- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Immunoprecipation Assay to Quantify the Amount of tRNAs associated with Their Interacting Proteins in Tissue and Cell Culture
Published: Vol 12, Iss 4, Feb 20, 2022 DOI: 10.21769/BioProtoc.4335 Views: 3577
Reviewed by: Pilar Villacampa AlcubierreDaniel Louis KissThirupugal Govindarajan
Original research article
The authors used this protocol in:

Sep 2021

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Transfer RNAs (tRNAs) are highly abundant species and, along their biosynthetic and functional path, they establish interactions with a plethora of proteins. The high number of nucleobase modifications in tRNAs renders conventional RNA quantification approaches unsuitable to study protein-tRNA interactions and their associated functional roles in the cell. We present an immunoprecipitation-based approach to quantify tRNA bound to its interacting protein partner(s). The tRNA-protein complexes are immunoprecipitated from cells or tissues and tRNAs are identified by northern blot and quantified by tRNA-specific fluorescent labeling. The tRNA interacting protein is quantified by an automated western blot and the tRNA amount is presented per unit of the interacting protein. We tested the approach to quantify tRNAGly associated with mutant glycyl-tRNA-synthetase implicated in Charcot-Marie-Tooth disease. This simple and versatile protocol can be easily adapted to any other tRNA binding proteins.
Graphic abstract:
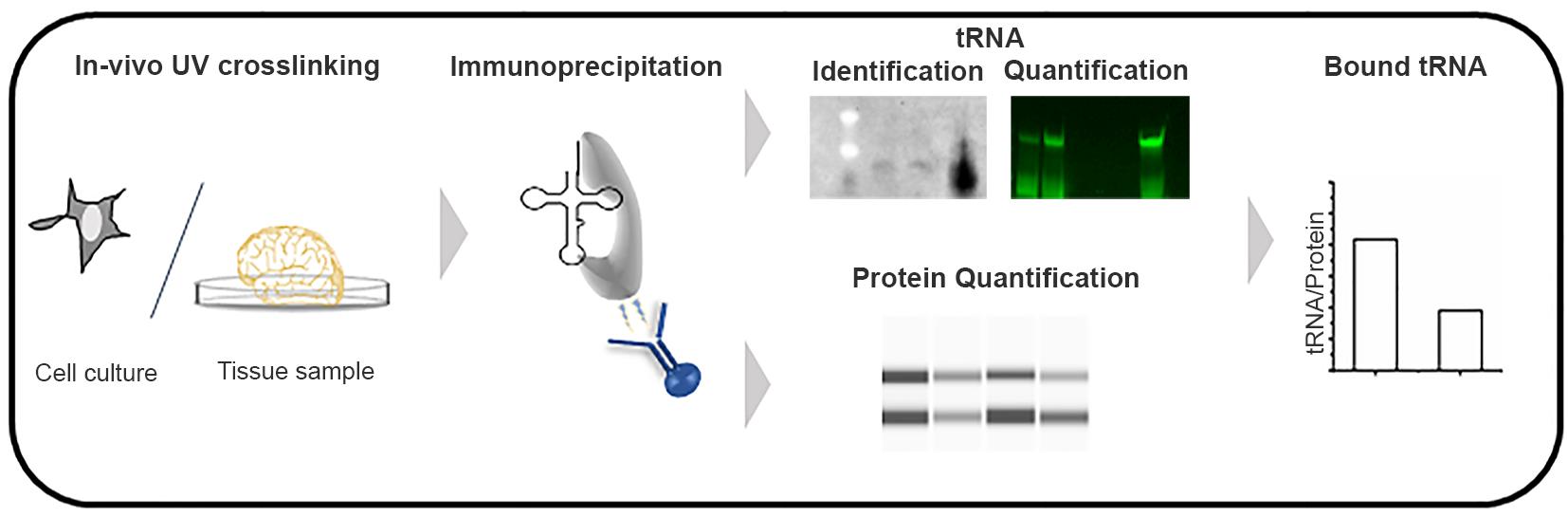
Figure 1. Schematic of the tRNA-Immunoprecipitation approach.
Background
tRNAs are ubiquitous molecules, representing 4–10% of all cellular RNAs. tRNAs undergo complex biogenesis during which they interact with different protein entities, including tRNA-splicing proteins, tRNA-base modifying enzymes, tRNA-charging enzymes, 3’-end modification and repair enzymes, and various nucleases, thereby generating active tRNA fragments or completely degrading tRNAs (Betat and Mörl, 2015; Kirchner and Ignatova, 2015; Fernández-Millán et al., 2016; Barciszewska et al., 2016; Schmidt and Matera, 2020; Tosar and Cayota, 2020). tRNAs are crucial components of the translation machinery and are charged at the 3’ ends with their cognate amino acid by an aminoacyl-tRNA-synthetase. Mutations in tRNAs or genes encoding tRNA-interacting partners are linked to complex human pathologies, with intricate heterogeneity at both the cell and tissue levels that modulates the disease (Abbott et al, 2014). Thus, a method for quantitative detection of tRNA-binding-protein interactions that can be widely used to study disease-related alterations of tRNA interactome in living cells and tissues is an urgent need.
Traditional methods to detect RNA-protein interactions include RNA immunoprecipitation (RIP) and crosslinking and immunoprecipitation (CLIP), both of which use antibodies to immunoprecipitate RNA-protein complexes, followed by identification of RNAs by sequencing. Unlike mRNA sequencing, despite recent advances (Zheng et al., 2015; Behrens et al., 2021) tRNA sequencing still has limited quantitative resolution towards many tRNA isoacceptors, likely because of their complex modification pattern (Orioli, 2017; Kimura et al., 2020; Warren et al., 2021). Combining immunoprecipitation (IP) of the RNA-protein complexes with tRNA-tailored detection (Figure 1), we have developed a new twist of the classic IP method that is suitable for quantifying tRNA-protein interactions in living cells. In a recent study, we have used this approach to quantify alterations in tRNA binding to mutated glycyl-tRNA-synthetase (GlyRS), which has been implicated in Charcot-Marie-Tooth (CMT) disease (Zuko et al., 2021). In a CMT-mouse model GarsC201R/+, we observed a stronger association of tRNAsGly with the mutant GlyRS, thus depleting the glycyl-tRNAGly pool and causing ribosome stalling at Gly codons (Zuko et al., 2021). The tRNA-IP methodology identifies and quantifies tRNAs bound to GlyRS in native conditions in tissues. This protocol is easily adapted to other aminoacyl-tRNA-synthetases or any tRNA-binding proteins, to quantify interactions in native conditions, in both cell culture and tissue.
Materials and Reagents
Pipette tips (filtered) (Sarstedt)
Syringe 1 mL with 26 G needles (BD Plastipak)
Cell Scraper, DNase/RNase free (Techno Plastic Products)
RNase-free 1.5 mL microtubes (Carl Roth, catalog number: CNT2.1)
RNase-free 1.5 mL amber microtubes (Carl Roth, catalog number: ENH0.2)
15 cm cell culture dishes (Sarstedt, catalog number: 83.3903)
3.5 cm cell culture dishes (Sarstedt, catalog number: 82.1135.500)
GarsC201R/+ mice (C3H.C-GarsC201R/H) (Achilli et al., 2009)
293 (HEK293) cell line 293 (HEK293) (ATCC CRL-1573TM)
Gibco Dulbecco's modified Eagle medium (DMEM) (Pan Biotech, catalog number: P04-03500)
Fetal bovine serum (FBS) (Pan Biotech, catalog number: P30-3302)
L-glutamine (Serva, catalog numbers: 22942)
PBS (GibcoTM, catalog number: 70011044)
0.25% Trypsin-EDTA (Pan Biotech, catalog number: P10-019500)
Triton® X-100 (Sigma, catalog number: T9284)
Tris Base (Sigma, catalog number: T6066)
cOmplete TM, EDTA-free protease inhibitor cocktail (Roche, catalog number: 11873580001)
Urea (Carl Roth, catalog number: 57-13-6)
NaCl (Carl Roth, catalog number: HN00.2)
Acid-phenol:chloroform, pH 4.5 (Invitrogen, catalog number:AM9720)
3 M sodium acetate, pH 5.2 (Carl Roth, catalog number: 6773.2)
Glycogen (Invitrogen, catalog number: AM9510)
Absolute ethanol (Carl Roth, catalog number: 9065.4)
Isopropanol (Carl Roth, catalog number: 7343.2)
40% polyacrylamide (Carl Roth,catalog number: A516.1 )
RNase inhibitor SUPERase-In (Invitrogen, catalog number: AM2694)
NP-40 (Sigma, catalog number: 6507)
DTT (Carl Roth, catalog number: 6908.2)
SDS (Carl Roth, catalog number: CN30.3)
Sodium deoxycholate detergent (SDC) (Thermo, catalog number: 89904)
EDTA (AppliChem, catalog number: A5097,1000)
BSA (Carl Roth, catalog number: T844.3)
Trisodium citrate (Carl Roth, catalog number: 3580.1)
Hybond-N membrane (GE Healthcare Life Sciences, RPN2250B)
T4 DNA ligase (NEB, catalog number: M0202)
DMSO (Sigma, catalog number: D8418)
Salmon sperm DNA (Invitrogen, catalog number: 15632011)
Anti-GARS antibodies (Abcam, catalog number: ab42905; Proteintech, catalog number: 15831-AP)
Pierce protein G magnetic beads (Thermo, catalog number: 88847)
RevertAid H Minus reverse transcriptase (Thermo, catalog number: K1631)
10 mM DNTPs (Thermo, catalog number: R0181)
T7 RNA polymerase (Thermo, catalog number: EP0113)
NTPs (Thermo, catalog number: R0481)
Crush and soak buffer (see Recipes)
2× RNA loading formamide dye (see Recipes)
20× saline sodium citrate buffer (SSC buffer; see Recipes)
Cell lysis buffer (see Recipes)
Tissue lysis buffer (see Recipes)
Wash buffer for cell culture (see Recipes)
Wash buffer for tissue (see Recipes)
1× SDS buffer (see Recipes)
Hybridization buffer (see Recipes)
Note: Except for the anti-GARS antibodies, all other reagents can be purchased from any other supplier.
Equipment
Standard molecular biology equipment
UV crosslinker (Analytik Jena, catalog number: CL-1000)
CellCrusher tissue pulverizer (Kisker, catalog number: 538004)
Eppendorf tube rotator (Eppendorf, catalog number: R5010)
Spectrophotometer UV-vis (DeNovix DS-C)
ChemiDocTM MP Imaging System multiplex fluorescence, chemiluminescence (Bio-Rad)
Magnetic bead separator (Thermo Fisher, catalog number: CS15000)
Jess automated western blots system (Protein Simple)
Software
Fiji ImageJ (ImageJ, https://imagej.net/software/fiji/)
Compass software for simple western (Protein Simple, a biotechne brand, https://www.proteinsimple.com/software_compass_simplewestern.html)
Procedure
Notes:
The starting material can be any tissue of interest, or mammalian cell culture endogenously expressing, stably transfected, or ectopically expressing the tRNA-interacting protein of interest. It is recommended to test and optimize the protocol with easily accessible material, e.g., cell culture, before performing experiments in tissue.
For quantitative assessment, it is important to perform the experiment in multiple independent biological replicates, i.e., at least ≥4 to enable statistical assessment.
All steps should be performed in an RNase-free environment.
Sample preparation
a. In-vivo UV crosslinking to stabilize transient tRNA-protein interactions in cell culture (here HEK293T cells, hereafter named only HEK)
Culture HEK cells in a 15 cm cell culture dish in DMEM supplemented with 10% FBS and 2.5 mM L-glutamine in 5% CO2 at 37°C.
Note: For one experiment, approximately 20 million cells are required. However, for scarcely available cell culture material, as little as 6 million cells can be used.
To prepare the cells for UV-crosslinking, aspirate the medium and gently add 5 mL of cold 1× PBS.
Note: The PBS volume should be adjusted in each experiment. It should minimal, but suffient to cover the specimen or the surface of the plate in adherent cell culture.
Place the cell culture plate on ice and illuminate with the UV light source (254 nm) of a crosslinker at 150 mJ/cm2 radiation.
Note: In parallel, cells treated the same way without UV crosslinking (i.e., omitting Step 3) could be used as a control.
Keep the cell culture dish on ice, gently aspirate the PBS solution, and add 800 µL of pre-cooled cell lysis buffer. Harvest the cells using a cell scraper and transfer them into a pre-cooled 1.5 mL Eppendorf tube.
Using a 1 mL syringe with a 26-gauge needle, pass the lysate through eight times, to further shear open the cells and facilitate lysis. To obtain a clear lysate, centrifuge at 16,000 × g and 4°C for 10 min. This is the starting material for the IP (Step C-a).
b. In-vivo UV Crosslinking to stabilize transient tRNA-protein interactions in tissue samples
Note: To choose the most appropriate tissue for the experiment, one can refer to the Human Protein Atlas. In our experiment, we use brain tissue from 3- to 6-week-old CMT model mice (GarsC201R/+; Achilli et al., 2009) and compare it to wild-type littermates (i.e., mice expressing WT GlyRS (C57Bl/6J). GarsC201R/+ mice carry an ENU-induced dominant point mutation that causes a cysteine to arginine substitution at residue 201 of the GlyRS protein. Heterozygous stock colonies of GarsC201R mice were maintained in the C57Bl/6J background. One hemisphere of the mouse brain tissue was enough to obtain a sufficient amount of tRNA and GlyRS for the IP. For scarcely expressed proteins of interest, organs from several animals (preferably littermates) can be pooled.
Flash freeze the freshly dissected brain tissue sample in liquid nitrogen and pulverize it in a pre-cooled CellCrusher tissue pulverizer. Transfer the pulverized tissue into a 3.5 cm cell culture dish placed on ice.
Note: Use homogenous tissue powder, as clumps and bigger tissue chunks would limit the crosslinking effectiveness.
Place the dish on an ice bath under the UV light source (254 nm) inside the UV crosslinker and apply 400 mJ/cm2 radiation.
Note: Dependent upon tissue availability, a non-crosslinked control should also be used.
Add 500 µL of cold tissue lysis buffer and mechanically shear by pipetting up and down, using pre-cooled wide-bore pipette tips (or 1,000 µL-pipette tips with the end cut out).
Add an additional 500 µL of cold lysis buffer to the lysed tissue and agitate at 4°C for 1 h. Clear the lysate by centrifugation at 16,000 × g and 4°C for 10 min. This is the starting material for the IP (Step C-b).
Preparation of magnetic beads and antibody coupling
Note: Select magnetic beads according to the immunoglobulin (Ig) type of the antibody to be used for the IP.
a. For the HEK cell lysate
Use protein G-coupled Dynabeads®. Use 20 µL of the bead slurry for each antibody coupling reaction.
Place the tube with the beads on the magnetic separator, wash twice with 500 µL of cold 1× PBS, and resuspend them in 50 µL of cell lysis buffer.
Add 2 µg of the antibody to the beads and incubate on a tube rotator for 45 min at room temperature. Leave on ice while preparing the lysates (Step A-a).
Use protein G-coupled Dynabeads®. Use 50 µL of the bead slurry for each coupling reaction.
Prepare the beads by washing them twice with 500 µL of cold 1× PBS solution, and resuspend them in 100 µL of tissue lysis buffer.
Add 4 µg of the antibody to the beads and incubate for 45 min on a tube rotator at room temperature. Leave on ice while preparing the lysates (Step A-b).
Note: For our experiment in both cell culture and mouse tissue, we used a mixture of two different anti-GlyRS antibodies, which we mixed in equal amounts (e.g., 1 or 2 µg each for the HEK sample or mouse brain lysate, respectively). Using a mixture of antibodies from different suppliers enhances the IP reproducibility between various supplier charges.
Immunoprecipitation (IP)
Note: To determine the efficiency of the antibodies, we suggest to first perform the pulldown with more accessible material (e.g., cell culture), thereby optimizing the amount of the beads with coupled antibody and the IP incubation time. For incubation time, we recommend starting with 1 h, or a few hours, up to overnight incubation. The optimal incubation time will be the one at which the antibodies maximally retain the desired target, with minimal to no non-specific RNA bands detectable on an ethidium bromide stained denaturing polyacrylamide gel.
a. For the HEK293 cell lysate
Add the cleared supernatant obtained from Step 5A-a (approximately 800–900 µL) to the prepared beads (Step B-a3).
Add 20 U of RNase inhibitor (SUPERase-IN) and rotate at 4°C for 2 h.
Place the tube on a magnetic separator to separate the immunoprecipitated tRNA-GlyRS bound to the antibody-coupled beads. Carefully discard the supernatant.
Wash the beads twice with 500 µL of 1× wash buffer for cell culture, and resuspend them in 500 µL of wash buffer for cell culture.
Withdraw 10 µL of the IP reaction (beads) and keep on ice for protein quantification in Step E.
Perform hot acid-phenol extraction of the remaining IP sample, directly on the beads, to denature the protein (here, GlyRS) and elute the bound tRNAs.
Note: Briefly, the procedure for hot-acid phenol extraction: add one volume of acid phenol:chloroform (5:1, pH 4.5) pre-heated to 65°C and 0.1 volume of 10% SDS to the reaction, incubate for 5 min, followed by incubation on ice for 5 min. Centrifuge at 21,000 × g for 5 min, remove the aqueous phase and add equal volume of acid phenol:chloroform. Incubate at room temperature, followed by centrifugation at 21,000 × g for 5 min. Collect the aqueous phase and add equal volume of chloroform:isoamyl alcohol (24:1) and 0.1 vol of 3 M NaOAc (pH 5.5). Centrifuge at 21,000 × g for 5 min to collect the final aqueuos phase. Precipiate the RNA in the aqueous phase with isopropanol.
Dissolve the recovered tRNA in 5 µL of sterile nuclease-free water.
b. For the brain tissue lysate:
Add the cleared supernatant obtained from Step A-b4 to the prepared beads (Step B-b3).
Add 20 U of RNase inhibitor and rotate at 4°C overnight.
Place the tube on a magnetic separator to separate the immunoprecipitated tRNA-GlyRS-coupled beads. Carefully discard the supernatant.
Wash twice with 500 µL of 1× wash buffer for tissue, and resuspend the beads in 500 µL of wash buffer for tissue.
Withdraw 10 µL of the IP reaction (beads) and keep on ice for protein quantification in Step E.
Perform hot acid-phenol extraction of the remaining IP to denature the protein (here, GlyRS) and elute bound tRNAs.
Dissolve the recovered tRNA in 5 µL of sterile nuclease-free water.
Identification and quantification of the bound tRNAs in the IP
a. Detection Method 1: tRNA identification by northern blot
Note: An in vitro transcribed tRNA of interest is required as a positive control. Here, tRNAGlyGCC was prepared by a standard T7-RNA polymerase run-off transcription reaction, using DNA template as described before (Albers et al., 2021). Two partly overlapping DNA primers were designed to cover the full-length tRNAGlyGCC, and the 5’ end of the forward primer bears the T7 promoter site (5’-TAATACGACTCACTATA-3’; Table 1). Both primers (100 µM each) were dissolved in 20 mM Tris-HCl (pH 7.5), denatured for 2 min at 95°C, and incubated for 3 min at room temperature, for their overlapping parts to anneal. Primer extension was performed for 40 min at 37°C, using 0.4 mM dNTPs and 4 U/μL RevertAid H Minus Reverse Transcriptase. The double-stranded DNA (dsDNA) was purified by phenol/chloroform and ethanol precipitation, and dissolved in DEPC-H2O. In vitro T7 promoter-based transcription of the dsDNA template was performed overnight at 37°C, with 0.6 U/μL T7 RNA polymerase in the presence of 2 mM NTPs, 5 mM GMP, and 1× transcription buffer. The transcribed tRNA was purified by denaturing polyacrylamide gel electrophoresis. tRNA was excised from the gel and eluted in crush and soak buffer rotating at 100 × g and 4°C overnight. Gel pieces were pelleted by centrifugation at 3500 × g for 5 min and tRNA was precipitated with ethanol.
Table 1. Example of DNA primers for in vitro T7 promoter-driven synthesis of tRNAGlyGCC. The forward primer contains 5’ upstream of the tRNA transcription start site the T7 promoter (underlined).
Forward 5’-TAATACGACTCACTATAGCATCGGTGGTTCAGTGGTAGAATGCTCGCCTGCCACGCGGGC-3’ Reverse 5’-TGGTGCATCGGCCGGGAATCGAACCCGGGCCGCCCGCGTGGCAGGCGAGCATTCTA-3’ To prepare the RNA recovered from the IP sample (Step C-a7 or C-b7) for northern blot analysis, mix the sample with RNA loading dye, heat at 95°C for 3 min, and place it on ice.
Load samples on a 10% denaturing polyacrylamide gel and run at 10 W for 30 min. In vitro synthesized tRNA is also loaded on the gel as a positive control (Figures 2A and 3A).
Transfer RNA from the gel onto Hybond-N blotting membrane in pre-cooled 0.5× TAE buffer at 10 V and 4°C overnight.
Immobilize the RNA to the membrane by illumiinating at 365 nm and 999.9 mJ/cm2 dosage.
Hybridize the membrane with an Atto565-labeled DNA oligo probe (5 µL of 100 µM probe), recognizing the tRNA of interest in hybridization buffer at 28°C overnight.
Note: If the tRNA-binding protein binds all tRNA isoacceptors of one tRNA family (that are all tRNAs recognizing different codons for a given amino acid, and thus aminoacylated with the same amino acid), we recommend using a mixture of probes to all isoacceptors. Here, we used two probes, including one with degenerate nucleotide sequence that recognizes all three tRNAGly isoacceptors (Table 2). Both probes were labeled with the same Atto-565 fluorophore. However, a mixture of probes labeled with separate fluorophores could have been chosen. Probes were fluorescently labeled at their 5’ ends by the manufacturer.
Wash blots thrice with 6x SSC supplemented with 0.1% SDS.
Wash once with 6× SSC.
Wash once with 2× SSC.
Wash once with 0.2× SSC.
Image the blot on a ChemiDoc TM MP Imaging system.
Table 2. Sequences of the Atto565-labeled DNA probes used in the northern blot experiment.
One probe contains degenerate bases, thus recognizing both tRNAGlyCCC and tRNAGlyGCC. The probes are labeled at their 5’ ends with Atto565.
Probe Sequence tRNAGlyTCC 5’-CCCGGGTCAACTGCTTGGAAGGCAGCTAT-3’ tRNAGlyCCC/GCC 5’-GYCTCCCGCGTGGSAGGCGAG-3’
b. Detection Method 2: Quantification of the tRNA bound to GlyRS by fluorescent tRNA labeling
Note: The IP (Step C) yields enough RNA for tRNA detection by northern blot and fluorescent quantification. Since the northern blot is performed only for tRNA identification, we recommend performing it in a single biological replicate, by loading the entire remaining extracted tRNA amount in multiple wells [e.g., 2 wells with equal amount of sample (Figure 2B)] onto the gels, and using them as multiple technical replicates, to average the signal in the fluorescent quantification step.
Label the tRNA from Step C-a7 or C-b7 with fluorescently labeled RNA: DNA hairpin oligonucleotide specific to tRNA with the following sequence: 5’-pCGCACUGCdTdTdTCy3dTdTdGdCdAdGdTdGdCdGdTdGdGdN-3’ or pCGCACUGCdTdTdTAtto647dTdTdGdCdAdGdTdGdCdGdTdGdGdN-3’ (d, denotes deoxyribose or DNA nucleotide, and p is the 5’ monophosphate).
For labeling, prepare the following labeling reaction: 1 µL of 10× T4 ligation buffer (NEB, #M0202), 1.5 µL of DMSO, 0.5 µL of Cy3-labeled 25-mer oligonucleotide (90 µM), 0.5 µL of T4 DNA ligase, and 1.5 µL of DEPC-H2O.
Note: The fluorescently labeled RNA:DNA hairpin oligonucleotide is designed to basepair to the unique unpaired 3’-NCCA end of the tRNAs, and is used to specifically label tRNAs as described previously (Kirchner et al, 2017).
Combine 5 µL of extracted tRNAs (Step C-a7 or C-b7) with 5 µL of the above labeling mix (Step D-a2 or D-b2) and incubate for 1 h at 25°C in the dark, to protect the fluorophores.
Heat the ligation mixture at 95°C for 3 min and place it on ice immediately.
Run it on a 10% denaturing-polyacrylamide gel at 10 W for 30 min in the dark.
Visualize the tRNA on a ChemiDocTM MP Imaging System in the respective fluorescent channel, and save a good quality image for further quantification of the fluorescent tRNA bands (a representative image is shown in Figures 2B and 3B).
Quantification of the GlyRS protein in the IP.
Use the 10 µL of IP sample reserved in Step C-a5 or C-b5. Use the magnetic rack to separate the beads with the bound antibody and protein-tRNA complexes from the liquid phase.
Add 10 µL of 1× SDS buffer to the beads and incubate at 50°C for 10 min by gentle shaking.
Use the magnetic rack to collect the beads, and transfer the solution to a fresh Eppendorf tube—this eluent consists of the bound tRNA-interacting protein.
Use the Jess automatic western blot system—a capillary-based automated western blot instrument—to quantify the protein (Figures 2C and 3C).
Note: Jess (Protein Simple) is an automated high-throughput western blotting system, that combines capillary electrophoresis to separate proteins by mass and immunodetection by antibodies in chemiluminescent or fluorescent detection mode. A conventional western blot can also be used instead, though the automated Jess system offers much higher sensitivity. We used purified human wildtype GlyRS protein at varying concentrations to establish a standard curve. Wildtype GlyRS was cloned into pET28 vector and expressed in the Escherichia coli Rosetta strain. The GlyRS sequence was extended by two purification tags, 6xHis and SUMO. The protein was purified to homogeneity using two consecutive chromatography steps, i.e., Ni-NTA-based affinity purification, followed by cleavage of both tags, and purification by size-exclusion chromatography. The detailed purification protocol is described in Zuko et al. (2021).
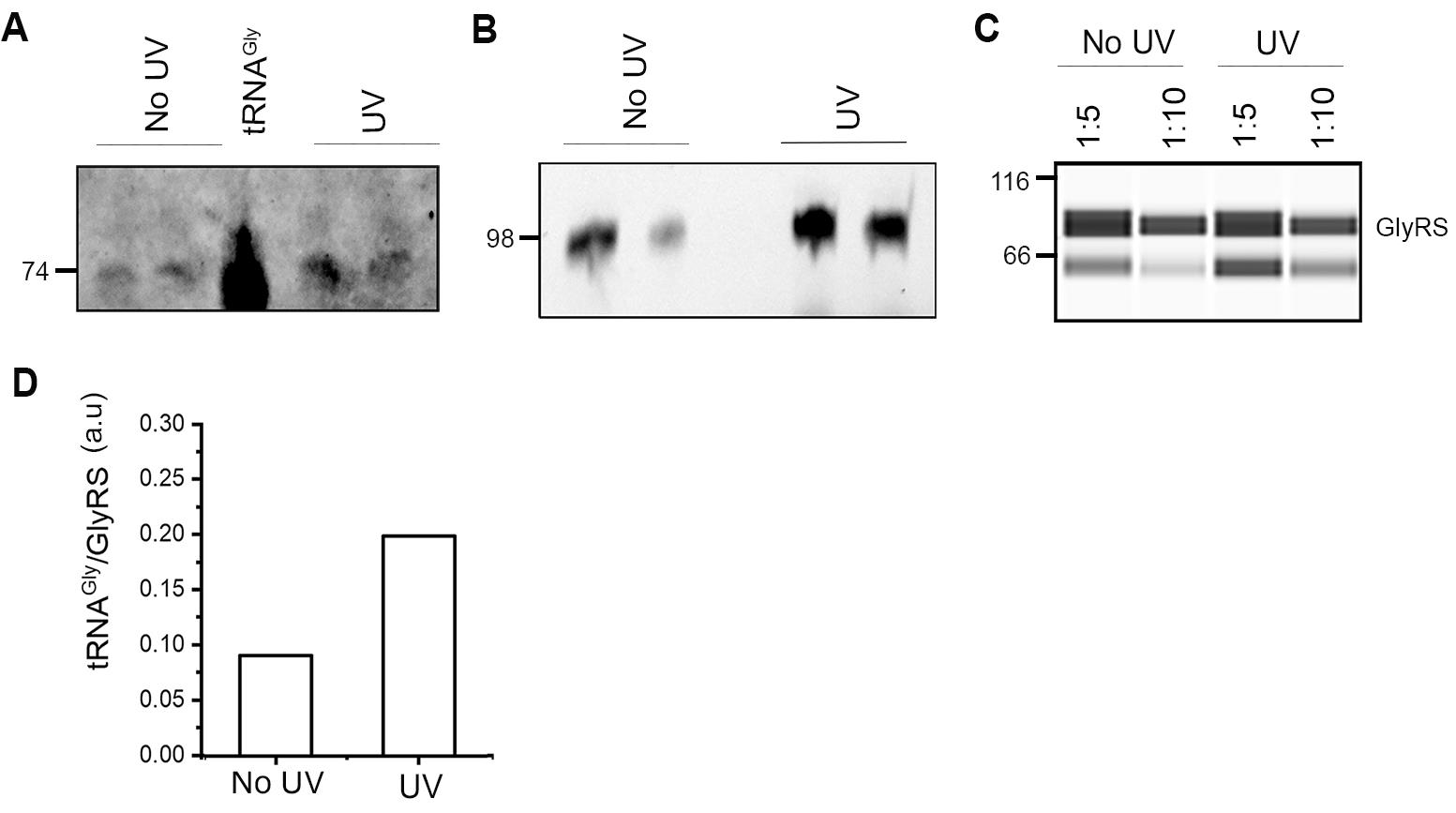
Figure 2. Establishing of the experimental conditions and quantification of tRNA bound to GlyRS in HEK cells. A. Detection of tRNAGly bound to GlyRS by northern blot using Atto565-labeled probes recognizing all three tRNAGly isoacceptors. In vitro transcribed tRNAGlyGCC was loaded as a positive control and has a size of 74 nt. UV and no UV denote cells treated with UV and with no UV treatment, respectively. B. Quantification of tRNAGly bound to GlyRS with Cy3-labelled fluorescent stem-loop RNA/DNA oligonucleotide. The ligated tRNA product was monitored on a 10% denaturing polyacrylamide gel. Fluorescently labeled extended tRNAs have a size of 98 nt. C. Immunoblot of of the IP analyzed by Jess automated western blot and probed with antibodies recognizing GlyRS. Different dilutions of the IP reactions were analyzed. Protein weight markers are shown on the left in kDa. D. Quantification of tRNA bound to GlyRS. Note that HEK cells were used to optimize the protocol and, hence, performed as a single experiment. UV crosslinking increased the yield of tRNAGly bound to GlyRS.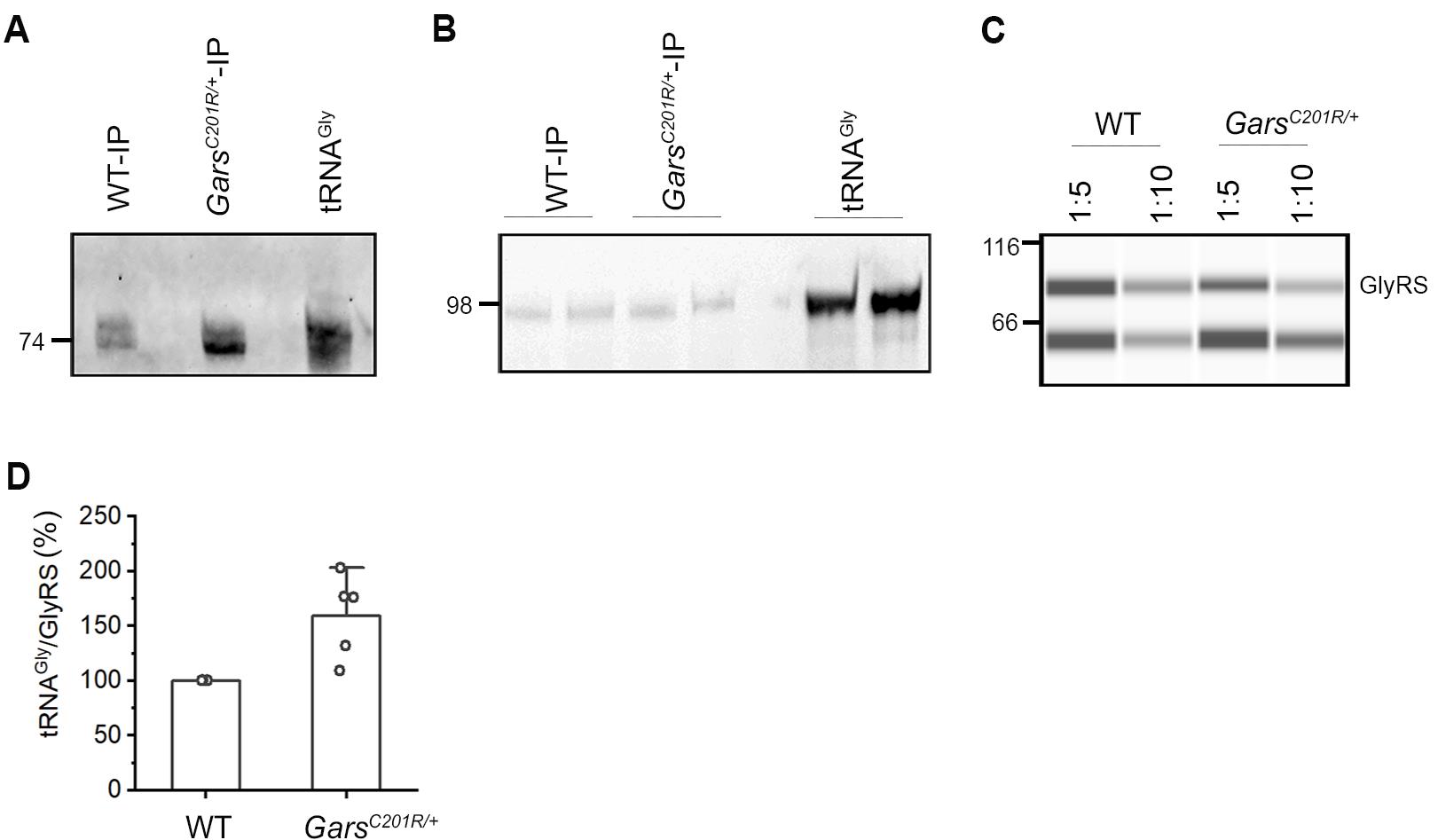
Figure 3. Quantification of tRNA bound to GlyRS in brain tissue from GarsC201R/+ mice (C201R-IP) and wildtype littermate (WT-IP). A. Detection of tRNAGly bound to mutant and wildtype GlyRS by northern blot using Atto565-labelled probes recognizing all three tRNAGly isoacceptors. In vitro transcribed tRNAGlyGCC was loaded as a positive control and has a size of 74 nt. B. Quantification of tRNAGly bound to GlyRS with Cy3-labelled fluorescent stem-loop RNA/DNA oligonucleotide. The ligated tRNA product was loaded on a 10% denaturing polyacrylamide gel. Fluorescently labeled extended tRNAs are with a size of 98 nt. C. Immunoblot of the Ips analyzed by the Jess automated western blot and probed with antibodies recognizing GlyRS. Different dilutions were analyzed. Protein weight markers are shown on the left in kDa. D. Quantification of tRNA bound to GlyRS and normalized to the tRNA/GlyRS ratio of wild-type mice, which is set as 100%. Data are shown as mean ± SEM (n=5 independent biological replicates).
Data analysis
The intensity of the tRNA band was quantified from the gel in Steps D-a6 and D-b6 (Figures 2A and 3A), using ImageJ software. Normalize the intensity of the tRNA band to that of the in vitro transcribed standard tRNA whose precise amount is known.
Note: Ensure that the samples to be compared are loaded onto the same gel and that the image is taken in grayscale. Use the same area when calculating the intensities and average it from multiple technical replicates. The intensity from a blank lane in the gel should be used to subtract the background signal.
From the Jess electrogram report, quantify the peak area corresponding to the protein of interest using the Compass software for Simple western (ProteinSimple). Determine the concentration using the standard curve with purified protein samples (see the note in Step E).
Divide the tRNA amount by that of the protein.
Note: If comparing two conditions or the effect of a mutation, normalize to the ratio to that of the wild-type control, whose ratio is set to 1. We used such additional normalization to enable assessment of the increase in the bound tRNA to CMT-mutant GlyRS (Figure 2E and 3E).
Recipes
Cell lysis buffer
20 mM Tris-HCl, pH 7.4
15 mM NaCl
1% NP-40
0.1% Triton® X-100
1× Protease Inhibitor
Tissue lysis buffer
20 mM Tris-HCl, pH 7.4
15 mM NaCl
1% NP-40
0.1% Triton® X-100
0.5% SDC
1× Protease inhibitor
Wash buffer for cell culture
20 mM Tris-HCl, pH 7.4
100 mM NaCl
1% NP-40
0.1% Triton® X-100
Wash buffer for tissue
20 mM Tris-HCl pH 7.4
100 mM NaCl
1% NP-40
0.1% Triton® X-100
2% SDC
1× SDS buffer
50 mM Tris-HCl pH 6.8
2% SDS
20× SSC buffer
3 M NaCl
0.3 M Trisodium citrate
Hybridization buffer
250 mM Na2HPO4, pH 7.2
1 mM EDTA
7% SDS
0.5% BSA
100 µg/mL salmon sperm DNA
Crush and soak buffer (1–5 mL per sample)
50 mM KOAc
200 mM KCl pH 7.0
2× RNA loading formamide dye
9.5 mL of formamide
330 mL of DEPC water
150 µL of 1% xylene cyanol
20 µL of 0.5 M EDTA
Note: All buffers are made fresh in DEPC-treated water.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, IG73/14-2) to Z.I. and the Donders Center for Neuroscience, the Muscular Dystrophy Association (MDA479773), the EU Joint Programme – Neurodegenerative Disease Research (JPND; ZonMW 733051075 (TransNeuro) and ZonMW 733051073 (LocalNMD), the Radala Foundation, AFM-Téléthon, ARSLA, and an ERC consolidator grant (ERC-2017-COG 770244) to E.S. This adpated protocol was orifginally published in our prevous manuscript (Zuko et al., 2021; Doi: 10.1126/science.abb3356).
Competing interests
The authors declare no competing interests.
Ethics
Mouse experiments were performed at the Radboud University (Nijmegen, Netherlands) and approved by the national Dutch ethics committee ‘Centrale Commissie Dierproeven’ (AVD1030020184826/2017-0067).
References
- Abbott, J. A., Francklyn, C. S. and Robey-Bond, S. M. (2014). Transfer RNA and human disease. Front Genet 5: 158.
- Achilli, F., Bros-Facer, V., Williams, H. P., Banks, G. T., AlQatari, M., Chia, R., Tucci, V., Groves, M., Nickols, C. D. and Seburn, K. L. (2009). An ENU-induced mutation in mouse glycyl-tRNA synthetase (GARS) causes peripheral sensory and motor phenotypes creating a model of Charcot-Marie-Tooth type 2D peripheral neuropathy. Dis Model Mech 2(7-8): 359-373.
- Albers, S., Beckert, B., Matthies, M. C., Mandava, C. S., Schuster, R., Seuring, C., Riedner, M., Sanyal, S., Torda, A. E., Wilson, D. N., et al. (2021). Repurposing tRNAs for nonsense suppression. Nat Commun 12: 3850.
- Barciszewska, M. Z., Perrigue, P. M. and Barciszewski, J. (2016). tRNA--the golden standard in molecular biology. Mol Biosyst 12(1): 12-17.
- Behrens, A., Rodschinka, G. and Nedialkova, D. D. (2021). High-resolution quantitative profiling of tRNA abundance and modification status in eukaryotes by mim-tRNAseq. Mol Cell 81(8): 1802-1815 e1807.
- Betat, H. and Morl, M. (2015). The CCA-adding enzyme: A central scrutinizer in tRNA quality control. Bioessays 37(9): 975-982.
- Fernández-Millán, P., Schelcher, C., Chihade, J., Masquida, B., Giege, P. and Sauter, C. (2016). Transfer RNA: From pioneering crystallographic studies to contemporary tRNA biology. Arch Biochem Biophys 602: 95-105.
- Kimura, S., Srisuknimit, V. and Waldor, M. K. (2020). Probing the diversity and regulation of tRNA modifications. Curr Opin Microbiol 57: 41-48.
- Kirchner, S. and Ignatova, Z. (2015). Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat Rev Genet 16(2): 98-112.
- Kirchner, S., Rauscher, R., Czech, A. and Ignatova, Z. (2017). Microarray-based quantification of cellular tRNAs. protocols.io. dx.doi.org/10.17504/protocols.io.hfcb3iw.
- Schmidt, C. A. and Matera, A. G. (2020). tRNA introns: Presence, processing, and purpose. Wiley Interdiscip Rev RNA 11(3): e1583.
- Tosar, J. P. and Cayota, A. (2020). Extracellular tRNAs and tRNA-derived fragments. RNA Biol 17(8): 1149-1167.
- Warren, J. M., Salinas-Giegé, T., Hummel, G., Coots, N. L., Svendsen, J. M., Brown, K. C., Drouard, L. and Sloan, D. B. (2021). Combining tRNA sequencing methods to characterize plant tRNA expression and post-transcriptional modification. RNA Biol 18: 64-78.
- Zheng, G., Qin, Y., Clark, W. C., Dai, Q., Yi, C., He, C., Lambowitz, A. M. and Pan, T. (2015). Efficient and quantitative high-throughput tRNA sequencing. Nat Methods 12(9): 835-837.
- Zuko, A., Mallik, M., Thompson, R., Spaulding, E. L., Wienand, A. R., Been, M., Tadenev, A. L. D., van Bakel, N., Sijlmans, C., Santos, L. A., et al. (2021). tRNA overexpression rescues peripheral neuropathy caused by mutations in tRNA synthetase. Science 373(6559): 1161-1166.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Das, S., Zuko, A., Thompson, R., Storkebaum, E. and Ignatova, Z. (2022). Immunoprecipation Assay to Quantify the Amount of tRNAs associated with Their Interacting Proteins in Tissue and Cell Culture. Bio-protocol 12(4): e4335. DOI: 10.21769/BioProtoc.4335.
- Zuko, A., Mallik, M., Thompson, R., Spaulding, E. L., Wienand, A. R., Been, M., Tadenev, A. L. D., van Bakel, N., Sijlmans, C., Santos, L. A., et al. (2021). tRNA overexpression rescues peripheral neuropathy caused by mutations in tRNA synthetase. Science 373(6559): 1161-1166.
Category
Neuroscience > Nervous system disorders > Neurodegeneration
Biochemistry > RNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.