- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Hypersensitive Genetically Encoded Fluorescent Indicator (roGFP2-Prx1) Enables Continuous Measurement of Intracellular H2O2 during Cell Micro-cultivation
Published: Vol 12, Iss 3, Feb 5, 2022 DOI: 10.21769/BioProtoc.4317 Views: 3338
Reviewed by: Alexandros AlexandratosGonzalo Durante-RodríguezKomuraiah MyakalaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2021

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Hydrogen peroxide (H2O2) is a toxic oxidant produced as a byproduct of several biological processes. At too high levels of hydrogen peroxide cells will experience oxidative stress, leading to a cellular response to decrease its levels and to protect the cells. Previously, methods used to study and quantify intracellular H2O2 have been limited by both sensitivity and specificity. However, an increasing number of genetically encoded fluorescent indicators (GEFIs) are becoming available, which can specifically detect low levels of intracellular hydrogen peroxide. In this study, we use such a biosensor designed to monitor cytosolic H2O2 levels in the budding yeast Saccharomyces cerevisiae during continuous cultivation and in the absence of a fluorescence microscope. The fluorescent biosensor contains a peroxiredoxin protein fused to an engineered GFP molecule expressed from a commonly used yeast plasmid (pRS416-TEF1). The peroxiredoxin-based fluorescent indicator reduces H2O2, ultimately resulting in a GFP signal being emitted by the sensor. Here, we apply this biosensor to study cytosolic H2O2 levels in S. cerevisiae strains with and without recombinant protein production.
Graphic abstract:
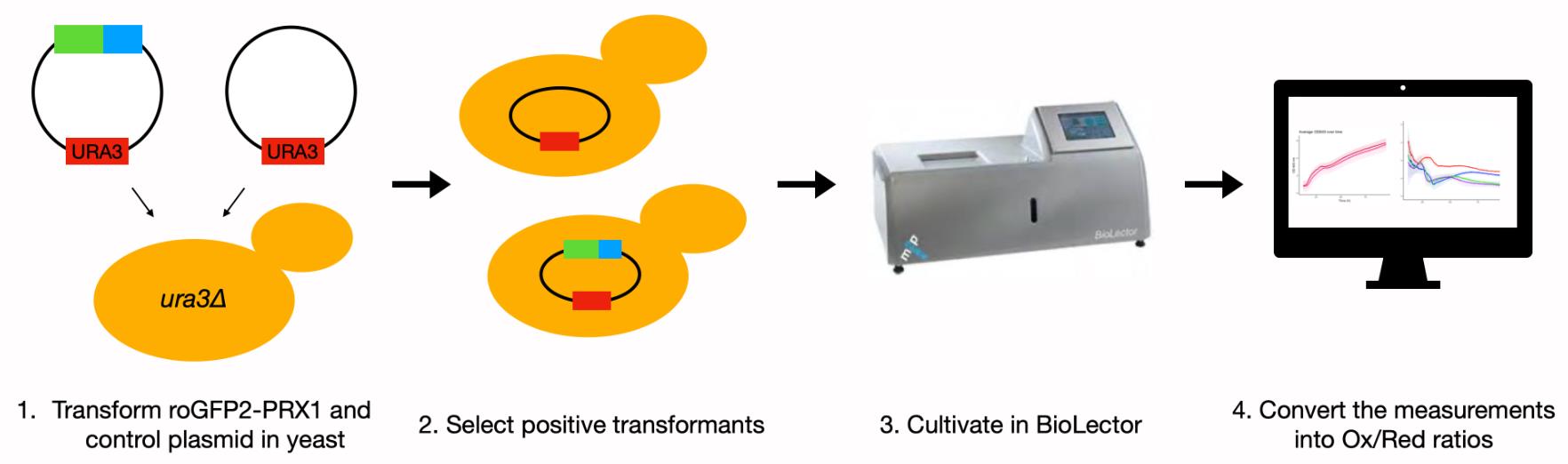
Schematic overview of experimental steps.
Background
In this protocol, we discuss the use of a hypersensitive hydrogen peroxide (H2O2) sensor, roGFP2-Prx1, in the yeast Saccharomyces cerevisiae. S. cerevisiae is a broadly used industrial yeast species but also a popular model organism to study eukaryotic cellular biology. Within biological systems, this includes the generation and buildup of unwanted toxins like H2O2. When oxygen is incompletely reduced to water, reactive oxygen species such as H2O2 are produced. If the intracellular level of H2O2 becomes too high, the cell will endure oxidative stress, which drains resources and increases maintenance costs (Jamieson, 1998; Malhotra et al., 2008; Dever et al., 2016). Oxidative phosphorylation in the mitochondria is responsible for a major part of the H2O2 generated in eukaryotic cells (Malhotra et al., 2008). However, another important source of H2O2 is the production of proteins destined for secretion, and more specifically their folding. The H2O2 resulting from secretory protein production is formed in the iterative process of the making and breaking of disulfide bonds before the proteins reach their final folded state. Especially for recombinant protein production, it is often the goal to achieve high protein titers and yields, which means a high flow through the secretory pathway and the folding machinery in the endoplasmic reticulum (ER). Increased secretory protein folding will lead to an elevated production of H2O2, and therefore potentially oxidative stress.
For many years, methods for measurement of intracellular reactive oxygen species (ROS), including H2O2, consisted of different ROS-sensitive dyes. These dyes are known for their undefined specificity and poor resolution, both temporally and spatially (Morgan et al., 2016). A new technology circumventing these problems was introduced in the form of genetically encoded fluorescent indicators (GEFIs) (Lukyanov and Belousov, 2014). GEFIs are fluorescent biosensors that enable real-time monitoring of several cellular processes but also of relevant compounds like H2O2 (Lukyanov and Belousov, 2014). The basis for the specific GEFI used in this protocol is a fusion of two proteins, which form a redox pair when together. One of these is a highly H2O2-reactive peroxiredoxin e.g., yeast Prx1, and the other a green fluorescent protein, roGFP2. RoGFP2 is an engineered enhanced GFP that has two cysteines in close proximity to the chromophore. Upon the making or breaking of a disulfide bond between those two cysteines, there will be a change in the protonation state of the chromophore, which changes the excitation wavelength (Schwarzländer et al., 2016). With the reduction of H2O2, the two cysteines in Prx1 form a disulfide bond that will next react into a mixed disulfide bond between Prx1 and the roGFP2 molecule; this finally resolves into an internal disulfide bond within roGFP2, thereby changing the excitation wavelength. A schematic overview of this mechanism is shown in Figure 1. RoGFP2 has excitation peaks at 488 nm in the reduced form and at 400 nm in the oxidized form that carries the internal disulfide bond (Schwarzländer et al., 2016). By dividing the signal measured at 400 nm by the signal measured at 488 nm, an oxidized:reduced ratio can be determined. At a basal level of endogenous H2O2 under non-stressed conditions, the sensor maintains an oxidized:reduced ratio near 1:1 (Schwarzländer et al., 2016). Furthermore, the sensor is ratiometric, and therefore any potential differences in sensor abundance between strains and/or conditions are expected not to affect measurements. The functionality and sensitivity of these sensors in cells have been tested by the direct addition of exogenous H2O2, but also a reducing agent (DTT), to the media (Morgan et al., 2016; Gast et al., 2021). Importantly, the roGFP2-Prx1 sensor can detect both oxidizing and reducing changes in the extracellular environment.
Here, we present a protocol on the use of the roGFP2-Prx1 sensor for in vivo measurements of cytosolic H2O2 during continuous micro-cultivation of S. cerevisiae in a BioLector (Mp2 Labs). This protocol was successfully used to detect changes in cytosolic H2O2 levels in S. cerevisiae due to recombinant protein production (Gast et al., 2021). The sensors are expected to be adequate to measure H2O2 produced from sources other than recombinant protein production as well.
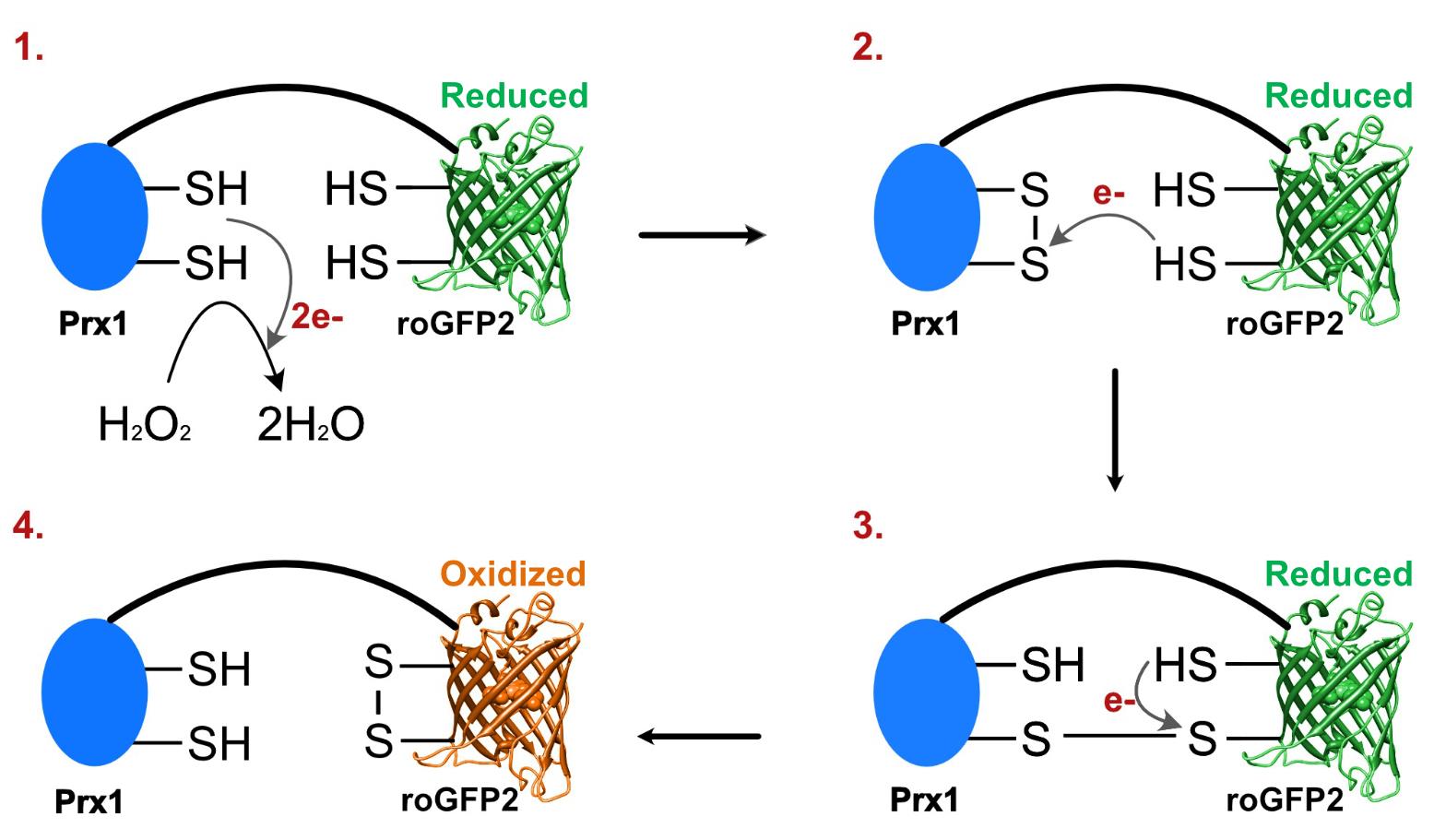
Figure 1. H2O2 oxidizes cysteines in Prx1-roGFP2 into a disulfide bond altering sensor fluorescence. (1) Prx1 in the genetically-encoded fluorescent indicator reduces H2O2 to water by making an internal disulfide bond. (2) One of the electrons of a cysteine in the roGFP2 reacts with one of the cysteines in the disulfide bond, resulting in a new disulfide bond between Prx1 and roGFP2. (3) The second cysteine in roGFP2 reacts with the other cysteine in the disulfide bond between Prx1 and roGFP2, resulting in a new disulfide bond between the two cysteines in roGFP2. (4) The disulfide bond between the cysteines in roGFP2 changes the equilibrium of different forms of the chromophore, resulting in a shift of the wavelength of maximal excitation of roGFP2 from 488 to 400 nm.
Materials and Reagents
Materials
48-well flowerplates (Mp2 Labs, catalog number: MTP-48-B)
Sealing foil reduced evaporation (Mp2 Labs, catalog number: F-GPR48-10)
Sterile 1.5 mL tubes (Eppendorf, catalog number: 0030120086)
Culture tubes (VWR, catalog number: 211-0082)
Cuvettes PS (VWR, catalog number: 634-0676)
50 mL Falcon tubes (Corning, catalog number: 352070)
Petri dishes (Sigma-Aldrich, catalog number: P5731)
Biological materials and reagents
S. cerevisiae strain B184 ura3Δ
Plasmid pRS416-TEF1-roGFP2-PRX1 (Morgan et al., 2016)
Plasmid pRS416M1-TEF1 (control) (Kaur and Bachhawat, 2009), but plasmid pRS416-TEF1 works equally well. pRS416M1-TEF1 was constructed by removing all the restriction sites in the cloning site of pRS416-TEF1, except for BamHI and EcoRI.
Boiled ss-carrier DNA
Sterile H2O (store at RT)
Sterile 0.1 M LiAc (store at RT)
YNB w/o AA (Formedium, catalog number: CYN0405)
NaH2PO4·2H2O (Merck, catalog number: 06342)
Na2HPO4 (Merck, catalog number: 06586)
Glucose·H2O (Merck, catalog number: 08342)
BSA (Sigma-Aldrich, catalog number: A7030)
Arginine (Sigma-Aldrich, catalog number: A5006)
Aspartic acid (Sigma-Aldrich, catalog number: A9256)
Glutamic acid (Sigma-Aldrich, catalog number: G1251)
Glycine (Sigma-Aldrich, catalog number: G7126)
Histidine (Sigma-Aldrich, catalog number: H8000)
Isoleucine (Sigma-Aldrich, catalog number: I2752)
Leucine (Sigma-Aldrich, catalog number: L8000)
Methionine (Sigma-Aldrich, catalog number: M9625)
Phenylalanine (Sigma-Aldrich, catalog number: P2126)
Threonine (Sigma-Aldrich, catalog number: T8625)
Tryptophan (Sigma-Aldrich, catalog number: T0254)
Tyrosine (Sigma-Aldrich, catalog number: T3754)
Valine (Sigma-Aldrich, catalog number: V0500)
Yeast extract (Fisher Scientific, catalog number: AC451120010)
LiAc (Sigma-Aldrich, catalog number: L6883)
PEG 3350 (Sigma-Aldrich, catalog number: 202444)
Agar (Sigma-Aldrich, catalog number: 05040)
Complete Supplement Mixture Single Drop-Out -Ura (Formedium, catalog number: DCS0169)
SD2xSCAA medium (store at 4°C) (see Recipes)
SD-URA agar plates (store at 4°C) (see Recipes)
Yeast extract peptone dextrose media (YPD) (store at RT) (see Recipes)
Sterile 50% PEG 0.1 M LiAc (store at RT) (see Recipes)
Equipment
Micropipettes (Eppendorf, model: Research® plus)
BioLector I (Mp2 Labs, BioLector)
Emission filters:
a. GFP filter: excitation 480 nm and emission 520 nm (Mp2 Labs, model: E-OP-404)
b. UV-GFP filter: excitation 405 nm and emission 508 nm (Mp2 Labs, model: E-OP-412)
c. Biomass filter: absorption 620 nm (Mp2 Labs, model: E-OP-401)
OD spectrophotometer (ThermoFisher, Genesys 20)
Temperature controlled environment at 30°C
Water bath at 42°C
Shaking platform with 96 culture tubes rack holders (VWR, Advanced orbital Shaker)
Laminar flow hood with UV sterilization
Centrifuge (Eppendorf, model: 5417 R)
Software
RStudio: integrated development environment for R. (2021) (Rstudio Team, Vienna, Austria, https://www.rstudio.com) (optional)
BioLection software (Mp2 Labs)
Procedure
Transformation of S. cerevisiae ura3Δ strain with pRS416-TEF1-roGFP2-PRX1 and pRS416M1-TEF1
Isolate the plasmid DNA of pRS416-TEF1-roGFP2-PRX1 and pRS416M1-TEF1 (if necessary).
Inoculate 2 mL of liquid YPD in a cultivation tube with the S. cerevisiae ura3Δ strain and shake (200 rpm at 30°C) overnight.
Inoculate 20 mL of fresh YPD in a 50 mL Falcon tube with 1 mL of the overnight culture and shake at 220 rpm at 30°C.
After 5-6 h of incubation, spin down the S. cerevisiae cells (3 min at 3,500 × g).
Remove the YPD in a sterile environment (e.g., in a laminar flow hood), wash the cells with sterile water, vortex, and spin down (3 min at 3,500 × g).
Remove the sterile water in a sterile environment, wash the cells with 0.1 M LiAc, vortex, and spin down (3 min at 3,500 × g).
Remove the 0.1 M LiAc in a sterile environment, add 200 μL of fresh 0.1 M LiAc, vortex, and keep the suspended cells like this.
Take three autoclaved Eppendorf tubes per strain, add 5 μL of the boiled ss-carrier DNA and the two plasmids (100-500 ng) to separate tubes, leaving one negative control (no DNA addition). Also add 25 μL of the yeast 0.1 M LiAc mixture, and finally 200 μL of 50 % PEG 0.1 M LiAc. Vortex the tubes and incubate in 30°C room from 35 min to 3 h, while shaking. The rest of the cells can be discarded.
Turn on the water bath to 42°C and incubate the tubes 20-30 min.
Spin down 20-30 s at 8,000 rpm and remove the supernatant.
Add 100-200 μL of sterile water and resuspend the pellet.
Plate the suspensions on marked SD-URA agar plates.
Incubate the plates for 72 h at 30°C.
Micro-cultivation in the BioLector
Before starting this experiment, determine a layout for the 48-well plate. Leave one or two wells containing only medium, which can serve as the ‘blank’ for the BioLector and as controls for sterile handling. We included three biological replicates, each including two technical replicates for the strain with the pRS416-TEF1-roGFP2-PRX1 plasmid, and two biological replicates with two technical replicates each of the strain carrying the pRS416M1-TEF1 control plasmid. If strains are to be compared regarding their intracellular H2O2, they should be examined in the same experiment since the exact profiles differ slightly between experiments. The BioLector plates are delivered sterile and are for single-time use, but they can be reused once or twice after cleaning by the following procedure. Collect the waste directly after the previous run and wash the plate twice with 70% ethanol. Rinse it multiple times with demineralized water to remove the ethanol. Let the plate air dry and store with a plastic layer on the bottom for protection. Before the next run, sterilize with UV light for at least 30 min.
Restreak three (or more) colonies per strain on selective solid SD-URA medium.
Inoculate three colonies isolated from the SD-URA selective plates per strain in separate cultivation tubes containing 1 mL of SD2xSCAA.
Incubate the cultures in an angle of approximately 45° and shake overnight (220 rpm at 30°C).
After overnight incubation, determine the OD600 for the precultures with the spectrophotometer.
Prepare 3 mL of SD2xSCAA media fresh in culture tubes and add the preculture until an OD600 of 0.01 or 0.005 (no washing step).
From these 3 mL tubes, two technical duplicates of 1 mL are added to wells in the 48-well flower-plate.
After all the wells are filled, cover the plate with the sealing foil for reduced evaporation and ensure the dots are in the middle of the wells.
Load the plate into the BioLector. Before, remove the protective layer at the bottom!
Make sure to refill the (demineralized) water up to above the 150 mL mark in the BioLector for humidity control.
Settings for the BioLector.
Temperature control on and set to 30°C.
Humidity control on and set to 85%.
Shaking on and set to 1200 rpm.
Filters and measurements: Three different filters were used to measure 1) biomass concentration (absorbance at 620 nm), 2) GFP fluorescence (excitation 488 nm), and 3) UV-GFP fluorescence (excitation 405 nm). The BioLector allows for six channels to be measured at the same time. Since this experiment only required three different filters, one can choose to measure two gains per filter. For the OD620, we selected gains 20 and 30, for GFP, gains 50 and 100, and for UV-GFP, gains 50 and 100. The cycle time of the BioLector to measure the six channels chosen is 20 min. An overview of the filters and gains used is shown in Table 1.
Table 1. Filters and gain settings used in the BioLector.
Excitation and emission wavelengths for the three BioLector filters and information on the corresponding gain settings used for each of them.Filter Excitation [nm] Emission [nm] Gain Biomass 620 620 20 Biomass 620 620 30 GFP 488 520 100 GFP 488 520 50 UV-GFP 405 508 100 UV-GFP 405 508 50
Start the measurement (double check that the plastic has been removed from the bottom of the plate). Check the cultivation frequently during the run.
When finished, remove the plate (and clean immediately).
Data analysis
The data analysis protocol is presented with the measurements of four example strains, here renamed strain 1, strain 2, strain 3, and strain 4, to direct the attention to the redox sensor measurements. A clarification of the strains used is shown in Table 2 and will be discussed briefly at the end of the protocol; this can also be found in the original article (Gast et al., 2021).
Export data from the BioLector software (Biolection) to Excel format (or any format of choice).
For the data analysis, we only used the data of gain 100 for UV-GFP and GFP, and of gain 20 for OD620.
Import data into R (or any other software of choice). The data includes the three measurements per well, the OD620, the UV-GFP, and GFP, each with their selected gain at each of the timepoints, and the time.
The first step, depending on your setup, is the removal of the background signal of the media from the measurement. This can be done by the BioLector or manually.
Table 2. Genotypes of the strains used in this protocol.
Strain genotype details helping to interpret data generated by the protocol in our experiments. For more details see the original study (Gast et al., 2021).
Filter Description Strain 1 B184 expressing amylase Strain 2 B184 gcn2Δ expressing amylase Strain 3 B184 with control plasmid Strain 4 B184 gcn2Δ with control plasmid Plot the OD620, the UV-GFP, and GFP against time, for each strain.
Compare the growth of each strain with the sensor plasmid to that of the same strain with the control plasmid. Check OD620 to ascertain whether the growth of the strain expressing the roGFP2-Prx1 sensor plasmid (pRS416-TEF1-roGFP2-PRX1) and the strain with the control plasmid (pRS416M1-TEF1) are comparable. In Figure 2A, we show comparable growth based on the OD620 measurements of strain 4 expressing the plasmid with roGFP2-Prx1 sensor to the control plasmid (Figure 2A). If the growth is not comparable, an additional step, which is outlined in the Notes section, needs to be taken.
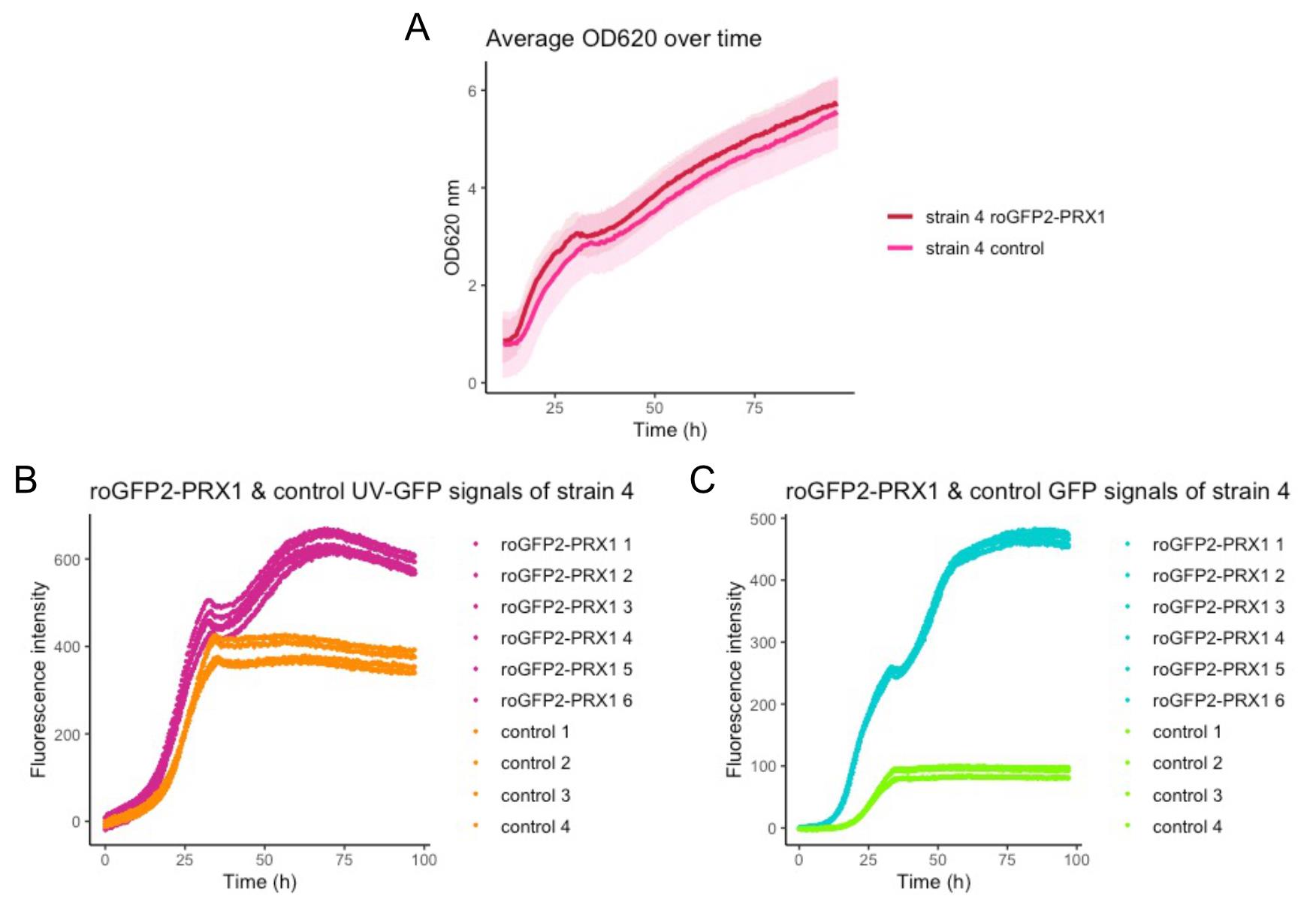
Figure 2. Measurements of OD620, UV-GFP, and GFP plotted against the time. (A) The average of OD620 measurements of strain 4 with the plasmid encoding the roGFP2-Prx1 sensor or the control plasmid. The light bars show the standard deviation of the independent OD620 measurements per strain, (B) UV-GFP measurements of strain 4 with roGFP2-Prx1 sensor (pink) or the control (orange), (C) GFP measurements of strain 4 with roGFP2-Prx1 sensor (cyan) or the control (light green).Verification of the UV-GFP and GFP measurements of the roGFP2-Prx1 sensor and the control for each strain.
Review the measurements of UV-GFP and GFP in each strain, paying special attention if there is a difference in fluorescence intensity between your roGFP2-Prx1 sensor and the control strain.
The raw measurements of UV-GFP in cells carrying the roGFP2-Prx1 sensor and in control cells are shown in Figure 2B, and the same for GFP in Figure 2C. In both plots, there is a visible difference between the strain with the roGFP2-Prx1 sensor and the control. During this step, you can verify if the sensors are working in your setup and control for outliers present within the replicates. Outliers may be removed, but ensure that you still have at least four measurements with the roGFP2-Prx1 sensor and three measurements with the control. The fluorescence measured in the strain with the control plasmid corresponds to the auto fluorescence of the respective strain. The UV-GFP and GFP fluorescence intensity of this control should be subtracted from the UV-GFP and GFP fluorescence measurements of the strain with biosensor in the next step.
Determination of UV-GFPcorr and GFPcorr by removing the autofluorescence from the roGFP2-Prx1 UV-GFP and GFP measurement values.
Subtract the auto-fluorescence (control measurements) from the UV-GFP and GFP values (roGFP2-Prx1 measurements). First, calculate average values of autofluorescence for each strain. Determine one average value per timepoint. Subtract the average autofluorescence value from the single UV-GFP and GFP values for the corresponding strain. The formulas for the calculations of UV-GFPcorr (1) and GFPcorr (2) are shown below. Average values of the UV-GFPcorr and GFPcorr are shown in Figure 3.
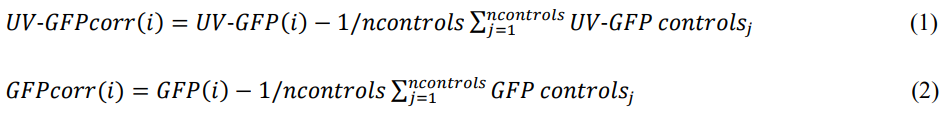
Determination of the final Ox/Red ratio.
In this final step, use the individual UV-GFPcorr and GFPcorr values determined for each well and time-point to determine an Ox/Red ratio per time-point and well. Use the available Ox/Red ratios from your experiment, and determine an average value per strain and timepoint, in addition to a corresponding standard deviation. The equation is shown below (3). Make a final plot in which you plot the average ratios of your strains against time, including information on standard deviations. An example is shown in Figure 4.

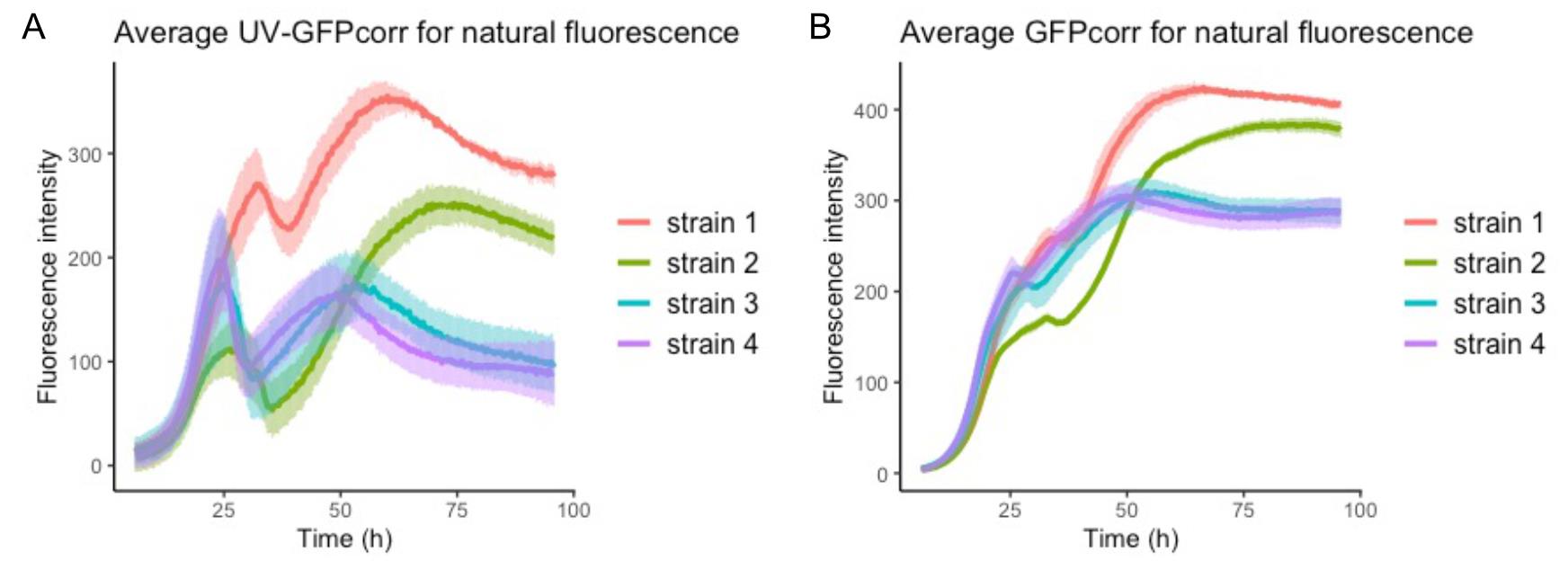
Figure 3. Average values of UV-GFPcorr and GFPcorr measurements of strain 1 (red), strain 2 (green), strain 3 (blue), and strain 4 (purple). (A) Average values of the UV-GFPcorr values. (B) Average values of the GFPcorr values. The light bars represent the standard deviation of all the samples in each strain.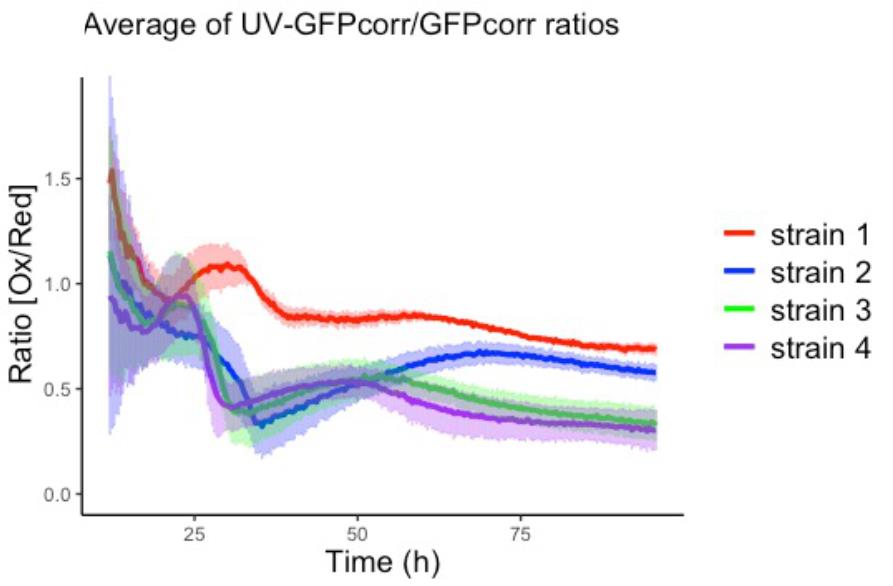
Figure 4. Average ratio [Ox/Red] for strain 1 (red), strain 2 (blue), strain 3 (green) and strain 4 (purple). The light bars represent the standard deviation of all the samples in each strain.Data interpretation
To put the results in Figure 4 in context, we hereby provide a brief background to the original study (Gast et al., 2021). Recombinant protein production is known to be a source of H2O2 and oxidative stress. It remains however complicated to study these phenomena due to limitations in H2O2 detection methods (Malhotra et al., 2008). In the original study, roGFP2-Prx1 biosensors were used to document the influence of recombinant protein production on cytosolic H2O2 levels in S. cerevisiae. Strain 1 is a S. cerevisiae strain that produces recombinant amylase, and strain 3 is the control strain not producing a recombinant protein. Figure 4 shows that strain 1 has a higher Ox/Red Ratio throughout most of the cultivation, indicating a higher level of H2O2 in the cytosol as a consequence of recombinant protein production. We were also interested in the role of the protein kinase Gcn2 within recombinant protein production and oxidative stress. The kinase Gcn2 is best known for its role within the general amino acid control pathway (Hinnebusch, 2005). When S. cerevisiae experiences amino acid starvation, the kinase Gcn2 is activated and phosphorylates the translation initiation factor eIF2a, leading to a reduction of translation initiation (Hinnebusch, 1984). Interestingly, the kinase Gcn2 can also be activated by the addition of H2O2 to the medium (Shenton et al., 2006). In our study, we examined if the kinase Gcn2 could also be activated by the endogenous H2O2 produced as a by-product of recombinant protein production, and if the removal of the kinase would affect cytosolic H2O2 and the efficacy of recombinant protein production. Strains 2 and 4 are similar to strains 1 and 3, respectively, but lacking the GCN2 gene. Interestingly, the removal of kinase Gcn2 lead to a reduction in cytosolic H2O2 in the strain that produces amylase (compare strain 1 to strain 2), despite boosting protein production two-fold (Gast et al., 2021). However, this is not seen in the strain without recombinant protein production (compare strain 3 to strain 4).
Notes
We found that the GFP measurements are quite sensitive, and we would suggest to use new 48-well plates if possible. We did obtain similar results when reusing the 48-well plates in the repetitions, but it seems that the sensitivity of the plate decreases slightly upon cleaning and reuse.
If the growth of strains carrying the plasmid with roGFP2-Prx1 sensor and the control plasmid are not similar, it is necessary to normalize the GFP and UV-GFP measurements to the biomass. This should be done before removing the autofluorescence from the GFP and UV-GFP measurements. However, we found that this normalization led to noisy Ox/Red ratios, particularly in the beginning of the experiments when cell numbers are low. Therefore, we decided to exclude this normalization, since the biomass growth of the strains carrying the plasmid with the roGFP2-Prx1 sensor and the control plasmid were very similar in our experiments. If growth is similar between the strains with the roGFP2-Prx1 sensor and the control plasmid, then normalization to biomass is not required.
Although here we grew cells in SD2xSCAA medium, we also performed experiments with the sensors in synthetic minimal medium for yeast (Verduyn et al., 1992; Gast et al., 2021). On the other hand, a strongly colored medium (e.g., rich YPD medium) might be more problematic because of its stronger fluorescence.
We expect that this method may be used in several prokaryotes and eukaryotes, if the species can grow in suspension in the BioLector, and suitable fluorescent reporters are available.
Recipes
We used a medium specific for high production of proteins called SD2xSCAA medium (Wittrup and Benig, 1994). To generate it, we make four separate solutions that, due to different sterilization methods, are mixed only in the end. The fractions of each of these solution to be used in the final medium are indicated in brackets.
SD2xSCAA
YNB + Sodium Buffer 0.05 M, pH 6.4 – add (7/10)
YNB w/o AA 6.9 g/L
NaH2PO4·2H2O 8.56 g/L
Na2HPO4 5.4 g/L
The combination of sodium phosphate buffer and YNB does not dissolve easily. We would suggest to initially make two solutions, one with only YNB and one with the sodium buffer, and mix these after the components have dissolved.
Filter sterilize and store at 4°C.
After sterilization, add the other three solutions within the day, since the sodium tends to precipitate; but, when all the components are added, the mixture remains rather stable.
20% Glucose – add (1/10)
Glucose·H2O 220 g/L
Filter sterilize and store at RT
1% BSA – add (1/10)
BSA 10 g/L
Filter sterilize and store at 4°C
Aminoacid solution – add (1/10)
Arginine 1.9 g/L
Aspartic acid 4 g/L
Glutamic acid 12.6 g/L
Glycine 1.3 g/L
Histidine 1.4 g/L
Isoleucine 2.9 g/L
Leucine 4 g/L
Methionine 1.08 g/L
Phenylalanine 2 g/L
Threonine 2.2 g/L
Tryptophan 0.4 g/L
Tyrosine 0.52 g/L
Valine 3.8 g/L
Filter sterilize and store at 4°C
We make the 10× stocks (Glucose, BSA, and amino acid solution) and the YNB+sodium buffer (10/7×) fresh, and add the three 10× stocks of the other components to the YNB+sodium buffer solution directly after preparation, to ensure that the sodium salts do not precipitate. Store the final SD2xSCAA media at 4°C and use within 6 months. Check for precipitation before use.
YPD
Yeast extract 10 g/L
Peptone from meat 20 g/L
Glucose·H2O 22 g/L
Autoclave and store at RT
SD-URA agar plates
To prepare SD-URA plates, two separate solutions should be made and combined to a final SD-URA agar solution that is poured into the Petri dishes. The recipe below is for making 1 L of SD-URA agar solution, which will make around 50 SD-URA agar plates.
Solution 1 (250 mL)
CSM Single drop out:
URA 0.77 g
YNB w/o AA 6.9 g
Glucose·H2O 22 g
Set pH to 5.5-6.0
Heat the solution to 60°C in a microwave oven
Filter sterilize
Solution 2 (750 mL)
Agar 20 g
Autoclave
Allow solution 2 to cool down until around 60°C. Add solution 1 to solution 2 and mix thoroughly before preparing the plates. Aim to add 20 mL of SD-URA agar solution to each petri dish.
50% PEG 0.1 M LiAc
Prepare 0.2 M LiAc and add PEG 3350 1:1
Autoclave and store at RT
Acknowledgments
We would like to thank Dr. Kate Campbell for performing the groundwork for this study. The work was supported by the VINNOVA center CellNova (2017-02105) and grants from Cancerfonden (2017-0778), ÅForsk and Vetenskapsrådet (2020-05422). This protocol is derived from the original research article Gast V et al (2021), doi: 10.1128/AEM.00301-21
Competing interests
The authors declare that they have no competing interests.
References
- Dever, T. E., Kinzy, T. G. and Pavitt, G. D. (2016). Mechanism and Regulation of Protein Synthesis in Saccharomyces cerevisiae. Genetics 203(1): 65-107.
- Gast, V., Campbell, K., Picazo, C., Engqvist, M., Siewers, V. and Molin, M. (2021). The Yeast eIF2 Kinase Gcn2 Facilitates H2O2-Mediated Feedback Inhibition of Both Protein Synthesis and Endoplasmic Reticulum Oxidative Folding during Recombinant Protein Production. Appl Environ Microbiol 87(15): e0030121.
- Hinnebusch, A. G. (1984). Evidence for translational regulation of the activator of general amino acid control in yeast. Proc Natl Acad Sci U S A 81(20): 6442-6446.
- Hinnebusch, A. G. (2005). Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol 59: 407-450.
- Jamieson, D. J. (1998). Oxidative stress responses of the yeast Saccharomyces cerevisiae. Yeast 14(16): 1511-1527.
- Kaur, J. and Bachhawat, A. K. (2009). Gln-222 in transmembrane domain 4 and Gln-526 in transmembrane domain 9 are critical for substrate recognition in the yeast high affinity glutathione transporter, Hgt1p. J Biol Chem 284(35): 23872-23884.
- Lukyanov, K. A. and Belousov, V. V. (2014). Genetically encoded fluorescent redox sensors. Biochim Biophys Acta 1840(2): 745-756.
- Malhotra, J. D., Miao, H., Zhang, K., Wolfson, A., Pennathur, S., Pipe, S. W. and Kaufman, R. J. (2008). Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci U S A 105(47): 18525-18530.
- Morgan, B., Van Laer, K., Owusu, T. N., Ezerina, D., Pastor-Flores, D., Amponsah, P. S., Tursch, A. and Dick, T. P. (2016). Real-time monitoring of basal H2O2 levels with peroxiredoxin-based probes. Nat Chem Biol 12(6): 437-443.
- Schwarzländer, M., Dick, T. P., Meyer, A. J. and Morgan, B. (2016). Dissecting redox biology using fluorescent protein sensors. Antioxid Redox Signal 24(13): 680-712.
- Shenton, D., Smirnova, J. B., Selley, J. N., Carroll, K., Hubbard, S. J., Pavitt, G. D., Ashe, M. P. and Grant, C. M. (2006). Global translational responses to oxidative stress impact upon multiple levels of protein synthesis. J Biol Chem 281(39): 29011-29021.
- Verduyn, C., Postma, E., Scheffers, W. A. and Van Dijken, J. P. (1992). Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast 8(7): 501-517.
- Wittrup, K. D. and Benig, V. (1994). Optimization of amino acid supplements for heterologous protein secretion in Saccharomyces cerevisiae. Biotechnol Tech 8: 161-166.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Gast, V., Siewers, V. and Molin, M. (2022). A Hypersensitive Genetically Encoded Fluorescent Indicator (roGFP2-Prx1) Enables Continuous Measurement of Intracellular H2O2 during Cell Micro-cultivation. Bio-protocol 12(3): e4317. DOI: 10.21769/BioProtoc.4317.
Category
Cell Biology > Cell signaling > Stress response
Microbiology > Microbial cell biology
Cell Biology > Cell signaling > Intracellular Signaling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.