library coverage is at least 500× (e.g., for a library containing 60,000 unique guides, at least 30 million calculated colony-forming units), extract plasmid library DNA from the pelleted E. coli in step 10. If library coverage is insufficient, plasmid library DNA can still be extracted and added to a repeat of the transformation (steps 7–10), to meet library coverage guidelines. Library coverage is additive for the purpose of determining whether several pooled plasmid extractions meet the necessary coverage. If pooling multiple plasmid library extractions from separate transformations, the extractions should be pooled in a manner ratiometric with their estimated library coverage (e.g., a plasmid library with 100× coverage and one with 400× coverage should be pooled in a 1:4 molar ratio, respectively).
Extract plasmid library DNA from frozen cell pellets, using a HiSpeed Plasmid Midi Kit according to the manufacturer's instructions (Qiagen, catalog number: 12643).
Confirm gRNA insertion by sequencing. Pick 20 individual colonies from the dilution plates, create liquid cultures, and purify plasmids according to plasmid miniprep kit instructions. Perform Sanger sequencing on these 20 plasmid purifications and on the purified plasmid pool, using custom sequencing primer RM377. Nearly all individual colonies (at least 18 out of 20) should contain a single, correct gRNA insertion, and the plasmid pool sequencing should report 20 “N” bases followed by properly phased vector sequence after the variable gRNA sequence.
Assemble a library of dual barcodes into the gRNA plasmid library, targeting an average of ~4 barcodes per gRNA
Anneal and extend oligos RM720 and RM721 to generate the dual barcode insertion. Figure 3 indicates how oligos RM720 and RM721 anneal to each other. Set up eight 50 µL of Q5 PCR reactions according to the manufacturer’s instructions, using a final concentration of 0.5 µM for oligos RM720 and RM721.

Figure 3. Construction of the dual barcode insert. The dual barcode insert is constructed by annealing and extending two oligos. The final product contains two 26-random nucleotide barcodes, an Asc1 restriction site for downstream insertion of divergent promoters, and Gibson homology arms.Use the following thermocycler conditions:
98°C 30 s
98°C 15 s
[68°C 15 s, 72°C 5 s] ×6
12°C hold
This unusual protocol deliberately excludes the denaturing step in the six cycles, to avoid heteroduplex formation and barcode mixing. The goal is to capture each barcode exactly once.
Purify the annealed and extended barcode PCR amplicon, using a DNA Clean and Concentrator kit. Elute with 13 µL of nuclease-free water. This PCR product contains the dual N26 barcode with flanking homology arms, as shown in Figure 3.
Determine which restriction enzymes will be used in downstream plasmid library preparation, any in vitro transcription reactions of the eventual sequencing library preparation (if preparing DNA barcode libraries), the PacBio library construction, and any other downstream manipulations. Because the random nucleotide barcode can contain restriction enzyme cleavage sites by chance, some barcodes will be lost from the population during these downstream restriction digestions. Thus, pre-digesting the dual barcode PCR amplicon with any restriction enzymes used in the downstream workflow will purge your final dual barcode library of restriction enzyme barcode artifacts. For this experimental set-up, suggested enzymes are HindIII-HF, PmeI, BglII, and XhoI (used for downstream long-read PacBio sequencing library prep), and MfeI-HF, or PvuII-HF (used to linearize plasmid during in vitro transcription step of DNA barcode library prep).
Digest the dual barcode PCR amplicon with the set of chosen restriction enzymes in a 200 µL reaction and suitable NEB digestion buffer that optimizes reaction conditions for the chosen set of restriction enzymes. Use the following quantities and buffer conditions for a digestion that uses HindIII-HF, PmeI, BglII, XhoI, MfeI-HF, and PvuII-HF. Digest the dual barcode PCR amplicon at 37°C for 2 h.
Component Volume
10× rCutSmartTM Buffer 20 µL
Column-purified dual barcode PCR amplicon 13 µL
HindIII-HF 0.5 µL
PmeI 0.5 µL
BglII 0.5 µL
XhoI 0.5 µL
MfeI-HF 0.5 µL
PvuII-HF 0.5 µL
Nuclease-free water to 200 µL
Purify the digestion reaction using a Zymo DNA Clean and Concentrator kit.
Digest 5 µg of gRNA plasmid library with AscI according to the manufacturer’s protocol, incubating at 37°C for 4 h, to ensure complete digestion. Purify the digested DNA using a Zymo DNA Clean and Concentrator kit.
Refer to steps 5 and 6 to assemble the dual barcode library into the digested gRNA plasmid library. Use a 12:1 molar ratio of dual barcode product to plasmid backbone in the DNA HiFi assembly reaction.
Set up a transformation using one vial of NEB® 10-beta Competent E. coli (High Efficiency) C3019H and 3 µL of the purified DNA assembly reaction from step 20, according to the manufacturer’s instructions. Follow the manufacturer’s instructions up to and including the 1 h outgrowth step at 37°C. Heat-shock transformation is used at this step instead of electroporation with the MegaX electrocompetent cells, because the lower transformation efficiency of the heat-shock transformation is more convenient for controlling library diversity. Plate two separate dilutions equal to 1:100,000 and 1:1,000,000 of the total cells represented in the transformation outgrowth on LB-carbenicillin selective plates, and incubate at 37°C.
The total library diversity is controlled at this step by aliquoting varying amounts of the transformation outgrowth into separate flasks, as shown in Figure 4. Inoculate separate flasks with the following fractions of the transformation: 4%, 6%, 8%, 22%, and 44%. For each fraction, add a proportional amount of LB-carbenicillin liquid media to the flask, 500 mL media for each 1 mL of transformation outgrowth. Thus, as an example, add 20 mL media to the 4% inoculation (40 µL of the 1 mL outgrowth). Incubate flasks with shaking at 250 rpm at 30°C, until cultures reach an OD600 close to 2.

Figure 4. Controlling total library size during the dual barcode insertion step. Transformation outgrowth fractions of variable library coverage are used to obtain a target library size that covers each gRNA with an average of four barcodes. Outgrowth fractions are pooled to reach the desired library size. As long as the target library size is less than the total number of unique plasmids within the full set of outgrowth fractions, the strategy depicted here can reach a selected library size to within 2%.Use the dilution plates from step 21 to estimate the number of total colony-forming units, and hence the total number of unique plasmids represented in each flask. As an example, a dilution plating that indicates 2 million total transformants in the transformation outgrowth means the 4% fraction flask contains 80,000 plasmids. Determine the desired total library diversity and pool fractions accordingly. For a plasmid library containing 60,000 unique gRNAs, an average of four barcodes per gRNA is suggested, to provide a sufficient number of barcode measurement replicates per gRNA while still maintaining a manageable total library size. In the example indicating 2 million total transformants, combining the 4% fraction flask (80,000 barcoded gRNA plasmids) and the 8% fraction flask (160,000 barcoded gRNA plasmids) reaches the target of 240,000 barcoded gRNA plasmids. Harvest the pooled liquid culture by centrifuging at 4,000 × g for 10 min. Discard media and freeze E. coli pellets at -80°C.
Extract plasmid library DNA from the cell pellet using a HiSpeed Plasmid Midi Kit according to manufacturer's instructions (Qiagen, catalog number: 12643).
Sequence the dual barcode insertion, both on a collection of ~20 individual colonies from one of the dilution plates, and on the overall extracted plasmid pool. At least 18 out of the 20 individual colonies should contain a correct dual barcode insertion, and the bulk plasmid sequencing should report 26 “N” bases at each dual barcode annotation, followed by the properly phased vector sequence after the barcode sequences. Use primer KS533 to sequence the dual barcode region.
Refer to "Barcode to gRNA assignment" protocol for details on preparing a PacBio sequencing library to assign barcodes to gRNAs. The plasmid library is designed in such a way that the divergent promoters can be modularly inserted, and the same barcode-gRNA assignment can be used for each divergent promoter plasmid library.
Insertion of divergent promoter reporter into barcoded gRNA plasmid library
Generate a DNA fragment that contains the divergent promoters and ORFs of interest with Gibson homology arms that match the barcoded gRNA library on either side of the AscI site (in between the dual barcodes). This can be done by PCR amplification, or restriction digest from a template plasmid. The P(PGK1)-P(HIS4) divergent promoter template plasmid is available on Addgene (Plasmid, catalog number: 164917, pNTI757 P[PGK1]-citrine P[HIS4]-mCherry) for this purpose, and digestion with BciVi will generate a fragment that contains the necessary Gibson homology arms at each end (Figure 5A). Restriction enzyme sites that flank each promoter in pNTI757 facilitate cloning in other regulatory DNA sequences of interest. The regulatory sequences should not contain the BciVI cut site, if planning to use BciVI restriction digestion to generate the DNA fragment insert. If amplifying an insert by PCR, make sure the reaction yields one band at the expected size. Primer dimers or PCR amplification of shorter products can still insert, if they have the same Gibson homology arms. As an optional additional consideration, the DNA fragment can include a 5-nt identifier at each end, placed just upstream of the 3′ UTR barcodes, within 50 nucleotides of TruSeq adapter sequence. This allows multiple reporter libraries to be assayed in the same pooled experiment. With different nucleotide identifiers, the single-end reads can sequence through the 26-nt barcode, as well as the 5-nt reporter ID.
AscI cuts in between the two 26-nt random nucleotide barcodes, such that an inserted divergent promoter will drive expression of each separately. Digest 5 µg of barcoded gRNA library pool with AscI, according to the manufacturer instructions with two modifications: scale the reaction to accommodate 5 µg of plasmid, and increase the digestion time to 3 h, to ensure complete digestion. After the AscI digestion, add 0.2 units of T5 exonuclease (NEB, catalog number: M0663S), and digest at 30°C for 20 min. AscI and T5 conveniently use the same buffering conditions, so the T5 enzyme can be added directly to the digestion reaction. The T5 exonuclease chews back the overhangs left by AscI and prevents re-ligation during the DNA assembly reaction. Clean and concentrate the backbone using a Zymo DNA clean and concentrator kit.
Assemble the prepared divergent reporter insert from step 27 into 1 µg of digested backbone from step 28, according to the manufacturer’s instructions for HiFi DNA assembly (NEB, catalog number: E2621L). Use a 5×-molar ratio of insert to backbone. Scale the DNA assembly to a 50 µL reaction. Do not substitute with standard Gibson master mix, as this lowers the efficiency of desired DNA assembly. Incubate at 50°C for 1 h.
Clean and concentrate the DNA assembly reaction using a Zymo DNA clean and concentrator kit. Elute in 6 µL of nuclease-free water to maximize concentration.
Refer to steps 7–12 to electroporate the DNA assembly reaction and purify the final plasmid library from liquid culture.
Sequence the divergent promoter insertion, both on a collection of ~20 individual colonies from your dilution plate, and on the overall extracted plasmid pool. At least 17 out of the 20 individual colonies should contain the divergent promoter insertion, and the bulk plasmid sequencing should report 26 “N” bases at each dual barcode annotation, followed by properly phased vector sequence after the barcode sequences. Use primers KS533 and RM336 to sequence the divergent promoter insertion at each end. The plasmid sequence should match the design shown in Figure 5B. This plasmid library is ready for library experiments with yeast expressing dCas9-Mxi1 (Muller et al., 2020).

Figure 5. Assembly and layout of the Final Plasmid Library. A) Schematic of barcoded gRNA plasmid library referred to in step 26 and pNTI 757, the template for divergent reporters. Restriction sites are noted that allow the divergent promoter to be cut from pNTI 757 and inserted by Gibson assembly into the barcoded gRNA plasmid library. B) Schematic of the final reporter plasmid library. Divergent promoters drive expression of a fluorescent protein – mCherry or Citrine – and a 26-nt barcode embedded into the 3′ UTR. Flanking TruSeq sequences facilitate downstream sequencing applications to quantify barcode expression.
2. PacBio Library Preparation
Because PCR amplification can introduce artifacts and PCR-mediated exchange, barcoded gRNA DNA fragments are generated by restriction digestion of the barcoded gRNA plasmid library. For libraries constructed in the pNTI743 backbone, digest 5 μg of dual barcoded gRNA plasmid library with HindIII-HF and PmeI, according to the manufacturer’s restriction digest protocol. Separately digest 5 μg of dual barcoded gRNA plasmid library with restriction enzymes BglII and XhoI, according to the manufacturer’s protocol. During plasmid library construction, the dual barcode sequence was pre-digested with HindIII-HF, PmeI, BglII, and XhoI, to avoid dropout of random barcodes that contain at least one of these restriction sites. (See "Plasmid library construction" protocol for additional details on the barcode pre-digestion). The gRNA library, however, still contains sequences that can be digested by these restriction enzymes. Two separate sets of restriction enzymes thus ensure that nearly all gRNA sequences can be recovered in at least one restriction digestion set.
Run the two restriction digest reactions on separate lanes of a 1% agarose TBE gel. Ignore the backbone DNA fragment that runs at 4,080 bp and extract the ~1,200-bp fragment for each lane, which corresponds to the digestion fragment containing the dual barcode and gRNA. Extract the DNA fragment from the gel pieces using a Zymoclean Gel DNA Recovery Kit (Zymo, catalog number: D4001) according to manufacturer instructions. Elute in 12 µL of nuclease-free water.
Quantify the concentration of the two purified DNA fragments by placing 1.5 µL of sample on a nanodrop spectrophotometer. Mix the two samples equimolarly. Use 1,000 ng of the equimolar mix as the input for the dumbbell adapter ligation reaction. Save 50 ng of each gel-purified fragment for downstream fragment analysis by TapeStation in step 5. Follow the manufacturer’s instructions using the SMRTbell Express Template Prep Kit 2.0 (#100- 938-900) to perform the dumbbell adapter ligation. The Version 02 Part Number 101-791-800 (April 2020) protocol was used for this study. The multi-step kit protocol will perform the DNA damage repair, the DNA end repair and A-tailing, and the dumbbell adapter ligation reactions.
Ampure XP beads bind nucleic acids within a size range that is determined by the bead ratio. Use Ampure XP bead size selection to purify the sequencing library, and remove unligated dumbbell adapters. Follow Ampure XP bead manufacturer’s instructions, which can be found in the SPRIselect user guidebook, version B24965AA. The Ampure XP/SPRI-select documentation recommends a left side size selection with a 0.6× bead ratio, to purify the sequencing library (>1,000bp) from the free adapters (<100bp). Perform the final elution in nuclease-free water.
Perform DNA fragment analysis on the input controls from step 3 and the PacBio library from step 4 using an Agilent TapeStation. A fragment size below 1000 bp should be analyzed with a High Sensitivity D1000 ScreenTape. Fragments over 1,000 bp should be analyzed using a Denomic DNA ScreenTape. Refer to the appropriate Agilent ScreenTape Quick Guide for step-by-step instructions. Samples will need to be diluted to a concentration within the suggested linear range specified in the Quick Guide. Compare the PacBio library from step 4 against the two input controls saved from step 3. A peak shift of about 150 bp between the input and final library indicates dumbbell adapter ligation as shown in Figure 6. The sample is ready for sequencing using the Sequel II system. See PacBio documentation for more details about sequencing.
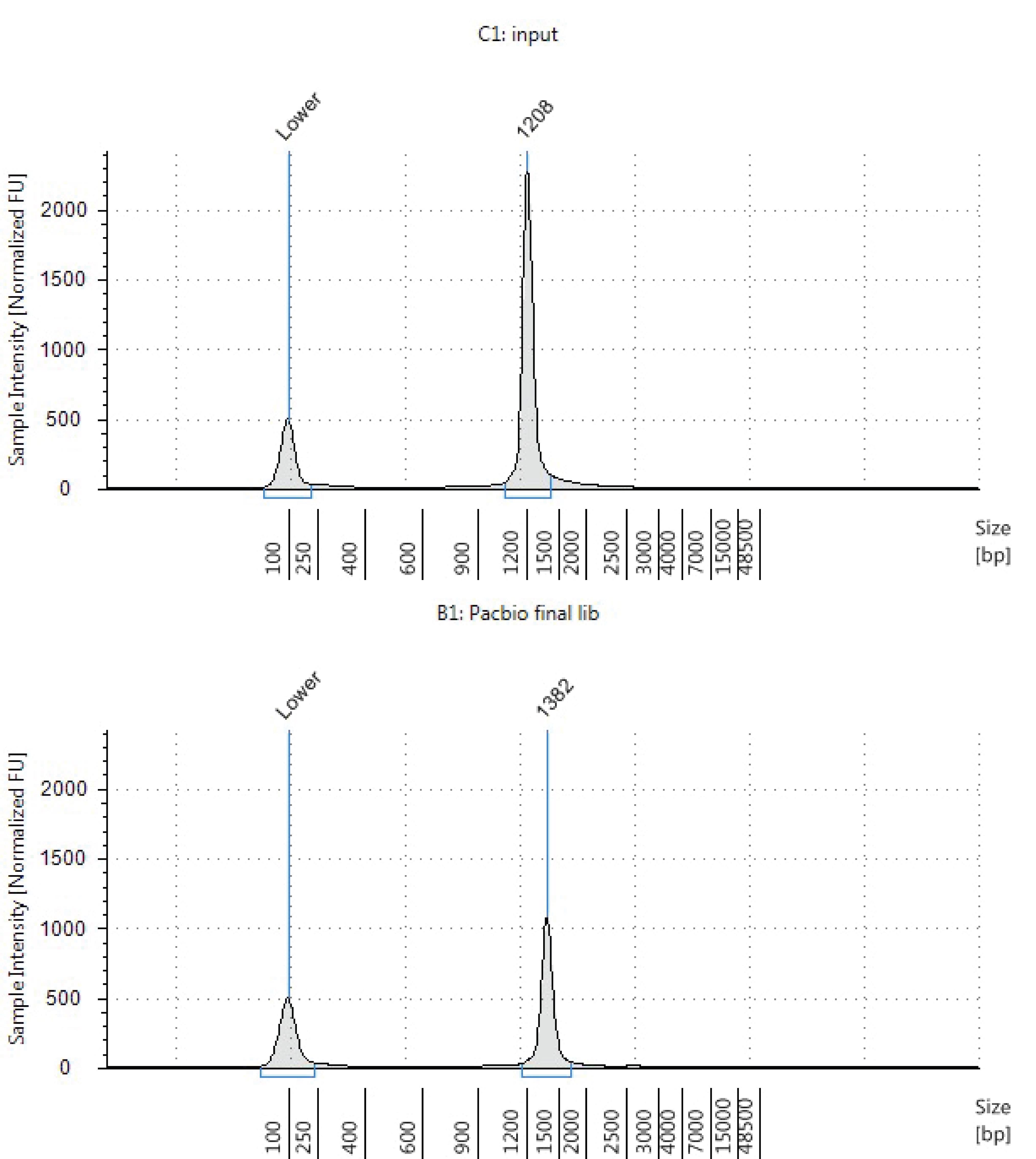
Figure 6. Fragment analysis of input library and assembled PacBio library. The shown example library was analyzed using a genomic DNA ScreenTape. The input library runs at 1,208bp and shows an upshift to 1,382bp after the dumbbell linker ligation.
3. Expressed RNA barcode library preparation
RNA Extraction:
Note: This protocol has been adapted from Cold Spring Harbor, RNA yeast extraction protocol (Ares, 2012).
Place Thermomixer in fume hood and set to 65°C.
Chill microcentrifuge to 4°C (all centrifugation steps should take place at 4°C).
Prepare 10 mL of AE buffer (see Recipes section for details).
Thaw frozen yeast pellets on ice. The pellets should each contain ~250–500 million yeast cells, corresponding to 25–50 mL of spun down haploid yeast at OD = 1 (1 mL of haploid yeast at OD600 = 107 cells). This is enough to cover a 240,000 barcoded guide library with at least 1,000× coverage.
Note: It is good to harvest, pellet, and freeze down (at -80°C) extra yeast pellets as back-up.
Resuspend the pelleted cells in 400 μL of AE buffer and transfer to a 1.5-mL RNase-free microcentrifuge tube.
Add 40 μL of 10% SDS and 400 μL of phenol-chloroform-isoamyl alcohol (PCA) (Ambion, catalog number: AM9730). Place caplock clip on tube. Vortex the mixture quickly and then place the tube into the thermomixer set at 65°C.
Incubate the cells for 15 min at 65°C on the thermomixer at 2,000 rpm.
Remove from thermomixer and incubate the cells for 5 min on ice with the caplock clip still on.
Centrifuge the sample at 20,000 × g at 4°C in a microcentrifuge for 5 min. After centrifugation, the organic phase will separate from the aqueous phase. The RNA will be in the upper aqueous phase.
Using a pipette, transfer the top aqueous phase to a separate RNase-free tube. Avoid transferring any of the interphase, which is the white layer in between the top aqueous phase and the bottom organic phase. The interphase contains DNA, which will interfere with downstream library preparation steps. It is preferable to get less aqueous phase than to contaminate the recovered material with DNA from the interphase. Discard the lower organic phase.
Add 400 μL of chloroform to this top (aqueous) phase. Mix by shaking for 10 s. This step removes water-dissolved organic solvents from the aqueous phase. Centrifuge again at 20,000 × g and 4°C for 5 min.
Add another 400 μL of chloroform to the tube and shake again. Centrifuge one last time at 20,000 × g and 4°C for 5 min.
Carefully transfer the top aqueous phase into a new 1.5 mL microcentrifuge tube and quantify the volume by micropipette.
RNA Precipitation:
Add 50 μL of 3 M NaOAc, pH 5.2, and 1 µL of Glycoblue to the aqueous phase.
Calculate the required volume of 100% ethanol needed to reach a final concentration of 70% ethanol when added to the aqueous phase. Do not overfill the tube. If the required volume exceeds the size of the tube, split the aqueous phase across multiple tubes before adding ethanol. Make sure that the cap fits and then mix well by inverting several times. The RNA should precipitate readily at this concentration, so it is not necessary to chill the ethanol (If needed, this is a good stopping point; RNA can be stored in ethanol at -20°C overnight).
Centrifuge at 20,000 × g at 4°C for 10 min. You should obtain an RNA pellet at the bottom of the tube. The pellet should be mostly white, with some light blue from the GlycoBlue. Due to the scale of this RNA extraction, you should have a sizable pellet, roughly the size occupied by 10–15 μL of water.
Pipette off the ethanol and rinse the pellet with 70% ethanol. Centrifuge at 20,000 × g at 4°C for 5 min.
Pipette away the 70% ethanol wash. Centrifuge at 20,000 × g for 30 s, to collect residual ethanol wash at the bottom of the tube using a smaller tip size to pipette off the remaining residual ethanol away from the RNA pellet. Open the tubes and place them sideways in a rack to prevent debris from falling in. Allow the pellet to air-dry at room temperature, which takes roughly 15 min. Do not dry the pellet for longer than 1 h, as this can lead to over-drying. Confirm that all the residual ethanol is gone from the pellet before proceeding to RNA resuspension.
RNA Resuspension:
Resuspend the pellet in 100 μL of nuclease-free H2O. Vortex or triturate with a pipette until fully dissolved. It may take some time for the RNA to dissolve, so be patient and keep the RNA on ice. Carefully inspect the sample to make sure that there are no transparent chunks of RNA precipitate.
Tip: Freezing the RNA solution once after this step can help to homogenize the solution.
Once the pellet is completely redissolved, take a Nanodrop measurement to quantify the RNA. If the RNA is pure, the A260:A280 ratio should be 1.8–2.2, the A260:A230 ratio should be above 2, and the A280 should never be higher than the A260 value; otherwise, there is potential phenol contamination.
Store the RNA in H2O at -80°C. It should be stable for several years.
Preparation of barcoded RNA sequencing libraries
This protocol is specifically designed for preparing RNA sequencing libraries from yeast transformed with dual barcoded-expression cassette gRNA libraries, such as the dual citrine/mCherry expression cassette used inMuller et al. (2020). This plasmid is constructed with 5-nt identifiers for both divergent expression cassettes, to allow pooling of multiple yeast strains and/or libraries within a single experiment. Expression cassettes that do not use citrine or mCherry will need custom primers not provided in this protocol to add the TruSeq adapters. The TruSeq R2 custom primer, as shown in Figure 7, should anneal to the chosen cassette roughly 300 nucleotides upstream of the 26-nt barcode and include the TruSeq R2 overhang.

Figure 7. Experimental workflow to prepare sequencing libraries from expressed barcodes. Expressed barcodes are reverse transcribed and amplified by PCR to introduce necessary adapters and TruSeq sequences for high-throughput sequencing.
Thaw extracted RNA on ice.
Treat 20 µg of RNA with Turbo DNase I according to the manufacturer’s instructions and purify the RNA using the Zymo RNA clean and concentrator kit. Elute the RNA in 30 µL of nuclease-free water. Quantify the RNA concentration, which should be approximately 500 ng/µL.
Reverse transcribe 4 µg of purified RNA for each sample, according to Protoscript II manufacturer’s protocol. (This will be a 4× scaled, 80 µL reverse transcription reaction, since the protocol recommends 1 µg of RNA per 20 µL reaction). Prime the reverse transcription reaction with oligo dT.
Following the heat denaturation step of the reverse transcription reaction, add 0.5 µL of RNase A and 0.5 µL of RNase H directly to the 80 µL reverse transcription reaction, and incubate at 37°C for 1 h. (This is important to get rid of RNA that wasn’t reverse transcribed, like rRNA and tRNA, since these can compete with column binding.) Purify the reverse-transcribed DNA with a Zymo DNA clean and concentrator kit according to the manufacturer’s instructions, using ssDNA 7:1 ratio binding buffer. Elute in 13 µL of nuclease-free water.
Set up a 50 µL PCR reaction using Q5 polymerase according to the manufacturer's instructions, to amplify a stretch of ~300-nt on the cDNA template, containing the expressed barcode. This step adds the i7 annealing site for downstream library preparation. Use standard Q5 buffering conditions with no additional high GC enhancer. Use six extension cycles to avoid PCR over-amplification, and half the purified reverse transcription product from step 25 as template. Save the other half of the column-purified reverse transcription product to repeat the procedure, if necessary. Use primers RM511 and RM810 to amplify barcodes expressed by the citrine expression cassette (annealing temperature 70°C). Set up a separate 50 µL PCR reaction, using primers RM812 and RM810, to amplify barcodes expressed by the mCherry expression cassette (annealing temperature of 68°C). Use an extension at 72°C for 10 s.
Thermocycler conditions:
Initial Denaturation 98°C 30 s 6 Cycles 98°C
68°C or 70°C
72°C
10 s
15 s
10 s
Final Extension 72°C 2 min Hold 4–10°C Column purify the PCR product according to the Zymo DNA clean and concentrator kit instructions, and elute in 13 µL of nuclease-free water.
The purified PCR products now have the primer annealing sites that will allow for subsequent PCR amplification with NEB i5/i7 dual index primers (NEB, catalog number: E7600S). Assign unique dual index primer pairs to each sample, making sure to choose a set of dual index primers that are compatible for pooled sequencing, and will allow the samples to be separated by an adapter sequence. Refer to the dual index primer manual for more information. Set up a 50 µL PCR reaction using Q5 polymerase according to the manufacturer's instructions, with half of the column-purified product from step 27 as template. Set aside the other half of the column-purified product to repeat the procedure if necessary. Use standard Q5 buffering conditions with no additional high GC enhancer. Use an annealing temperature of 72°C, and a 10 s extension time. Aim for a low number of PCR cycles, to avoid overamplification of PCR product. For highly-expressed genes, like those driven by the PGK1 promoter, 8–10 cycles is sufficient, but lower-expressed transcripts may need more cycles. An appropriate cycle number for a given transcript can be estimated by determining its abundance relative to PGK1 mRNA transcript. For example, a transcript that is expressed at 1/16th the abundance of PGK1 mRNA should be amplified with four additional cycles (24 = 16), for a total of 12–14 cycles.
Thermocycler conditions:
Initial Denaturation 98°C 30 s 8–14 Cycles 98°C
72°C
72°C
10 s
15 s
10 s
Final Extension 72°C 2 min Hold 4–10°C Column purify the PCR product according to the Zymo DNA clean and concentrator kit instructions, and elute in 13 µL of nuclease-free water.
Run a 1:10 dilution of the PCR product on a Tape Station/Bioanalyzer, to determine the concentration and sizing of each amplified barcode library. One sharp peak at the expected amplicon size, ~330bp, indicates a successfully-prepared sample. An additional peak that runs at a high molecular weight may indicate overamplification, as amplicons that melt and re-anneal form bubbles at the mismatched barcode sequence tend to run slower on the Tape Station (Figure 8). This over-amplification peak should be <10% the molarity of the ~330bp peak. Over-amplified samples can be re-prepared by decreasing the number of PCR cycles and repeating steps 28 and 29, using the saved remaining PCR product from step 27 as the template. Free primers and primer dimers in the sample should be removed using AMPure XP size selection or a similar size selection strategy, as they affect the quality of downstream sequencing.

Figure 8. Fragment analysis of RNA barcode library. The final barcode sequencing library runs at 359 bp in the example TapeStation. The peak at 534 bp represents barcode library PCR amplicons that have melted and re-annealed to mismatched barcodes. The mismatched duplexes run at a higher molecular weight on the TapeStation.Successfully-prepared samples can be pooled and submitted for HiSeq or NovaSeq. Barcode libraries should be sequenced at a depth of at least 100 sequence reads per barcode per sample. For example, an experiment spanning four samples that contain 240,000 barcodes each should be sequenced with at least 96 million reads.
4. DNA library prep from yeast pellets
Thaw frozen, pelleted yeast library samples on ice. It is good to harvest, pellet, and freeze down (at -80°C) extra yeast so that you can repeat the library preparation in case the process fails. For reference, 1 mL of haploid yeast at OD600 of 1 corresponds to 107 cells, so a pellet from 50 mL of culture at OD600 of 1 contains approximately 500 million cells. Such a 500 million cell pellet should cover a library with 240,000 barcodes, with approximately 2,000 cells per barcode. Note that the yeast miniprep yield may be poor, so aiming for 500 million cells should still give sufficient coverage in the event of inefficient plasmid recovery.
Perform the Zymo Yeast miniprep according to the manufacturer's instructions for extracting plasmid from yeast liquid culture, with the following protocol adjustments. Resuspend the pellet in 1 mL of solution 1 (digestion buffer), and add 30 µL of zymolase. Digest for 3 h and mix in a thermomixer at 900 rpm, to ensure efficient plasmid extraction. Scale up volumes of solution 2 and solution 3 by five times, to match the 5× scale of solution 1. Distribute across multiple microcentrifuge tubes, so the entire reaction can be centrifuged, and sequentially pass the supernatants from each tube across the same column. To maximize DNA recovery, perform the final plasmid elution using 20 µL of nuclease-free water warmed to 37°C.
Digest all extracted plasmid with MfeI-HF (if using a citrine expression cassette in your library), PvuII-HF (if using mCherry expression cassette in your library), or an appropriate restriction enzyme (if using a custom expression cassette) to linearize the plasmid. If using a custom expression cassette, choose a restriction enzyme that does not cut in between the embedded T7 promoter and the annealing site for the TruSeq R2 primer, as depicted in Figure 9. Perform a 3 h digestion to ensure complete digestion has occurred. Purify digested DNA using a Zymo DNA clean and concentrator according to the manufacturer’s instructions, and elute using 20 µL nuclease-free water warmed to 37°C to maximize DNA recovery.

Figure 9. Experimental workflow for preparing barcode sequencing libraries from extracted plasmid. Sequencing libraries are prepared by linearizing extracted plasmid, in-vitro transcribing the barcodes, reverse transcribing the in-vitro transcription product, and PCR amplifying the prepared cDNA, to add the necessary adapters and TruSeq sites for downstream sequencing applications.Perform an in vitro transcription (IVT) reaction, using the T7 HiScribe kit according to the manufacturer's recommendations for small length product (30 µL reaction, short transcripts, at 37ºC overnight). Incubate the reactions at 37°C in an incubator to prevent evaporation/condensation on the top of the lid.
Use the following amounts of reagents.
Reagent Amount
NTP Buffer Mix 10 µL
Purified plasmid digest 18 µL
T7 RNA Polymerase Mix 2 µL
Total 30 µL
Use the T7 HiScribe suggested DNase I treatment to remove template DNA. Add 18 µL of nuclease-free water and 2 µL DNase I from the kit and digest at 37°C for 15 min. Purify the RNA using a Zymo RNA clean and concentrator kit (Zymo, catalog number: R1017) according to the manufacturer’s instructions.
Quantify the in vitro transcribed RNA via Nanodrop and reverse transcribe 2 µg of IVT product according to Protoscript II (NEB, catalog number: M0368L) manufacturer’s protocol. This will be a 2× scaled reverse transcription reaction, since the protocol recommends 1 µg of RNA per reaction. Use the primer corresponding to your plasmid library (511 DNA_IVT_RT for the citrine expression cassette, 546 mcher IVT_RT for the mCherry expression cassette, or custom primer) to perform the RT reaction. This primer adds the i7 annealing site for downstream library preparation. The plasmid already contains the i5 (TruSeq R1) priming sequence adjacent to the random nucleotide barcode.
Note: T7 has non-specific polymerase activity (especially for overnight and low input reactions) so only roughly 10% (estimated) of the IVT RNA product is the intended template. The rest is arbitrary RNA. These non-specific RNA species generally don't cause issues as they won't be reverse transcribed or amplified by PCR in downstream steps.
Following the reverse transcription heat denaturation step, add 0.5 µL RNase A and 0.5 µL RNase H to the reverse transcription reaction. Incubate at 37°C for 1 h. Purify the cDNA using a DNA Clean & Concentrator-5, with a 7:1 ratio of binding buffer to sample, according to the manufacturer’s instructions for ssDNA purification (Zymo, catalog number: D4013).
The purified cDNA now contains both sequences required for amplification with NEB i5/i7 dual index primers. Assign distinct primer pairs to each sample, making sure to choose a set of dual index primers that are compatible for pooled sequencing, and will allow the samples to be separated according to their i5/i7 adapter sequences. Refer to the dual index primer manual for more information. Perform Q5 PCR amplification with half of the column-purified product from step 7 as template, and NEB i5/i7 dual index primers according to the manufacturer's instructions. Save the other half of the column-purified product to repeat amplification in case this is necessary. Use an annealing temp of 72°C, and a 10 s extension. Aim for a low number of amplification cycles, to avoid overamplification of the PCR product—ideally 6–10 cycles, but more may be needed if the yield is insufficient).
Column purify the PCR product according to the Zymo DNA clean and concentrator kit instructions, and elute in 13 µL of nuclease-free water.
Run a 1:10 dilution of the amplified library samples from step 9 on a Tape Station or Bioanalyzer, to determine the concentration and sizing of each amplified barcode library. Refer to step 30 of the RNA library preparation protocol for details on assessing library quality. Refer to Figure 8 for an example TapeStation of the barcode library.
Samples can be pooled and sequenced on an Illumina HiSeq or NovaSeq. Barcode libraries should be sequenced at a depth of at least 100 sequence reads per barcode per sample. For example, an experiment spanning four samples that contain 240,000 barcodes each should be sequenced with at least 96 million reads.
Recipes
AE buffer
Reagent Quantity for 10 mL Final Concentration Sodium Acetate (NaOAc; 3 M pH5.5) 0.167 mL 50 mM EDTA (O.5M, pH 8.0) 0.2 mL 10 mM Nuclease-Free H2O 9.63 mL
Acknowledgments
This work was funded by NIH grants DP2 CA195768, R01 GM130996, and R01 GM135233. This work used the Vincent J. Coates Genomics Sequencing Laboratory at the University of California, Berkeley, supported by NIH S10 OD018174 instrumentation grant. These set of protocols were derived from Muller et al. (2020). All source code is available at https://github.com/ingolia-lab/CiBER_seq and archived at Zenodo doi: 10.5281/zenodo.4035711. Plasmids have been made available through Addgene and plasmid libraries will be available pending internal AddGene processing.
Competing interests
The authors report no competing financial or non-financial interests.
References
- Alford, B. D., Tassoni-Tsuchida, E., Khan, D., Work, J. J., Valiant, G. and Brandman, O. (2021) ReporterSeq reveals genome-wide dynamic modulators of the heat shock response across diverse stressors. eLife 10: e57376.
- Aranda-Díaz, A., Mace, K., Zuleta, I., Harrigan, P. and El-Samad, H. (2017). Robust Synthetic Circuits for Two-Dimensional Control of Gene Expression in Yeast. ACS Synth Biol 6(3): 545-554.
- Ares, M. (2012). Isolation of total RNA from yeast cell cultures. Cold Spring Harb Protoc 2012(10): 1082-1086.
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Gietz, R. D. and Schiestl, R. H. (2007). High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2(1): 31-34.
- Gilbert, L. A., Larson, M. H., Morsut, L., Liu, Z., Brar, G. A., Torres, S. E., Stern-Ginossar, N., Brandman, O., Whitehead, E. H., Doudna, J. A., et al. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154(2): 442-451.
- Jinek, M., East, A., Cheng, A., Lin, S., Ma, E. and Doudna, J. (2013). RNA-programmed genome editing in human cells. Elife 2: e00471.
- Kampmann, M. (2020). CRISPR-based functional genomics for neurological disease. Nat Rev Neurol 16(9): 465-480.
- McGeachy, A. M., Meacham, Z. A. and Ingolia, N. T. (2019). An Accessible Continuous-Culture Turbidostat for Pooled Analysis of Complex Libraries. ACS Synth Biol 8(4): 844-856.
- McGlincy, N. J., Meacham, Z. A., Reynaud, K. K., Muller, R., Baum, R. and Ingolia, N. T. (2021). A genome-scale CRISPR interference guide library enables comprehensive phenotypic profiling in yeast. BMC Genomics 22(1): 205.
- Muller, R., Meacham, Z. A., Ferguson, L. and Ingolia, N. T. (2020). CiBER-seq dissects genetic networks by quantitative CRISPRi profiling of expression phenotypes. Science 370(6522).
- Myint, L., Avramopoulos, D. G., Goff, L. A. and Hansen, K. D. (2019). Linear models enable powerful differential activity analysis in massively parallel reporter assays. BMC Genomics 20(1): 209.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Muller, R. Y., Meacham, Z. A. and Ingolia, N. T. (2022). Plasmid and Sequencing Library Preparation for CRISPRi Barcoded Expression Reporter Sequencing (CiBER-seq) in Saccharomyces cerevisiae. Bio-protocol 12(7): e4376. DOI: 10.21769/BioProtoc.4376.
- Muller, R., Meacham, Z. A., Ferguson, L. and Ingolia, N. T. (2020). CiBER-seq dissects genetic networks by quantitative CRISPRi profiling of expression phenotypes. Science 370(6522).
Category
Molecular Biology > RNA > Transcription
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.