- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Fluorescence-based Assay for Measuring Phospholipid Scramblase Activity in Giant Unilamellar Vesicles
Published: Vol 12, Iss 6, Mar 20, 2022 DOI: 10.21769/BioProtoc.4366 Views: 3091
Reviewed by: Gal Haimovichsujan kumar mondalPhilipp A.M. Schmidpeter
Original research article
The authors used this protocol in:

Jul 2021
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






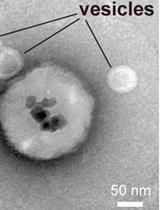
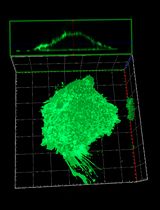

Abstract
Transbilayer movement of phospholipids in biological membranes is mediated by a diverse set of lipid transporters. Among them are scramblases that facilitate rapid bi-directional movement of lipids without metabolic energy input. In this protocol, we describe the incorporation of phospholipid scramblases into giant unilamellar vesicles (GUVs) formed from scramblase-containing large unilamellar vesicles by electroformation. We also describe how to analyze their activity using membrane-impermeant sodium dithionite, to bleach symmetrically incorporated fluorescent ATTO488-conjugated phospholipids. The fluorescence-based readout allows single vesicle tracking for a large number of settled/immobilized GUVs, and provides a well-defined experimental setup to directly characterize these lipid transporters at the molecular level.
Graphic abstract:

Giant unilamellar vesicles (GUVs) are formed by electroformation from large unilamellar vesicles (LUVs) containing phospholipid scramblases (purple) and trace amounts of a fluorescent lipid reporter (green). The scramblase activity is analyzed by a fluorescence-based assay of single GUVs, using the membrane-impermeant quencher dithionite. Sizes not to scale. Modified from Mathiassen et al. (2021).
Background
Biogenesis and maintenance of cellular membranes require dynamic regulation of transbilayer lipid movement. Specific membrane proteins, termed lipid transporters, play a central role in this process. Among these are lipid scramblases that facilitate rapid bi-directional movement of lipids without metabolic energy input. Lipid scramblases are either constitutively active or regulated by physiological stimuli, e.g., a rise in intracellular Ca2+, or proteolytic cleavage (Bevers and Williamson, 2016; Pomorski and Menon, 2016). Constitutively active scramblases are found in the bacterial cytoplasmic membrane and the endoplasmic reticulum (ER), and promote uniform growth of the membranes after synthesis of lipids on the cytoplasmic side. The molecular identity of specific ER scramblases is not known, but at least two proteins contribute to overall scramblase activity in the ER (Chang et al., 2004; Vehring et al., 2007). Constitutive phospholipid scramblase activity is also an unexpected property of Class A G-protein-coupled receptors (Menon et al., 2011; Goren et al., 2014; Morra et al., 2018). In the plasma membrane of eukaryotic cells, regulated scramblases are responsible for a controlled loss of lipid asymmetry, and the appearance of anionic phospholipids, such as phosphatidylserine, at the cell surface. Two major families of proteins are implicated in regulated scrambling: the TMEM16 family (Suzuki et al., 2010; Malvezzi et al., 2013; Brunner et al., 2014), and the Xk-related (Xkr) family (Suzuki et al., 2014). Despite their physiological importance, the precise mechanism of phospholipid scrambling, and how it is regulated by the membrane lipid environment remains to be established.
In vivo analysis of lipid scramblases at the molecular level is hampered by the complexity of the cell. Therefore, phospholipid translocation studies frequently rely upon reconstituted systems involving synthetic large unilamellar vesicles (LUVs, ~200 nm in diameter). However, a significant drawback of ensemble LUV measurements arises from the compositional heterogeneity of protein reconstitutions, which hampers quantitative analysis of vesicle properties (including stoichiometry of lipids, sterols, and transmembrane proteins), and correlation with protein activity (Larsen et al., 2011).
Giant unilamellar vesicles (GUVs, 10–100 µm in diameter) are model membrane systems with dimensions comparable to that of a cell, allowing single vesicle analysis directly by microscopy techniques, such as fluorescence microscopy, fluorescence correlation spectroscopy, or atomic force microscopy (García-Sáez et al., 2010; Bagatolli and Needham, 2014; Velasco-Olmo et al., 2019; Aden et al., 2021).
Here, we describe the incorporation of phospholipid scramblases into GUVs, and how to measure phospholipid flip-flop in single vesicles by fluorescence microscopy (Mathiassen et al., 2021). The first step is based on LUV formation and protein reconstitution (Chalat et al., 2012). We used a composition of egg phosphatidylcholine (eggPC), trace amounts of a fluorescent labeled reporter lipid ( ATTO488-phosphatidylethanolamine, ATTO488-PE), and biotinylated phosphatidylethanolamine (biotin-PE), in combination with Triton X-100 extracted ER membrane proteins (Triton extract, TE), which are known to contain constitutive active phospholipid scramblases (Chalat et al., 2012; Sanyal et al., 2008). The TE was fluorescently labeled with an amine-reactive dye to track the presence of membrane proteins and mixed with detergent-solubilized phospholipids; afterward, detergent was removed using polystyrene Bio-Beads, to generate sealed scramblase-containing LUVs with diameters in the range of 150–300 nm. The scramblase-containing LUVs serve as the basis for the formation of GUVs in the second step (Girard et al., 2004), via electroformation under low salt conditions in an indium tin oxide (ITO)-coated chamber. In the third step, fluorescence microscopy is used to detect the fluorescently labeled TE proteins in individual GUVs, and to quantify the fluorescence intensity of the lipid reporter (ATTO488-PE) prior to versus after dithionite treatment, as an indicator of scramblase activity in the GUVs. The method described here enables fast and easy quantification of a large data set of single GUVs. The GUV preparation and analysis is easily applicable for non-experts, and is anticipated to be applied for the study of lipid scramblases.
Materials and Reagents
Biological Materials
Triton X-100 extracted endoplasmic reticulum (ER) membrane proteins (TE) were prepared according to Chalat et al. (2012) and Verchère et al. (2021) resulting in ~1 µg µL-1, in 10 mM HEPES/NaOH, pH 7.4, 100 mM NaCl, 0.5 mM phenylmethylsulfonyl fluoride, 1× protease inhibitor cocktail (Calbiochem, Merck Millipore Sigma, Darmstadt, Germany, catalog number: 539131), 1% (weight/volume) Triton X-100, stored at -80°C.
Note: The protocol can also be applied to purified protein preparations in combination with reconstitution procedures not based on Triton X-100, as described here. In all cases, samples should be prepared in phosphate-free buffers, to allow for lipid phosphate determination.
Materials and Reagents
Aluminium foil
Clamps (e.g., office paper clamps, Lomax, catalog number: 9801110)
Copper tape (cut to ~70 mm long, 50 mm wide, 0.07 mm thick, Conrad Electronik, catalog number: 545614-62)
Cover glass slides (26 × 76 mm, #1.5, Thermo Fisher Scientific, Life Technologies Corporation Eugene, OR, USA)
Detergent for cleaning the ITO glass slides
Disposable glass Pasteur pipettes (150 mm; VWR, catalog number: 612-1701)
Filters (SAMPLE DISCS, SS-033, ELITechGroup, BIOMEDICAL SYSTEMS, Logan, UT, USA)
Forceps (e.g., VWR, catalog number: 232-0032)
Glass jar(s) (e.g., Fisher Scientific, catalog number: 15603977)
Glass pipettes (e.g., graduated pipettes BLAUBRAND® Type 3 Class AS, 10 mL, Graduation: 10 mL; Carl Roth, catalog number: HXT8.1)
Glass vials (Rotilabo®-screw neck ND8 vials, Brown glass, 1.5 mL; Carl Roth, catalog number: KE30.1) with screw caps (without borehole, without septum, PP, black, ND8; Carl Roth, catalog number: KE39.1) for lipid aliquotation
Ice bucket (e.g., Magic Touch 2TM ice bucket with lid; Sigma-Aldrich, catalog number: BAM168072002)
ITO-coated glass slides (50 × 60 mm, 1 mm thick; Präzisions Glas & Optik GmbH, Iserlohn, Germany)
Microcentrifuge tubes of 1.5 mL capacity (SARSTEDT AG & Co. KG, catalog number: 72.690.001)
Microcentrifuge tubes of 2 mL capacity (SARSTEDT AG & Co. KG, catalog number: 72.691)
Pyrex flask (Fisher Scientific, catalog number: 15446113)
Round bottom glass tubes (KIMBLE® Disposable Screw Thread Culture Tube, Round Bottom, 16 × 100 mm, DWK Life Science, catalog number: 73770-16100) with screw caps (PP screw cap with seal, black, DURAN WHEATON KIMBLE, DWK Life Science, catalog number: 299901307)
Silicone rubber spacer (cut to 50 × 60 mm, 1 mm thick, Deutsch & Neumann GmbH, catalog number: 510 1001)
Sticky-slide 8 Well (Ibidi, catalog number: 80828)
Wipes (Precision Wipes, KIMTECH Science, Kimberly-Clark® Professional, catalog number: 7552)
Acetic acid, glacial (VWR, catalog number: 20104)
Acetone (Sigma-Aldrich, catalog number: 904082)
Alexa Fluor 647 NHS-Ester (Succinimidyl Ester) (Thermo Fisher Scientific, Life Technologies, catalog number: A20006), dissolved in DMSO and stored at -20°C
Ammonium molybdate tetrahydrate (Carl Roth, catalog number: 3666.1)
(3-Aminopropyl)triethoxysilane (APTES, Sigma-Aldrich, catalog number: 440140)
L-ascorbic acid (Carl Roth, catalog number: 3525.2)
Avidin (Thermo Fisher Scientific, Life Technologies, catalog number: 434401), stored at -20°C
Bio-Beads SM-2 Resin (Bio-Rad Laboratories Inc., catalog number: 1523920)
Biotin-Poly (Ethylene Glycol)-Succinimidyl carbonate ((Ethylene Glycol)-(biotin-PEG-SC, 5,000 Da, Laysan Bio Inc., Alabama, USA), stored at -20°C
Bio-Gel P-6 (Bio-Rad Laboratories Inc., catalog number: 1504130), dissolved in deionized water and stored at 4°C
Chloroform, ethanol-stabilized and certified for absence of phosgene and HCl (VWR, catalog number: 22711.290)
Deionized water
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine labeled with ATTO488 (ATTO488-PE) was purchased as powder (ATTO-TEC GmbH, catalog number: 67335), dissolved in chloroform, and stored at -20°C
DMSO (Sigma-Aldrich, catalog number: 276855)
Egg L-α-phosphatidylcholine (EggPC, catalog number: 840051) and 1,2-dioleoyl-sn-glycero-3-phosphoetholamine-N-(cap biotinyl, 18:1) (biotin-PE, catalog number: 870273) were purchased as powders (Avanti Polar Lipids, Birmingham, AL, USA), dissolved in chloroform:methanol (1:1, volume/volume), and stored at -20°C
70% ethanol
Glucose (Fisher Scientific, catalog number: 10122730)
HEPES (Carl Roth, catalog number: 6763.3)
Ice
Methanol (VWR PROLABO® CHEMICALS; Fontenay-sous-Bois, catalog number: 20834.291)
Methoxy-Poly (Ethylene Glycol)-Succinimidyl carbonate (mPEG-SC, 5,000 Da, Laysan Bio Inc., Alabama, USA), stored at -20°C
N2 gas (ALPHAGAZ 1 N2, 99.999%, Air Liquide, Düsseldorf, Germany)
NaOH (Sigma-Aldrich, catalog number: 30620)
NaCl (AnalaR NORMAPUR, VWR PROLABO® CHEMICALS; Fontenay-sous-Bois, catalog number: 27810.295)
Norland Optical Adhesive 60 (Norland Products, Inc. catalog number: 6016)
Osmolarity standards of 290 mmol kg-1 (OPTIMOLETM, 290 mmol kg-1, ELITechGroup, BIOMEDICAL SYSTEMS, Logan, UT, USA)
Parafilm (PARAFILM® M; Sigma-Aldrich, catalog number: P7793-1EA)
Perchloric acid (VWR, catalog number: 20589)
Pierce spin column (Bio-Rad Laboratories Inc., catalog number: 7326008)
Polypropylene tubes of 15 mL and 50 mL capacity (e.g., Falcon tubes, SARSTEDT AG & Co. KG, catalog numbers: 62.554.502 and 62.547.254)
Polyethersulfone membrane with a pore size of 0.2 μm (Filtropur, SARSTEDT AG & Co. KG, catalog number: 83.1826.001)
Potassium hydroxide (Fisher Scientific, catalog number: P250-1)
Sodium bicarbonate (Fisher Scientific, catalog number: S233-500)
Sodium dithionite (Fisher Scientific, catalog number: 10274490)
Di-Sodium hydrogenphosphate dihydrate (VWR, catalog number: 28029)
Sucrose (Fisher Scientific, catalog number: 10638403)
Triton X® 100 (Carl Roth, catalog number: 3051.3)
Low ionic labeling buffer (see Recipes)
Bio-Gel P-6 in deionized water (see Recipes)
Low ionic buffer (see Recipes)
Low ionic buffer with Triton X-100 (see Recipes)
Phosphate standard solution (4 mM) (see Recipes)
Molybdate solution (12 mg mL-1) (see Recipes)
Ascorbic acid solution (50 mg mL-1) (see Recipes)
250 mM sucrose solution (osmolality 250 mmol kg-1) (see Recipes)
250 mM glucose solution (osmolality 250 mmol kg-1) (see Recipes)
Saturated sodium chloride solution (see Recipes)
5.0 M potassium hydroxide (see Recipes)
1.0 M potassium hydroxide (see Recipes)
Sodium bicarbonate (10 mM, pH 8.5) (see Recipes)
Avidin 0.2 mg mL-1 in deionized water (see Recipes)
Equipment
Analytical balance (e.g., Sartorius Entris-I II, 220 g/0.1 mg; Buch Holm, catalog number: 4669128)
Confocal laser scanning microscope. For this protocol, a Leica TCS SP8 (Leitz, Wetzlar, Germany) equipped with 63×/1.20, NA water objective was used. Images were acquired using a 400 Hz unidirectional scanner, a pixel size of 246.27 × 246.27 μm, a pinhole of 100 μm (1 AU) with Leica HyD detectors.
End-over-end rotator (INTELLI-MIXER, neoLab®, Heidelberg, Germany, catalog number: 7-0045)
Flow cabinet to work with organic solvents
Freezer (-20°C)
Freezer (-80°C)
Function generator (e.g., TG315 Function generator 3 MHz; Telemeter Electronic, catalog number: 29788)
Glass desiccator (Boro 3.3 with socket in lid, 20 cm, including stopcock; BRAND GmbH, catalog number: 65238)
Glass beaker (e.g., Aldrich® Essentials beaker, Griffin; Sigma-Aldrich, catalog number: Z740572)
Glass tubes (Carl Roth, catalog number: DURAN C208.1)
Glass marbles (e.g., Fisher Scientific, catalog number: S04581)
Hamilton 700 Series Syringes of 10 µL, 100 µL, 250 µL, 500 µL, and 1,000 µL (Hamilton® syringe, 700 and 1000 Series, Nevada, USA)
Heating block (Rotilabo®-Block-Heater H 250; CARL ROTH, catalog number: Y264.1)
Ice machine
Magnetic stirrer (e.g., IKAMAG®, DREHZAHL ELECTRONIC, IKA, Staufen im Breisgau, Germany)
Magnets
pH-meter (pH-Meter 761 Calimatic, Knick, Berlin, Germany)
Pipettes P20, P200, P1000 (GILSON®, catalog numbers: FD10001, FD10005, and FD10006)
Pipette tips 2 µL, 20 µL, 200 µL, and 1,000 µL (SARSTEDT AG & Co. KG, catalog numbers: 70.1130.212, 70.3021, 70.760.002, and 70.3050.020)
Polystyrene cuvettes (SARSTEDT AG & Co. KG, catalog number: 67.742)
Quartz cuvette (10 mm wide, QS, High Precision Cell, Quartz SUPRASIL®; Hellma® Analytics, catalog number: HL109004F-10-40)
Refrigerator (4°C)
Rotoevaporator (BUCHI, Vacuum Pump V-100, Interface I-100, Rotavapor R-100, Flawil, Switzerland)
Scissors
Sonicator (130 W, ULTRASONIC CLEANER; ALLPAX, catalog number: 10015895;0)
Spectrophotometer (e.g., UV-VIS Spectrophotometer, UV-1280, SHIMADZU, Kyoto, Japan)
Tabletop centrifuge (Eppendorf 5810 R, rotor A-4-62, Hamburg, Germany)
Ultra-Violet/Ozone Probe and Surface Decontamination Unit (Novascan Technologies Inc., Boone, IA, USA)
Vapor Pressure Osmometer (WESCOR®, Model 5600, ELITechGroup, BIOMEDICAL SYSTEMS, Logan, UT, USA)
Vortexer (Vortex Genie 2 TM, BENDER & HOBEIN AG, Zurich, Switzerland)
Software
GraphPad Prism version 9.1.0 for Windows (GraphPad Software, 2021, La Jolla California, USA, www.graphpad.com)
ImageJ (Wayne, Rasband, S., U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/index.html)
Inkscape (Inkscape Project., 2020, Inkscape, Team, 2021, https://inkscape.org)
Leica Application Suite X (LAS X, Leitz, Wetzlar, Germany)
Microsoft Excel (Microsoft Corporation, 2018, https://office.microsoft.com/excel)
Considerations before starting
Choice of lipid reporter
The fluorescent lipids employed in the microscopy-based assay need to fulfill three requirements: they should be (i) substrates for the ER scramblases under low salt conditions, (ii) rendered non-fluorescent by chemical reduction with the membrane-impermeant dianion dithionite, and (iii) be photostable for microscopy. These requirements are met by lipid analogues tagged with fluorescent ATTO488 (Andra et al., 2018; Mathiassen et al., 2021). Phospholipid scramblases display a relatively low specificity and transport ceramide-based lipids and glycerophospholipids, including the ATTO488-modified phospholipids, equally well within the limits of the time resolution of the assay (Herrmann et al., 1990; Buton et al., 2002; Vishwakarma et al., 2005; Sahu and Gummadi, 2008; Sanyal et al., 2008; Chalat et al., 2012; Wang et al., 2018; Bushell et al. 2019; Mathiassen et al., 2021). We verified requirements (i) and (ii) for each scramblase-containing LUV reconstitution prior to GUV formation. Requirement (iii) was verified by a series of confocal fluorescence imaging of individual GUVs containing ATTO488-PE, and by quantifying the membrane ATTO488 fluorescence intensity at the first image versus the subsequent images (Mathiassen et al., 2021). Note that the majority of ATP-dependent lipid transporters show headgroup specificity for phospholipid transport, which in this case excludes the use of headgroup-labeled lipid analogues (Theorin et al., 2019).
Formation of giant unilamellar vesicles
We reconstituted TE proteins into LUVs under low salt conditions to facilitate their subsequent use in electroformation for GUV formation by applying an oscillating electric field in an ITO coated chamber. The electroformation technique under low salt conditions was effective in incorporating and maintaining the activity of the sarcoplasmic reticulum Ca2+-ATPase and the H+-pump bacteriorhodopsin in GUVs (Girard et al., 2004). However, activity may not be preserved for all membrane transporters under these conditions. In this case, we note that GUVs can be prepared in solutions containing physiological salt concentrations, by applying a voltage with a higher frequency (~500 Hz) (Pott et al., 2008; Méléard et al., 2009; Shaklee et al., 2010; Stein et al., 2017). Under these conditions, the yield of GUVs is lower and protein incorporation into GUVs is more heterogeneous, mainly because of the electric field being shielded by the ions of the solution (Girard et al., 2004; Politano et al., 2010; Aimon et al., 2011; Garten et al., 2015). In addition, negatively charged lipids can give rise to an asymmetric lipid distribution over the bilayer directly after electroformation. Under such conditions, the GUV suspension should be stored at room temperature for several hours and the lipid distribution tested, by using dithionite-reactive fluorescent lipid probes for example (Steinkühler et al., 2018). Furthermore, various dimensions of ITO-coated glass slides are available, which may alter the amount, morphology, and size distribution of the GUVs formed. Hence, the GUV output should be analyzed to ensure reproducibility. We note that studies of other transporters may require other reconstitution procedures into LUVs for subsequent generation of protein-containing GUVs. We recommend verification of protein incorporation into the GUVs (e.g., by using fluorescently labeled proteins), and assaying the ATTO488-PE distribution over the bilayer in protein-free/empty GUVs using dithionite, as described in the next sections.
Handling of giant unilamellar vesicles
GUVs are fragile and rupture easily on glass. Thus, it is important to passivate the cover glass slides with e.g. β-Casein, to prevent bursting/attachment when observing free-floating GUVs (Streicher et al., 2009). In this protocol, we aimed to immobilize a fraction of the biotinylated GUVs to ease single vesicle tracking over time. The cover glass slides were cleaned using a protocol adapted from Chandradoss et al. (2014) , followed by aminosilanization using (3-Aminopropyl)triethoxysilane (APTES). Surface PEGylation was performed using a protocol adapted from Lamichhane et al. (2010) , which employed a mixture of functionalized PEG (mPEG-SC and biotin-PEG-SC), introducing biotinylated PEG molecules. To increase GUV immobilization using avidin as the immobilization-linker, adjustments to the protocol may be required, as most GUVs settled rather than immobilized in our hands.
Procedure
When utilizing other protein labeling techniques, e.g., SNAP labeling, and/or reconstitution procedures not based on Triton X-100 as described here, skip steps 1–4, and start at step 5.
Preparation of large unilamellar vesicles (see Figure 1)
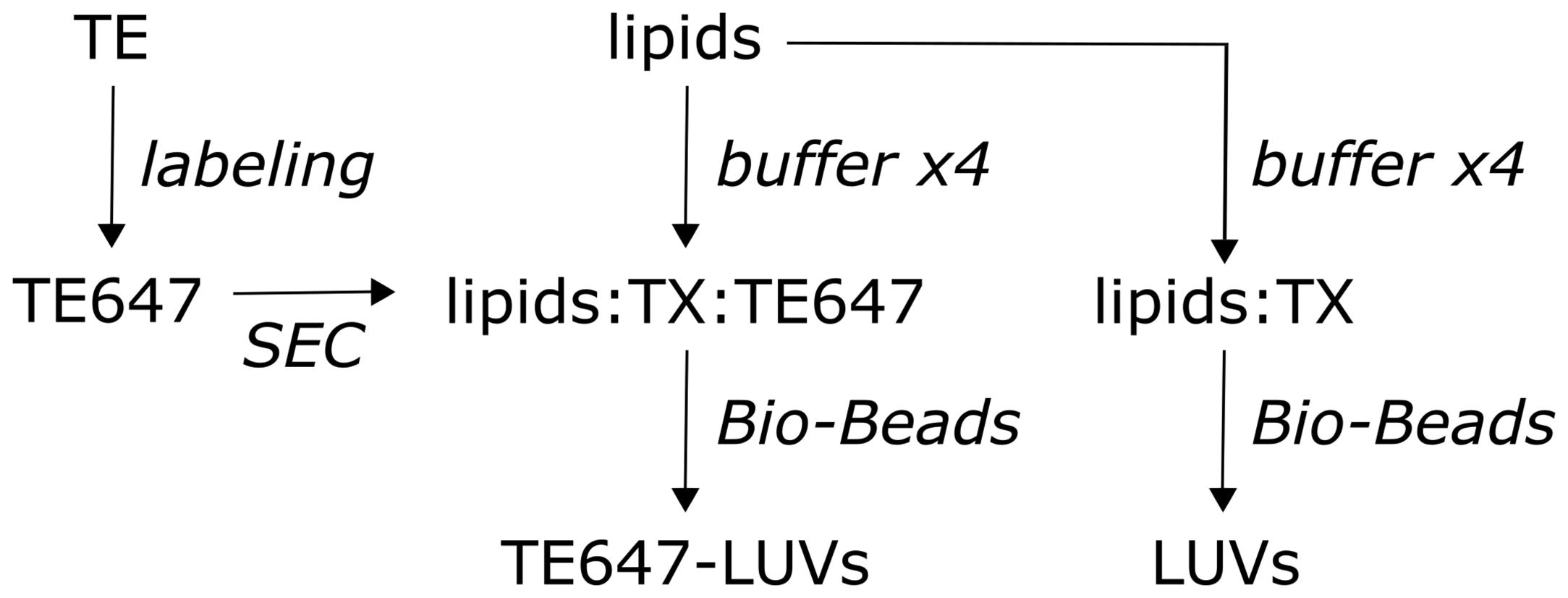
Figure 1. Protein reconstitution and preparation of large unilamellar vesicles. Experimental workflow. Triton-extracted ER membrane proteins (TE) from yeast are fluorescently labeled with Alexa Fluor 647 succinimidyl ester (left), yielding Alexa Fluor 647 fluorescent labeled TE (TE647). Unreacted fluorophore is removed by size-exclusion chromatography (SEC), and the eluate is subsequently reconstituted together with the lipids, eggPC, ATTO488-PE, and biotin-PE into large unilamellar vesicles (LUVs), using Triton X-100-containing buffer (TX). Pre-washed Bio-Beads are used to remove the detergent, generating sealed TE647-LUVs (middle). Protein-free/empty LUVs are generated in parallel (right).Amine labeling of the TE membrane proteins:
Thaw the TE on ice.
Mix the TE with 3-fold molar excess of Alexa Fluor 647 succinimidyl ester in 1.8-fold volume low ionic labeling buffer (see Recipe 1) in a 2 mL tube.
Note: We typically label 95 µg TE per reconstitution.
Incubate the ‘labeling mixture’ at room temperature for 1 h, under end-over-end rotation, covered in aluminum foil.
Place a spin column into a 15 mL Falcon tube with a cut-up open lid.
Add Bio-Gel P-6 (see Recipe 2) to the spin column, centrifuge in a tabletop centrifuge at 453 × gav and 4°C for 3 min.
Wash the P-6 column twice in low ionic labeling buffer (see Recipe 1), centrifuge in a tabletop centrifuge at 453 × gav and 4°C for 3 min.
Place the P-6 column into a clean 15 mL Falcon tube with the cut-up open lid.
Remove unreacted fluorophore using the equilibrated P-6 column, by placing the ‘labeling mixture’ on top of the P-6 column, waiting for ~2 min, and centrifuging using a tabletop centrifuge at 453 × gav and 4°C for 3 min.
SM-2 Bio-Bead wash:
Weigh slightly more SM-2 Bio-Beads than needed into a glass beaker, as some beads will be lost during washing.
Add a magnet and wash in methanol under the flow cabinet, twice for 15 min each. Use a glass pipette to remove the methanol.
Wash in deionized water, under gentle stirring at room temperature, twice for 15 min each. Use a glass pipette to remove the solution.
Wash in low ionic buffer (see Recipe 3), under gentle stirring at room temperature, for 15 min. Use a glass pipette to remove the low ionic buffer.
Store the washed SM-2 Bio-Beads in low ionic buffer (see Recipe 3) at 4°C, until use.
Note: Use freshly or within a week.
Reconstitute the eluted Alexa Fluor 647 fluorescent labeled TE (TE647) into LUVs of desired protein-to-phospholipid ratio (PPR), according to Chalat et al. (2012) . We typically reconstituted TE647 with a PPR of 35 (weight/weight), as follows:
Clean the Hamilton syringes three to five times in chloroform:methanol (1:1, volume/volume) under a flow cabinet.
Note: Distinguish between Hamilton syringes used for unlabeled and fluorescently labeled lipids. Avoid any use of plasticware when handling organic solvents.
Mix the desired amount of unlabeled (4.32 µmol eggPC and 43.2 nmol biotin-PE) and labeled (4.42 nmol ATTO488-PE) lipids (dissolved in chloroform:methanol) using Hamilton syringes, into a round bottom glass tube covered in aluminum foil to protect the fluorophore, on ice.
Evaporate the solvent under a gentle stream of N2 gas at room temperature for ~15 min, to form a thin lipid film.
Note: We slide the large end of a disposable glass Pasteur pipette into laboratory tubing that is attached to the regulator onto the N2 gas cylinder. Using the outlet valve on the regulator, the flow is adjusted for a gentle stream of N2 gas coming through the pipette. The pipette can be held with a ring stand clamp to hold it in a fixed position.
Place the lipid film in a vacuum desiccator at ~10 mbar and room temperature for 1 h, to remove the last traces of the solvent.
Note: We use a glass desiccator connected to a chemically resistant vacuum pump, reaching a final vacuum of 10 mbar (±2 mbar) and a suction capacity of 1.5 m3 h-1.
Add aliquots of 200 µL of low ionic buffer with Triton X-100 (see Recipe 4). Vortex gently and allow the sample to sit on ice for ~5 min, before adding the next aliquot.
For protein-free/empty LUVs, add up to 1 mL of low ionic buffer with Triton X-100 (see Recipe 4). For TE647-containing LUVs, after adding four times 200 µL of low ionic buffer with Triton X-100, add appropriate amounts of TE647, and fill up to 1 mL with low ionic buffer with Triton X-100.
Note: For PPR of 35 (weight/weight), we add 95 µg of TE647.
Gently and shortly vortex the samples, and allow the suspension to incubate while continuing to the next step.
Note: The mixture should be as clear as water before adding SM-2 Bio-Beads.
Add 100 mg washed and dried SM-2 Bio-Beads per 1 mL of sample. Place on an end-over-end rotator at room temperature for 3 h.
Note: Dried SM-2 Bio-Beads are defined by the removal of low ionic buffer in which the SM-2 Bio-Beads are stored using a disposable glass Pasteur pipette, just before weighting them.
Add an additional 200 mg of washed and dried SM-2 Bio-Beads per 1 mL of sample. Place on an end-over-end rotator at 4°C overnight.
Transfer the LUV suspension to a 1.5 mL tube without the SM-2 Bio-Beads, using a disposable glass Pasteur pipette and store at 4°C until use.
Determine the efficiency of detergent removal after reconstitution with Triton X-100, according to Hrafnsdóttir and Menon (2000). All the described steps are performed in a flow cabinet to avoid direct exposure to organic solvents.
Transfer 0, 7.5, 15, 30, and 45 µL of low ionic buffer with Triton X-100 (see Recipe 4) into separated round bottom glass tubes (corresponding to 0, 0.05, 0.1, 0.2, and 0.3% Triton X-100) and add low ionic buffer (see Recipe 3) to a final volume of 150 µL. We recommend preparing each standard in duplicates.
Transfer 50 µL of the TE647-LUVs and protein-free/empty LUVs into separate glass tubes, and add low ionic buffer (see Recipe 3) to a final volume of 150 µL. We recommend preparing duplicates.
Add 600 µL of methanol and 300 µL of chloroform to all the samples, including the standards, and vortex briefly.
Measure the absorbance at 275 nm (A275) on a spectrophotometer using quartz cuvettes.
Average the duplicate readings for each standard and sample.
Subtract the mean absorbance value of the blank standard (0% Triton X-100) from all standard and sample readings. This is the corrected absorbance.
Plot a standard curve (% Triton X-100 versus mean absorbance A275) using linear regression analysis with e.g., Microsoft Excel.
Use the linear regression equation to calculate the Triton X-100 concentration of the sample, by comparing the sample absorbance to the standard curve obtained.
Note: More than 99.95% of the initial Triton X-100 amount is typically removed.
Determine the phospholipid concentration of the generated protein-free/empty and TE647-LUVs according to Bartlett (1959) and Ploier and Menon (2016), with small modifications to the protocol.
Note: All steps described below should be conducted in a fume hood using appropriate personal protection and following lab safety guidelines.
Turn on the electrical heating block in a folw cabine and set it at 145°C.
Dilute the phosphate standard solution.
Transfer 0, 5, 12.5, and 20 µL of phosphate standard solution (see Recipe 5, corresponding to 0, 20, 50, and 80 nmol phosphate) into separate glass tubes, and add deionized water to a final volume of 50 µL. Additional standards are obtained from 10-fold diluted phosphate standard solution, e.g., 5, 12.5, and 25 µL of 0.4 M phosphate standard solution (corresponding to 2, 5, and 10 nmol phosphate). We recommend preparing each standard in duplicates.
Transfer 12 µL (about 50 nmol) of the LUV solutions into separate glass tubes and add deionized water to a final volume of 50 µL. We recommend preparing duplicates and using different amounts of the LUV solutions, to ensure the readings are within the standard value range.
Note: An aliquot of the detergent-solubilized LUVs prior to SM-2 Bio-Bead addition (step 3e) can be included in the analysis to determine phospholipid recovery. We typically achieve a recovery of 70–90% phospholipid after reconstitution.
Add 300 µL of perchloric acid to all samples including standards, close the glass tube loosely with glass marbles to prevent evaporation, and heat at 145°C for 1 h.
Remove the glass tubes from the heating block and let them cool to room temperature.
Set the temperature on a heating block to 100°C.
Add 1 mL of deionized water to all samples and vortex.
Add 400 µL of freshly prepared molybdate solution (see Recipe 6) to all samples and vortex briefly.
Add 400 µL of freshly prepared ascorbic acid solution (see Recipe 7) to all samples and vortex briefly.
Close the glass tube loosely with glass marbles, and heat at 100°C for 10 min.
Let the samples cool to room temperature and measure the absorbance at 797 nm (A797) on a spectrophotometer using disposable polystyrene cuvettes.
Average the duplicate readings for each standard and sample.
Subtract the mean absorbance value of the blank standard (0 nmol phosphate) from all standard and sample readings. This is the corrected absorbance.
Plot a standard curve (nmol phosphate versus mean absorbance [A797] using linear regression analysis) with e.g., Microsoft Excel.
Use the linear regression equation to calculate the phosphate concentration of the sample, by comparing the sample absorbance to the standard curve obtained.
Preparation of giant unilamellar vesicles (see Figure 2)

Figure 2. Preparation of giant unilamelar vesicles from large vesicles. A. Equipment for a giant unilamellar vesicle (GUV) formation chamber. B. Schematic workflow of GUV formation by electroformation. Deposition of diluted large unilamellar vesicles (LUVs) on two indium tin oxide (ITO)-coated glass slides, followed by dehydration under saturated sodium chloride (NaCl) conditions overnight, to form a thin lipid film. The two ITO-coated slides are sandwiched to assemble an ITO chamber separated by a silicone rubber spacer with copper tape on each side, sealed by Parafilm, and held together by clamps. A syringe is used to inject 1 mL of 250 mM sucrose solution (not illustrated), which is then sealed with Parafilm, and the chamber is immediately connected to a function generator for GUV formation. Modified from Mathiassen et al. (2021) .UV/ozone clean the indium tin oxide (ITO) coated glass slides:
Clean four ITO-coated glass slides with detergent, deionized water, and 70% ethanol.
Note: Two ITO slides are required to assemble one ITO chamber. Scramblase-containing and protein-free/empty GUVs are always made in parallel for comparison (sGUVs and eGUVs, respectively).
Dry the slides with wipes.
UV/ozone clean the conducting side using a UV/ozone cleaner for 10 min.
Turn on the Surface Decontamination Unit for 15 min before opening the chamber.
Adjust the osmolarity of the solutions used for giant unilamellar vesicle handling:
Turn on the Vapor Pressure Osmometer and let it equilibrate for ~30 min before use.
Place a filter with forceps at the Vapor Pressure Osmometer.
Equilibrate the Vapor Pressure Osmometer using osmolarity standards of 290 mmol kg-1, by applying a volume of 10 µL onto the filter.
Measure the osmolarity of the 250 mM sucrose solution (see Recipe 8) and the 250 mM glucose solution (see Recipe 9).
Adjust the osmolarity of the solutions with deionized water, if required.
Giant unilamellar vesicle formation by electroformation:
Dilute the TE647-LUVs and protein-free/empty LUVs to a lipid concentration of 0.8 mg mL-1 in deionized water to a total volume of 100 µL in separate 1.5 mL tubes.
For each sample, two cleaned ITO-coated glass slides are prepared by depositing a volume of 50 µL in 2 µL droplets on each side of the conducting and cleaned ITO-coated glass slides.
Dehydrate the ITO-coated glass slides in a desiccator containing saturated sodium chloride (see Recipe 10) overnight, to form a lipid film.
Assemble the two ITO-coated glass slides into a chamber separated by a silicone rubber spacer with copper tape facing the conducting sides. Seal with Parafilm and clamps to prevent leakage from the ITO chamber.
Inject 1 mL of 250 mM sucrose solution (see Recipe 8) into the chamber using a 1,000 µL Hamilton syringe. Seal the opening with Parafilm to prevent evaporation.
Immediately connect the ITO chambers to the function generator by applying a sinusoidal voltage for 4 h, 20 Hz, 0.2–1.3 V, increasing every 6 min. Cover the chamber in aluminium foil to protect the fluorophore.
Shear off the formed GUVs, by changing the electrical settings to 4 Hz and 1.3 V for 30 min.
Carefully invert the ITO chamber and remove the clamps and the Parafilm, while opening the chamber from one corner to transfer/drop the GUVs into a 1.5 mL tube.
Store the GUVs, covered in aluminium foil at room temperature, until microscopically observation.
Clean the disassembled ITO-coated glass slides, as described above.
Preparation of the giant unilamellar vesicle scramblase assay (see Figure 3)
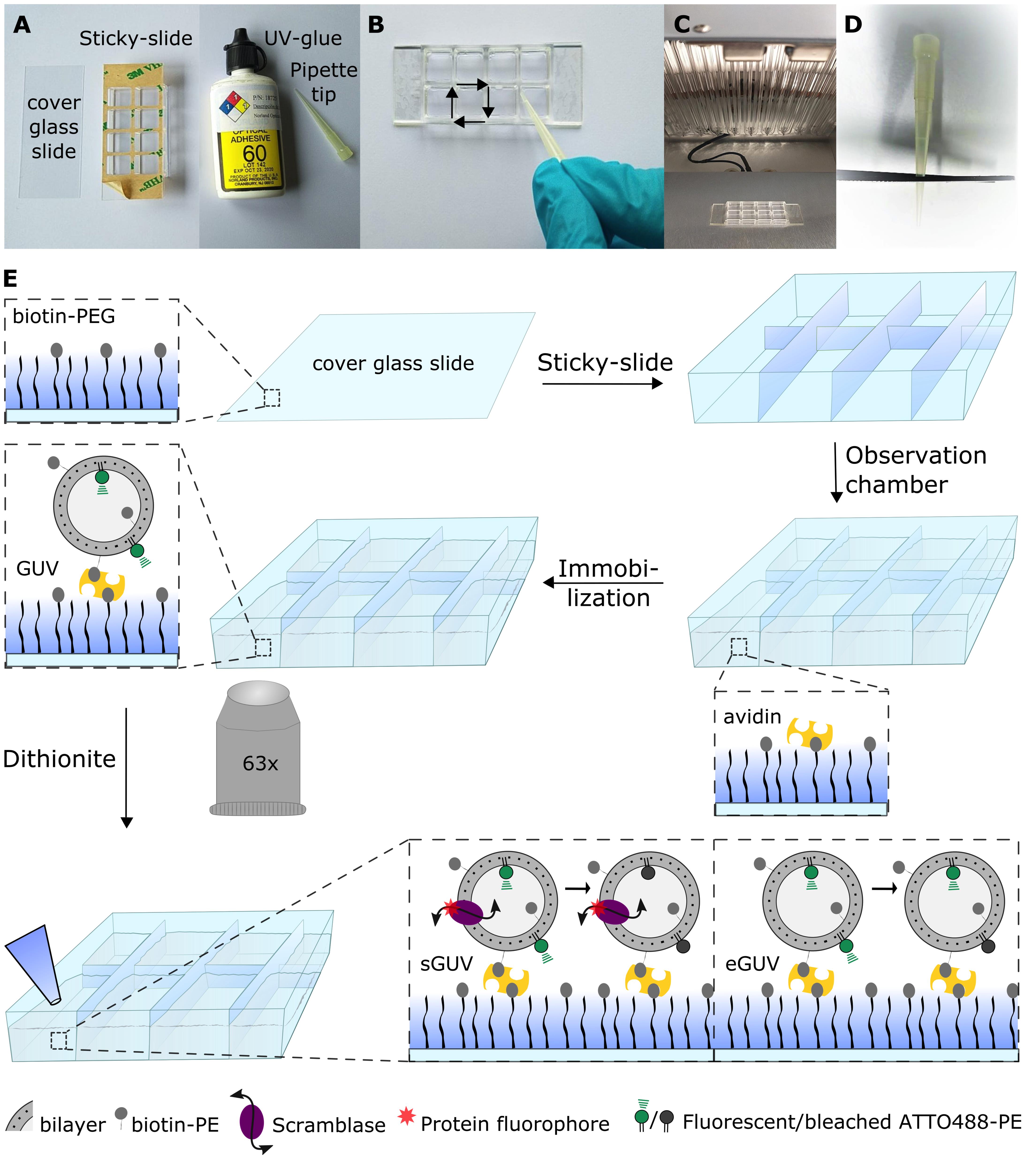
Figure 3. Preparing the giant unilamellar vesicle scramblase assay setup. A. Equipment for assembling the microscope observation chamber. B. UV-gluing the corners of the Sticky-slide, using a pipette tip with the PEGylated side facing upwards. C. UV-drying the UV-glue. D. Scissor-cut a pipette tip for giant unilamellar vesicle (GUV) handling. E. Schematic workflow of the fluorescence-based assay for measuring phospholipid scramblase activity in GUVs. The biotin-PEGylated cover glass slide is glued unto a Sticky-slide for assembling of the microscope observation chamber. Avidin is added to bind to the biotinylated PEG molecules, followed by biotinylated-GUV immobilization. The fluorescent lipid probe ATTO488-PE is observed using a confocal fluorescence microscope with a 63× water objective. Images are acquired prior to and after dithionite addition. Scramblase-containing GUVs (sGUVs) are expected to result in complete ATTO488 bleaching, whereas only outer leaflet ATTO488 is bleached in empty GUVs (eGUVs), generating a ~50% fluorescence reduction. Modified from Mathiassen et al. (2021).Clean cover glass slides and Pyrex flask according to Chandradoss et al. (2014) with modifications of the protocol, as outline below. All described steps are performed in a flow cabinet to avoid direct exposure to organic solvents.
Place the cover glass slides in a glass jar and rinse five times with deionized water.
Note: Any glass jar where the cover glass slides are fully covered in solution can be used.
Sonicate in fresh deionized water for 10 min to remove dirt.
Dispose of the deionized water and rinse three times with fresh deionized water.
Rinse with methanol and sonicate for 10 min.
Sonicate in acetone for 20 min.
Discard the acetone and rinse three times with deionized water, to remove any acetone residue.
Sonicate in 5.0 M potassium hydroxide (see Recipe 11) for 20 min.
Rinse six times with deionized water.
Sonicate in methanol for 2 min, and keep the cover slides in methanol until step 2 (see below).
Rinse a Pyrex flask several times with deionized water.
Sonicate the Pyrex flask in 1.0 M KOH (see Recipe 12) for 30 min.
Sonicate the Pyrex flask in methanol for 30 min.
Surface functionalization of the clean glass slides with amine groups via 3-aminopropylthriethoxysilane (APTES), according to Lamichhane et al. (2010) , using Methoxy-Poly (Ethylene Glycol)-Succinimidyl Carbonate (mPEG-SC) and biotin-Poly (Ethylene Glycol)-Succinimidyl Carbonate (biotin-PEG-SC), is described in the following:
Prepare the amino-silanization solution by mixing 300 mL of methanol, 15 mL of glacial acetic acid, and 3 mL of APTES in the cleaned Pyrex flask.
Replace the methanol in the glass jar containing the cleaned cover glass slides and incubate for 20 min, sonicate for 1 min, and further incubate for 10 min.
Rinse four times with methanol, once with deionized water, and once with methanol.
Dry the cover glass slides with a stream of N2 gas, before PEGylating them.
Place the dried cover glass slides in a clean container or a pipette tip box filled with ~10% DMSO or deionized water. Close the box to prevent dust accumulation.
Note: Remember to label the sides of the cover glass slides to distinguish the PEGylated side later.
Prepare a PEG solution (for ~35 slides) by dissolving 10 mg biotin-PEG-SC and 200 mg mPEG-SC in 1.6 mL of 100 mM sodium bicarbonate (see Recipe 13). Vortex gently to mix.
Note: Equilibrate the PEG molecules to room temperature before use. Before returning the PEG molecules to -20°C, flush the vial with N2 gas, seal with Parafilm, wrap in aluminum foil, and desiccate.
Add 90 µL of the PEG solution onto one cover glass slide at a time. Gently place another cleaned cover glass slide on top. Close the box and incubate in the dark at room temperature for 5–6 h.
Note: Make sure no air bubbles are trapped between the slides. Use a clean pipette tip to reposition the slides if they move during the incubation time.
Gently slide to separate the cover glass slides, rinse with deionized water, and dry with N2 gas.
Store the PEGylated cover glass slides in separate 50 mL Falcon tubes flushed with N2 gas, sealed with Parafilm, and wrapped in aluminum foil, at -20°C until use.
Note: We typically used the PEGylated cover glass slides within 3 months.
Assembly of the microscope observation chamber:
Allow a PEGylated cover glass slide to equilibrate to room temperature.
Glue an Ibidi Sticky-slide 8 well onto the PEGylated cover glass slide.
Seal the chamber’s corners with UV-glue.
Dry the UV-glue under the UV/ozone cleaner for 2 min.
Turn on the Surface Decontamination Unit for 5 min, before opening the chamber.
Giant unilamellar vesicle immobilization:
Add 200 µL of avidin (0.2 mg mL-1 in deionized water, see Recipe 14) to each chamber, incubate for 10 min or longer.
Gently wash away any unbound avidin with 400 µL of 250 mM glucose solution (see Recipe 9), using a pipette.
Cut a 200 µL pipette tip with scissors.
Add 100 µL of GUVs with the scissor-cut 200 µL pipette tip, and incubate for 10 min or longer.
Note: The final volume is 500 µL.
Assaying phospholipid scramblase activity in giant unilamellar vesicles:
Prepare 1.5 mL tubes with 20 mg dithionite powder and store on ice.
Turn on the confocal microscope, computer, and open the LAS X software. We use the following settings: 63×/1.20 water objective, image acquisition using a 400 Hz unidirectional scanner, a pixel size of 246.27 × 246.27 μm, and a pinhole of 100 μm (1 AU) with Leica HyD detectors. The λex/λem used for imaging are as follows: 507/517–550 nm for ATTO488, 645/655–780 nm for Alexa647.
Obtain images of several marked positions using a motorized specimen on the microscope slide before dithionite treatment (corresponding to t = 0 min).
Dissolve the dithionite in 600 µL of 250 mM glucose solution (see Recipe 9), and vortex just before use.
Note: Prepare a fresh dithionite solution for each experimental series. Dithionite in solution is not stable and disintegrates fast!
Add 5 µL of dithionite (for a 2 mM final concentration) and immediately obtain images over a time series (corresponding to t > 0 min).
A similar procedure is carried out for 250 mM glucose solution (see Recipe 9) addition, as a control.
Data analysis
ImageJ was used to analyze the membrane fluorescence intensity of individual GUVs prior to and after dithionite treatment (see Figure 4). Only unilamellar giant vesicles are used for analysis. Scramblase-containing and empty GUVs (sGUVs and eGUVS, respectively) should always be prepared and assayed on the same day.
Export data (series of images) as .tif files from the LAS X software and import into the ImageJ software.
A region of interest (ROI) is placed around the GUV of interest to measure the ATTO488 fluorescence intensity of the membrane, by measuring the integrated density value per pixel.
Another ROI is placed within the GUV lumen and subtracted from the membranal density value per pixel, which is then subtracted from the membrane signal, by multiplying with the area of the membrane ROI. The background signal was typically insignificant, but nevertheless used as an offset correction as part of our standard procedure.
Repeat the procedure for each time frame and save the data as an Excel (xls) file.
We used GraphPad software to analyze and characterize the fluorescence trace of the individual GUVs over time after dithionite treatment. The fluorescence intensity of an individual GUV prior to dithionite addition can be compared with the fluorescence intensity over time after dithionite addition, or with the last frame obtained. For each selected GUV, we normalized each data point to the initial fluorescence intensity prior to dithionite treatment, and analyzed the fluorescence over time by a “one phase decay model” with a nonlinear fit: y = (y0-plateau)*exp(-k*x) + plateau, where y0 and the plateau are same units as y, and y0 is constrained to 1.0, y starts at y0 and decays (with one phase) down to plateau, x is time in seconds, and the rate constant k equals the reciprocal of the x axis units, which is constrained to k > 0. The bleaching kinetics allow to extract the number of scramblase-competent GUVs (GUV that lost >80% of the initial fluorescence within 10 minutes). Analysis of the kinetics of scrambling as a function of the number of reconstituted scramblases is best carried out with fluorescently labeled purified scramblases, rather than the crude TE preparation that we use here as a source of scramblase activity.
Repeat the analysis of 250 mM glucose treated GUVs to ensure the ATTO488 fluorescence is not photobleached over the duration of the selected time frames. This experiment should ideally be the first analysis performed.
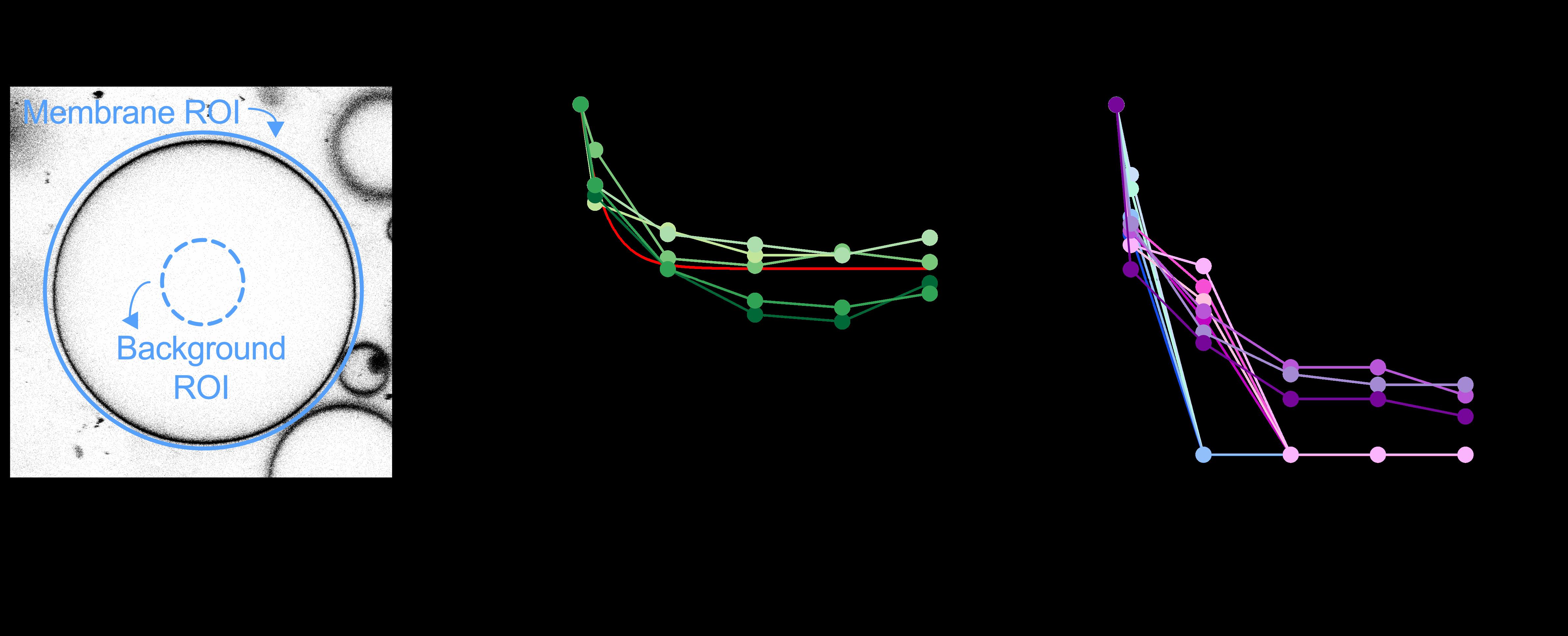
Figure 4. Quantification of ATTO488-PE membrane fluorescence. A. For giant unilamellar vesicle (GUV) analysis, an outer region of interest (ROI) is placed around the GUV membrane and its total ATTO488 fluorescence is determined (blue circle). Another ROI is defined within the GUV lumen to quantify background fluorescence (punctured blue circle). The average fluorescence per pixel of the lumenal ROI is determined and scaled to the pixel area of the outer ROI for background subtraction. B. Dithionite bleaching of ATTO488-PE fluorescence in five empty GUVs (eGUVs, coded in different shades of green). The arrowhead indicates dithionite addition. The red line is a monoexponential fit of the combined data (t½ = 0.56 min, span = 0.47). C. Dithionite bleaching of ATTO488-PE fluorescence in eleven individually tracked scramblase-containing GUVs (sGUVs, coded in different colors), normalized to the fluorescence value at t = 0 min. The arrowhead indicates dithionite addition. Four of the traces show complete loss of fluorescence by t = 3 min, four show complete loss of fluorescence by t = 6 min, and three still show detectable fluorescence at t = 12 min. The horizontal dashed line corresponds to 50% loss of fluorescence. Modified from Mathiassen et al. (2021).
Notes
Aliquots of the TE prior to, during, and after fluorescent amine labeling can be analyzed by SDS-PAGE in-gel fluorescence with aliquots after protein-reconstitution into LUVs. The presence of protein can by visualized by silver or colloidal staining (Mathiassen et al., 2021).
Recipes
All solutions were filter-sterilized over a 0.2 µm Acrodisc® Syringe Filter and stored at 4°C unless stated otherwise, and equilibrated to room temperature before use.
Low ionic labeling buffer
1 mM NaCl
2 mM HEPES/NaOH, pH 8.3
1% (weight/volume) Triton X-100
Adjust the pH with a 1 M NaOH solution.
Bio-Gel P-6 in deionized water
Swell P-6 gel in deionized water under gentle stirring at 4°C overnight, then store at 4°C.
Low ionic buffer
1 mM NaCl
2 mM HEPES/NaOH, pH 7.4
Adjust the pH with a 1 M NaOH solution.
Low ionic buffer with Triton X-100
1 mM NaCl
2 mM HEPES/NaOH, pH 7.4
1% (weight/volume) Triton X-100
Adjust the pH with a 1 M NaOH solution.
Phosphate standard solution (4 mM)
Dissolve 35.598 mg di-sodium hydrogenphosphate dihydate to a final volume of 50 mL in deionized water.
Molybdate solution (12 mg mL-1)
Dissolve 144 mg of fresh ammonium molybdate tetrahydrate to a final volume of 12 mL in deionized water. Do not store.
Ascorbic acid solution (50 mg mL-1)
Dissolve 600 mg of fresh L-ascorbic acid to a final volume of 12 mL in deionized water. Do not store.
250 mM sucrose solution (osmolality 250 mmol kg-1)
250 mM glucose solution (osmolality 250 mmol kg-1)
Saturated sodium chloride solution
Add approximately 35–40 g NaCl to 100 mL deionized water under stirring.
5.0 M potassium hydroxide
Dissolve 14 g of KOH to a final volume of 50 mL in deionized water.
1.0 M potassium hydroxide
Dissolve 2.8 g of KOH to a final volume of 50 mL in deionized water.
Sodium bicarbonate (10 mM, pH 8.5)
Dissolve 8.4 mg of sodium bicarbonate to a final volume of 10 mL in deionized water. There is no need of adjusting the pH.
Avidin 0.2 mg mL-1 in deionized water
Acknowledgments
We gratefully acknowledge Anant Menon (Weill Cornell Medical College, New York, USA), Robert Tampé, and Tim Diederichs (Goethe University, Frankfurt, Germany) for helpful discussions and technical advice on the project. This work was supported by DAAD (Grant 57386621 to T.G.P.) and the German Research Foundation (GU 1133/11-1; INST 213/886-1 FUGG to T.G.P). P.P.M.M. gratefully acknowledge support by a Gateway fellowship of the Ruhr University Bochum. This protocol is based on our previous publication Mathiassen et al. (2021).
Competing interests
The authors declare no competing financial interests.
References
- Aden, S., Snoj, T. and Anderluh, G. (2021). The use of giant unilamellar vesicles to study functional properties of pore-forming toxins. Methods Enzymol 649: 219-251.
- Aimon, S., Manzi, J., Schmidt, D., Poveda Larrosa, J. A., Bassereau, P. and Toombes, G. E. (2011). Functional reconstitution of a voltage-gated potassium channel in giant unilamellar vesicles. PLoS One 6(10): e25529.
- Andra, K. K., Dorsey, S., Royer, C. A. and Menon, A. K. (2018). Structural mapping of fluorescently-tagged, functional nhTMEM16 scramblase in a lipid bilayer.J Biol Chem 293(31): 12248-12258.
- Bagatolli, L. A. and Needham, D. (2014). Quantitative optical microscopy and micromanipulation studies on the lipid bilayer membranes of giant unilamellar vesicles. Chem Phys Lipids 181: 99-120.
- Bartlett, G. R. (1959). Phosphorus assay in column chromatography. J Biol Chem 234(3): 466-468.
- Bevers, E. M. and Williamson, P. L. (2016). Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol Rev 96(2): 605-645.
- Bushell, S. R., Pike, A. C. W., Falzone, M. E., Rorsman, N. J. G., Ta, C. M., Corey, R. A., Newport, T. D., Christianson, J. C., Scofano, L. F., Shintre, C. A., et al. (2019). The structural basis of lipid scrambling and inactivation in the endoplasmic reticulum scramblase TMEM16K. Nat Commun 10(1): 3956.
- Buton, X., Herve, P., Kubelt, J., Tannert, A., Burger, K. N., Fellmann, P., Muller, P., Herrmann, A., Seigneuret, M. and Devaux, P. F. (2002). Transbilayer movement of monohexosylsphingolipids in endoplasmic reticulum and Golgi membranes. Biochemistry 41(43): 13106-13115.
- Chalat, M., Menon, I., Turan, Z. and Menon, A. K. (2012). Reconstitution of glucosylceramide flip-flop across endoplasmic reticulum: implications for mechanism of glycosphingolipid biosynthesis. J Biol Chem 287(19): 15523-15532.
- Chandradoss, S. D., Haagsma, A. C., Lee, Y. K., Hwang, J. H., Nam, J. M. and Joo, C. (2014). Surface passivation for single-molecule protein studies. J Vis Exp(86): 50549.
- Chang, Q. L., Gummadi, S. N. and Menon, A. K. (2004). Chemical modification identifies two populations of glycerophospholipid flippase in rat liver ER. Biochemistry 43(33): 10710-10718.
- García-Sáez, A. J., Carrer, D. C. and Schwille, P. (2010). Fluorescence correlation spectroscopy for the study of membrane dynamics and organization in giant unilamellar vesicles. Methods Mol Biol 606: 493-508.
- Garten, M., Aimon, S., Bassereau, P. and Toombes, G. E. (2015). Reconstitution of a transmembrane protein, the voltage-gated ion channel, KvAP, into giant unilamellar vesicles for microscopy and patch clamp studies. J Vis Exp(95): 52281.
- Girard, P., Pecreaux, J., Lenoir, G., Falson, P., Rigaud, J. L. and Bassereau, P. (2004). A new method for the reconstitution of membrane proteins into giant unilamellar vesicles. Biophys J 87(1): 419-429.
- Goren, M. A., Morizumi, T., Menon, I., Joseph, J. S., Dittman, J. S., Cherezov, V., Stevens, R. C., Ernst, O. P. and Menon, A. K. (2014). Constitutive phospholipid scramblase activity of a G protein-coupled receptor. Nat Commun 5: 5115.
- Herrmann, A., Zachowski, A. and Devaux, P. F. (1990). Protein-mediated phospholipid translocation in the endoplasmic reticulum with a low lipid specificity. Biochemistry 29(8): 2023-2027.
- Hrafnsdóttir, S. and Menon, A. K. (2000). Reconstitution and partial characterization of phospholipid flippase activity from detergent extracts of the Bacillus subtilis cell membrane. J Bacteriol 182(15): 4198-4206.
- Lamichhane, R., Solem, A., Black, W. and Rueda, D. (2010). Single-molecule FRET of protein-nucleic acid and protein-protein complexes: surface passivation and immobilization. Methods 52(2): 192-200.
- Larsen, J., Hatzakis, N. S. and Stamou, D. (2011). Observation of inhomogeneity in the lipid composition of individual nanoscale liposomes. J Am Chem Soc 133(28): 10685-10687.
- Mathiassen, P. P. M., Menon, A. K. and Pomorski, T. G. (2021). Endoplasmic reticulum phospholipid scramblase activity revealed after protein reconstitution into giant unilamellar vesicles containing a photostable lipid reporter. Sci Rep 11(1): 14364.
- Méléard, P., Bagatolli, L. A. and Pott, T. (2009). Giant unilamellar vesicle electroformation from lipid mixtures to native membranes under physiological conditions. Methods Enzymol 465: 161-176.
- Menon, I., Huber, T., Sanyal, S., Banerjee, S., Barré, P., Canis, S., Warren, J. D., Hwa, J., Sakmar, T. P. and Menon, A. K. (2011). Opsin is a phospholipid flippase. Current Biol 21(2): 149-153.
- Morra, G., Razavi, A. M., Pandey, K., Weinstein, H., Menon, A. K. and Khelashvili, G. (2018). Mechanisms of Lipid Scrambling by the G Protein-Coupled Receptor Opsin. Structure 26(2): 356-367 e353.
- Ploier, B. and Menon, A. K. (2016). A Fluorescence-based Assay of Phospholipid Scramblase Activity. J Vis Exp(115): 54635.
- Politano, T. J., Froude, V. E., Jing, B. and Zhu, Y. (2010). AC-electric field dependent electroformation of giant lipid vesicles. Colloids Surf B Biointerfaces 79(1): 75-82.
- Pomorski, T. G. and Menon, A. K. (2016). Lipid somersaults: Uncovering the mechanisms of protein-mediated lipid flipping. Prog Lipid Res 64: 69-84.
- Pott, T., Bouvrais, H. and Meleard, P. (2008). Giant unilamellar vesicle formation under physiologically relevant conditions. Chem Phys Lipids 154(2): 115-119.
- Sahu, S. K. and Gummadi, S. N. (2008). Flippase activity in proteoliposomes reconstituted with Spinacea oleracea endoplasmic reticulum membrane proteins: evidence of biogenic membrane flippase in plants. Biochemistry 47(39): 10481-10490.
- Sanyal, S., Frank, C. G. and Menon, A. K. (2008). Distinct flippases translocate glycerophospholipids and oligosaccharide diphosphate dolichols across the endoplasmic reticulum. Biochemistry 47(30): 7937-7946.
- Shaklee, P. M., Semrau, S., Malkus, M., Kubick, S., Dogterom, M. and Schmidt, T. (2010). Protein incorporation in giant lipid vesicles under physiological conditions. Chembiochem 11(2): 175-179.
- Stein, H., Spindler, S., Bonakdar, N., Wang, C. and Sandoghdar, V. (2017). Production of Isolated Giant Unilamellar Vesicles under High Salt Concentrations. Front Physiol 8: 63.
- Steinkühler, J., De Tillieux, P., Knorr, R. L., Lipowsky, R. and Dimova, R. (2018). Charged giant unilamellar vesicles prepared by electroformation exhibit nanotubes and transbilayer lipid asymmetry. Sci Rep 8(1): 11838.
- Streicher, P., Nassoy, P., Bärmann, M., Dif, A., Marchi-Artzner, V., Brochard-Wyart, F., Spatz, J. and Bassereau, P. (2009). Integrin reconstituted in GUVs: A biomimetic system to study initial steps of cell spreading. Biochim Biophys Acta 1788(10): 2291-2300.
- Suzuki, J., Umeda, M., Sims, P. J. and Nagata, S. (2010). Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468(7325): 834-838.
- Suzuki, J., Imanishi, E. and Nagata, S. (2014). Exposure of phosphatidylserine by Xk-related protein family members during apoptosis. J Biol Chem 289(44): 30257-30267.
- Theorin, L., Faxén, K., Sørensen, D. M., Migotti, R., Dittmar, G., Schiller, J., Daleke, D. L., Palmgren, M., López-Marqués, R. L. and Günther Pomorski, T. (2019). The lipid head group is the key element for substrate recognition by the P4 ATPase ALA2: a phosphatidylserine flippase. Biochem J 476(5): 783-794.
- Vehring, S., Pakkiri, L., Schroer, A., Alder-Baerens, N., Herrmann, A., Menon, A. K. and Pomorski, T. (2007). Flip-flop of fluorescently labeled phospholipids in proteoliposomes reconstituted with Saccharomyces cerevisiae microsomal proteins. Eukaryot Cell 6(9): 1625-1634.
- Velasco-Olmo, A., Ormaetxea Gisasola, J., Martinez Galvez, J. M., Vera Lillo, J. and Shnyrova, A. V. (2019). Combining patch-clamping and fluorescence microscopy for quantitative reconstitution of cellular membrane processes with Giant Suspended Bilayers. Sci Rep 9(1): 7255.
- Verchère, A., Cowton, A., Jenni, A., Rauch, M., Häner, R., Graumann, J., Bütikofer, P. and Menon, A. K. (2021). Complexity of the eukaryotic dolichol-linked oligosaccharide scramblase suggested by activity correlation profiling mass spectrometry. Sci Rep 11(1): 1411.
- Vishwakarma, R. A., Vehring, S., Mehta, A., Sinha, A., Pomorski, T., Herrmann, A. and Menon, A. K. (2005). New fluorescent probes reveal that flippase-mediated flip-flop of phosphatidylinositol across the endoplasmic reticulum membrane does not depend on the stereochemistry of the lipid. Org Biomol Chem 3(7): 1275-1283.
- Wang, L., Iwasaki, Y., Andra, K. K., Pandey, K., Menon, A. K. and Butikofer, P. (2018). Scrambling of natural and fluorescently tagged phosphatidylinositol by reconstituted G protein-coupled receptor and TMEM16 scramblases. J Biol Chem 293(47): 18318-18327.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Mathiassen, P. P. M. and Pomorski, T. G. (2022). A Fluorescence-based Assay for Measuring Phospholipid Scramblase Activity in Giant Unilamellar Vesicles. Bio-protocol 12(6): e4366. DOI: 10.21769/BioProtoc.4366.
Category
Biological Engineering > Synthetic biology
Biochemistry > Lipid > Membrane lipid
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.