- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Flow Cytometry Analysis of Planarian Stem Cells Using DNA and Mitochondrial Dyes
(*contributed equally to this work) Published: Vol 12, Iss 2, Jan 20, 2022 DOI: 10.21769/BioProtoc.4299 Views: 3696
Reviewed by: Giusy TornilloPrasad AbnaveAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2021
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Planarians are free-living flatworms that emerged as a crucial model system to understand regeneration and stem cell biology. The ability to purify neoblasts, the adult stem cell population of planaria, through fluorescence-activated cell sorting (FACS) has tremendously increased our understanding of pluripotency, specialization, and heterogeneity. To date, the FACS-based purification methods for neoblasts relied on nuclear dyes that discriminate proliferating cells (>2N), as neoblasts are the only dividing somatic cells. However, this method does not distinguish the functional states within the neoblast population. Our work has shown that among the neoblasts, the pluripotent stem cells (PSCs) are associated with low mitochondrial content and this property could be leveraged for purification of the PSC-enriched population. Using the mitochondrial dye MitoTracker Green (MTG) and the nuclear dye SiR-DNA, we have described a method for isolation of PSCs that are viable and compatible with downstream experiments, such as transplantation and cell culture. In this protocol, we provide a detailed description for sample preparation and FACS gating for neoblast isolation in planaria.
Background
Fluorescent dyes that stain the nucleus in live cells are used for fluorescence-activated cell sorting (FACS) proliferating stem cells from planaria. In a seminal work, Hayashi et al. used Hoechst for staining planarian cells and categorized the cells into three populations, termed as X1, X2, and Xins population (Figure 2) and (Hayashi et al., 2006). This allowed sorting of the proliferating cells (X1 cells) in the planarian field of research (Hayashi et al., 2006). Later, Hoechst stain was found to be toxic so, for downstream applications where viable cells are required, a different FACS gate X1(fs) based on forward scatter (FSC) had to be used (Wagner et al., 2011). This population was obtained by back-gating the X1 population in an FSC vs SSC plot (Wang et al., 2018). However, X1(fs) is heavily contaminated by differentiated cells. Subsequently, the nuclear dye SiR-DNA was proposed as an alternative to Hoechst for obtaining proliferating stem cells with high viability (Lei et al., 2019; Niu et al., 2021). However, this flow cytometry method using a nuclear dye could not distinguish between the pluripotent and the specialized stem cell populations.
Methods that could distinguish pluripotent from specialized stem cells could advance our understanding of planarian stem cell biology and function. Since neoblasts have scant cytoplasm and reduced organelles, we reasoned that organelle complexity could be way to distinguish different cellular states (Higuchi et al., 2007). For this purpose, we used mitochondria, an organelle that plays an important role in metabolism and signaling. Studies in mammalian stem cells have suggested that the mitochondrial state, including the mitochondrial activity, content, morphology, and number varies between different cellular states. We have recently reported the use of MitoTracker Green (MTG) staining in combination with nuclear dyes, such as Hoechst and SiR-DNA, for the purification of pluripotent stem cells (Mohamed Haroon et al., 2021). Our data suggest that the population of proliferating cells with low MTG is enriched in pluripotent cells compared to the high MTG cells, as indicated by higher transplantation efficiency (~65% for MTG Low cells vs ~30% for MTG High cells). Further, this method could be used to enrich the 2N stem cells from the X2 population, which is a mixture of stem cells and mitotic progenitors. To this end, the X2 gate was divided into four populations based on MTG and FSC, which indicates size. We found that the 2N PSCs are enriched in the low MTG and high FSC gate (Mohamed Haroon et al., 2021). In contrast, X2 cells with low MTG and low FSC were predominantly early post-mitotic progenitors. The X2 high MTG cells, regardless of their size, were observed to be late progenitors (Mohamed Haroon et al., 2021). Here, we provide a step-by-step protocol for planarian dissociation, staining, and FACS gating of various MTG populations.
Materials and Reagents
50 mL centrifuge tubes (Tarsons, catalog number: 546021)
15 mL centrifuge tubes (Tarsons, catalog number: 546041)
35 mm culture dish
60 mm culture dish
Falcon 5 mL round bottom FACS tubes (BD, Falcon, catalog number: 352054)
SpheroTM Rainbow fluorescent particles (BD Biosciences, catalog number: 556291)
BD FACS Accudrop beads (BD, catalog number: 345249)
0.45 μm Millex-HV syringe filter unit (Merck Millipore, catalog number: SLHVR33RS)
0.22 μm Millex-GV syringe filter unit (Merck Millipore, catalog number: SLGVR33RS)
40 µm cell strainer (BD Falcon, catalog number: 352340)
Surgical blade No. 23 (Lister)
Bovine serum albumin (Sigma-Aldrich, catalog number: A2153)
Hoechst 33342 (Invitrogen, catalog number: H3570)
MitoTracker Green FM (Invitrogen, catalog number: M7514)
SiR-DNA (Cytoskeleton, Inc. catalog number: CY-SC007)
DAPI (Sigma-Aldrich, catalog number: D9542)
Propidium iodide (Sigma-Aldrich, catalog number: P4864)
MEM essential amino acids solution (50×) (ThermoFisher Scientific, GibcoTM, catalog number: 11130051)
MEM non-essential amino acids solution (100×) (ThermoFisher Scientific, GibcoTM, catalog number: 11140050)
MEM Vitamine solution (100×) (ThermoFisher Scientific, GibcoTM, catalog number: 11120052)
Sodium pyruvate (100×) (ThermoFisher Scientific, GibcoTM, catalog number: 11360070)
100× Penicillin streptomycin (ThermoFisher Scientific, GibcoTM, catalog number: 15070063)
L-glutamine (ThermoFisher Scientific, GibcoTM, catalog number: 25030081)
Fetal bovine serum (Himedia, catalog number: RM9955)
NaCl
CaC3
MgSO4
MgCl2
KCl
NaHCO3
NaH2PO4
KCl
Glucose
HEPES (free acid)
HEPES (sodium salt)
MnCl2
KH2PO4
D-biotin
D-glucose
D-trehalose
Tricine
Montjuïc salts (see Recipes)
Calcium, magnesium free buffer with BSA (CMFB) (see Recipes)
IPM + 10% FBS (see Recipes)
Equipment
Refrigerated centrifuge (Eppendorf, model: 5810R)
FACS sorter (BD ARIA III or BD ARIA Fusion)
Software
FlowJo
BD FACS DIVA
Procedure
Preparation of cell suspension
Separate the required number of worms that have been starved for at least 7 days and wash with fresh 1× Montjuïc salts.
Note: This protocol is optimized with the planarian species, Schmidtea mediterranea, and both sexual and asexual biotypes can be used. The number of worms required depends on the number of cells required for the downstream applications. In our experience, 35 worms of ~0.7 cm size will yield ~100,000 to 150,000 X1 cells.
Transfer the worms to the cap of a 60 mm or 35 mm culture dish. Remove the 1× Montjuïc salts completely.
Immediately add ~100 μL of CMFB to the planarians (Figure 1A).
Note: The volume of CMFB is approximate, as it should be enough to cover all the planarians. The volume could be scaled up or down based upon the specific requirement.
Using a scalpel, slice the worms into pieces as small as possible (Figure 1B). Intermittently wipe the scalpel with tissue paper, to remove the mucous. Ensure that no large fragments remain.
Once the tissues are diced into small fragments, add 1 mL of CMFB.
Make a wide bore 1 mL pipette tip by cutting its end using a sterile scalpel (Figure 1C). Using this wide-bore pipette, transfer the tissue fragments to a fresh 50 mL centrifuge tube.
Note: The tissue fragments may stick to the surface. If so, flush the tissue with excess CMFB and transfer it to the centrifuge tube. In our lab, we take the tissue fragments in ~5 mL of CMFB.
Mechanically dissociate the cells using a 1 mL pipette by gently pipetting multiple times. After ~20-30 times of pipetting, let the centrifuge tube sit for ~2-3 min. The undissociated tissue fragments will settle down. Transfer the supernatant containing cells to a fresh 50 mL tube without disturbing the settled tissue fragments. Leave some supernatant, so that the undissociated fragments don’t come along with the supernatant (Figure 1D).
Add another 3-4 mL of CMFB solution to the undissociated fragments and dissociate again by gently pipetting.
Repeat Steps A7 and A8 till all the tissue fragments are completely dissociated.
Note: Steps A7 and A8 are to reduce cell death as much as possible, since pipetting of already dissociated cells is unnecessary and may induce cell death.
Collect all the cell suspensions and strain through a 40 μm strainer into a fresh 50 mL centrifuge tube.
Centrifuge the strained cell suspension at 290 × g at 4°C for 10 min in a swinging bucket rotor. A dark cell pellet is observed at this stage (Figure 1E).
Discard the supernatant and resuspend the cell pellet in IPM + 10% FBS media. The volume of media depends on the number and size of the worms used. For ~40 worms of ~0.7 cm, 10 mL of media could be used.

Figure 1. Preparation of cell suspensions. A. Planarians are taken in the cap of a 35 mm culture dish and ~100 μL of CMFB is added. B. Representative image of diced planarian fragments. C. Representative image of the 1 mL pipette with the tips cut (right) compared to a normal pipette (left). D. After mechanical disruption by pipetting the cell suspension ~20-30 times, the solution is allowed to settle down (left centrifuge tube) and the supernatant is transferred to a fresh centrifuge tube (right) and fresh CMFB is added to the undissociated fragments. See Steps A7-A9 for details. E. Cell pellet after cell straining and centrifugation.
Staining of the cell suspensions
Add 40 μg/mL Hoechst 33342 to the cell suspension and incubate at room temperature in the dark for 40 min. Mix the solution intermittently at an interval of ~10 min.
After 40 min, add 100 nM MTG to the same cell suspension and incubate for another 20 min at room temperature in the dark.
Post incubation with MTG, centrifuge the cell suspension at 290 × g for 10 min at 4°C in a swinging bucket rotor. Discard the supernatant and resuspend the cell pellet in IPM + 10% FBS media.
The final volume could be adjusted based on the efficiency of the sorting machine. In our laboratory, for ~40 worms of ~0.7 cm, we use 3 mL of IPM + 10% FBS media.
Finally, add 1 µg/mL of propidium iodide to the cell suspension to discriminate live/dead cells.
For FACS sorting of cells for transplantation or in vitro culture experiments, SiR-DNA (1 μM) should be used instead of Hoechst 33342 at Step B1. At Step B4, for live/dead cell discrimination, use 10 μg/mL of DAPI instead of propidium iodide. The rest of the staining protocol remains the same.
FACS gating for X1 and X2 MTG populations
For sorting, use a 100 μm tip at 20 psi sheath pressure. Use the proprietary BD Sheath fluid.
Note: Before every sorting session, we routinely calibrate the instrument by setting up the appropriate laser delay with the SpheroTM Rainbow fluorescent particles and the drop delay with the BD FACS Accudrop beads. This step should be done according to the instrument used and the information is generally available with the instrument manual.
Run the Hoechst 33342 and MTG stained cells in the FACS machine. Adjust the forward scatter (FSC) and side scatter (SSC) voltage and make a gate (P1) to exclude debris and dead cells.
Next, gate the singlets using an FSC A (area) vs FSC W (width) dot plot (P2).
Within this singlet population, select the live cells indicated as propidium iodide negative events (P3).
Set the gates for X1, X2, and Xins population as represented in Figure 2 and Mohamed Haroon et al. (2021).
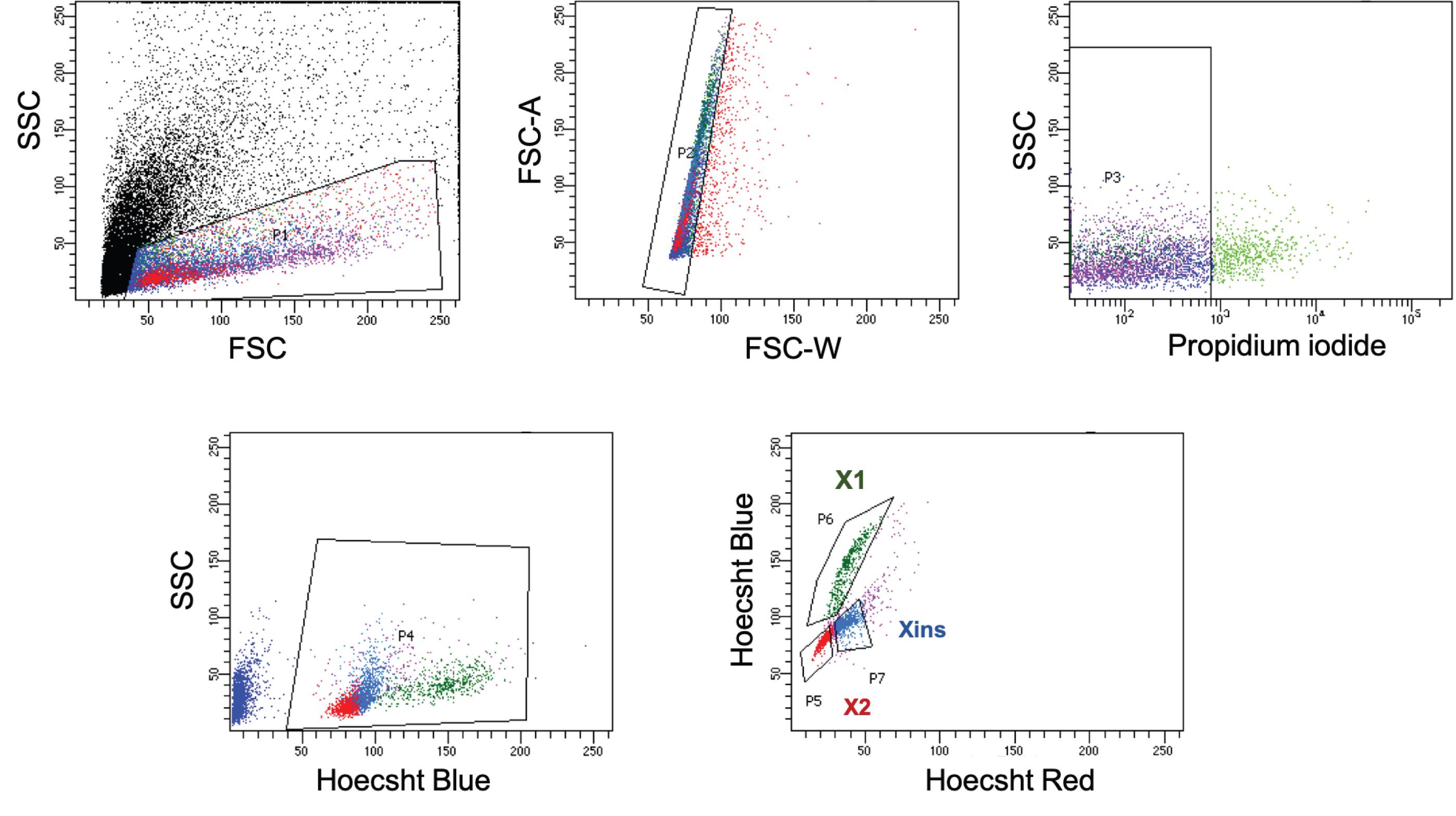
Figure 2. Representative FACS gating for X1, X2, and Xins populations. See Steps C2-C5 for details. FSC- forward scatter, SSC- side scatter, FSC-W width, FSC-A area. The blue (Hoechst Blue) emission of Hoechst 33342 can be collected using a 450/20 bandpass filter and for the red emission (Hoechst red), use 630/40 bandpass filter.To obtain a population with enriched pluripotent cells, gate MTG Low cells within X1. Similarly to enrich the lineage primed population gate corresponding to MTG High within the X1 cells (Figure 3A).
The X2 gate could also be separated into MTG Low and MTG High cells. To enrich the 2N stem cells within X2, make four gates based on MTG and FSC parameters (Figure 3B).
Note: C6-C7: With the current understanding, it is impossible to mark the exact fluorescent intensity or percentage of MTG Low cells that exactly represent stem cells while excluding the differentiating progenies in both X1 and X2 populations. Hence, for consistency, the population can be divided into equal percentages of MTG Low and MTG High.
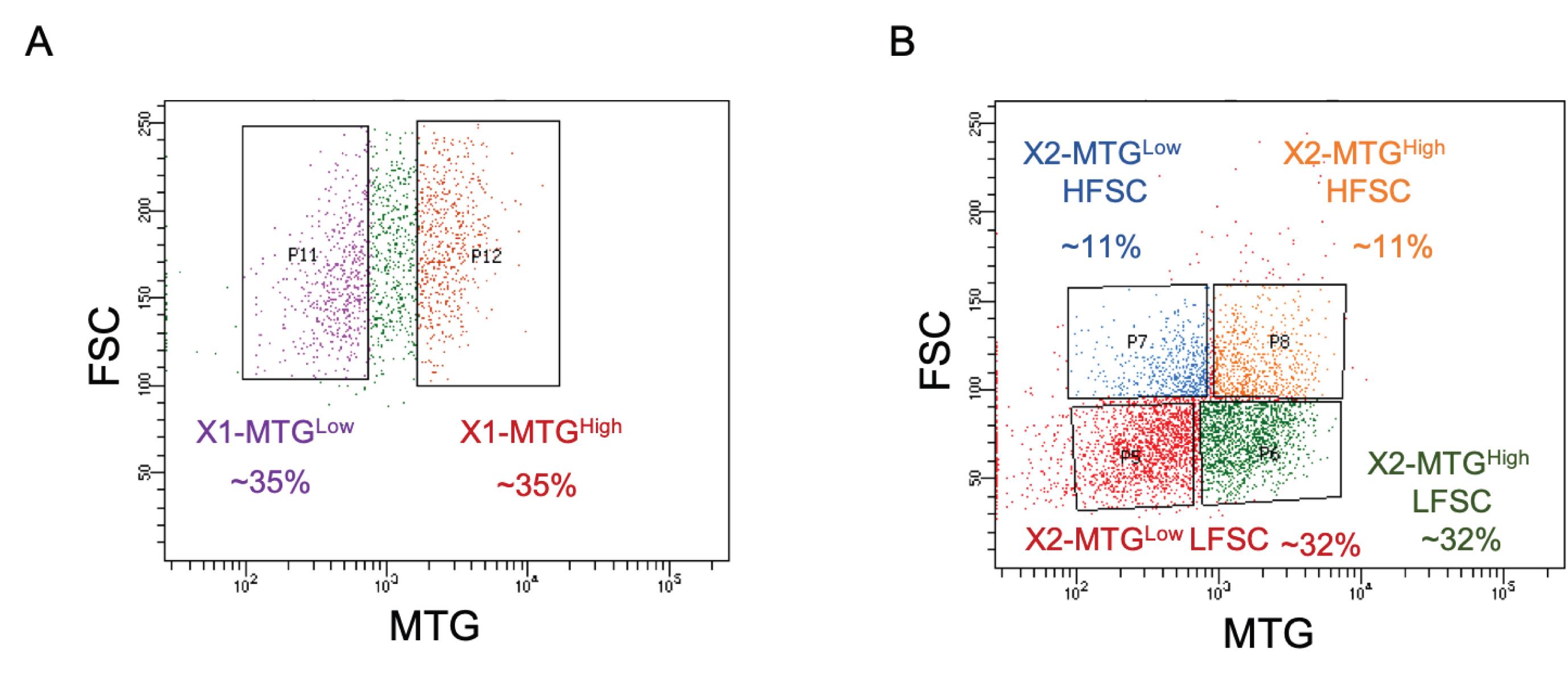
Figure 3. Gating strategy for MitoTracker Green (MTG) populations within (A) X1 and (B) X2 cells. For consistency, an equal percentage of MTG Low and High populations should be gated. HFSC-High FSC, LFSC- Low FSC.
FACS gating for SiR-DNA 2N and 4N MTG populations
Run SiR-DNA stained cells and set gates P1 and P2 as described above, to eliminate debris and doublets. Next, gate DAPI negative cells (P3) to obtain the live cells.
Within the live cells, gate SiR-DNA 2N (P4) and 4N (P5) populations (Figure 4).
Note: Unlike Hoechst staining, SiR-DNA staining does not separate 2N and 4N cells well in a FACS plot. Hence, to ensure accurate gating, use the reference plots in Figure 4 and other Sir-DNA plots (Lei et al., 2019). Alternatively, one could use 6000 rads irradiated animals (>2 days post irradiated animals) as a control that is devoid of proliferating stem cells (Wagner et al., 2011).
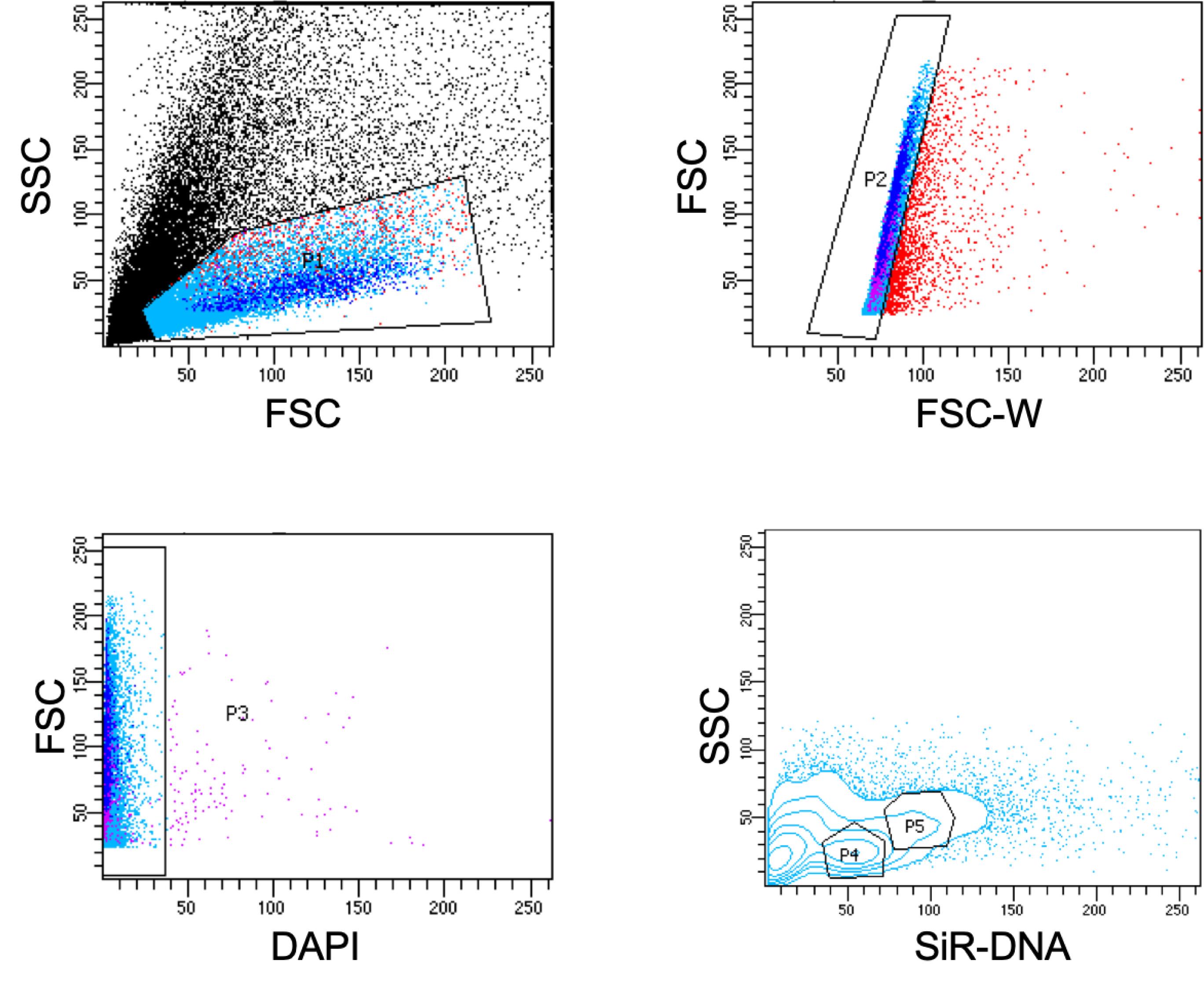
Figure 4. Representative FACS gating for 2N (P4-gate) and 4N (P5-gate) cells using SiR-DNA nuclear dye. See Steps D1-D2 for details.To obtain the population that is X1 equivalent, first determine X1(fs) gate by back-gating X1 cells in an FSC vs SSC plot (Figure 5) (Wagner et al., 2011). Within the X1(fs) population, gate the SiR-DNA 4N population as determined in the earlier Step D2 (Figure 5).
Similarly, determine X2(fs) gate by back-gating X2 cells in FSC vs SSC plot, and gate the SiR-DNA 2N population within the X2(fs) gate (Figure 5).
Set gates for MTG Low and High, as well as FSC low and high within 4N and 2N gates, similarly to the Hoechst stained cells as explained in Steps C7-C8.
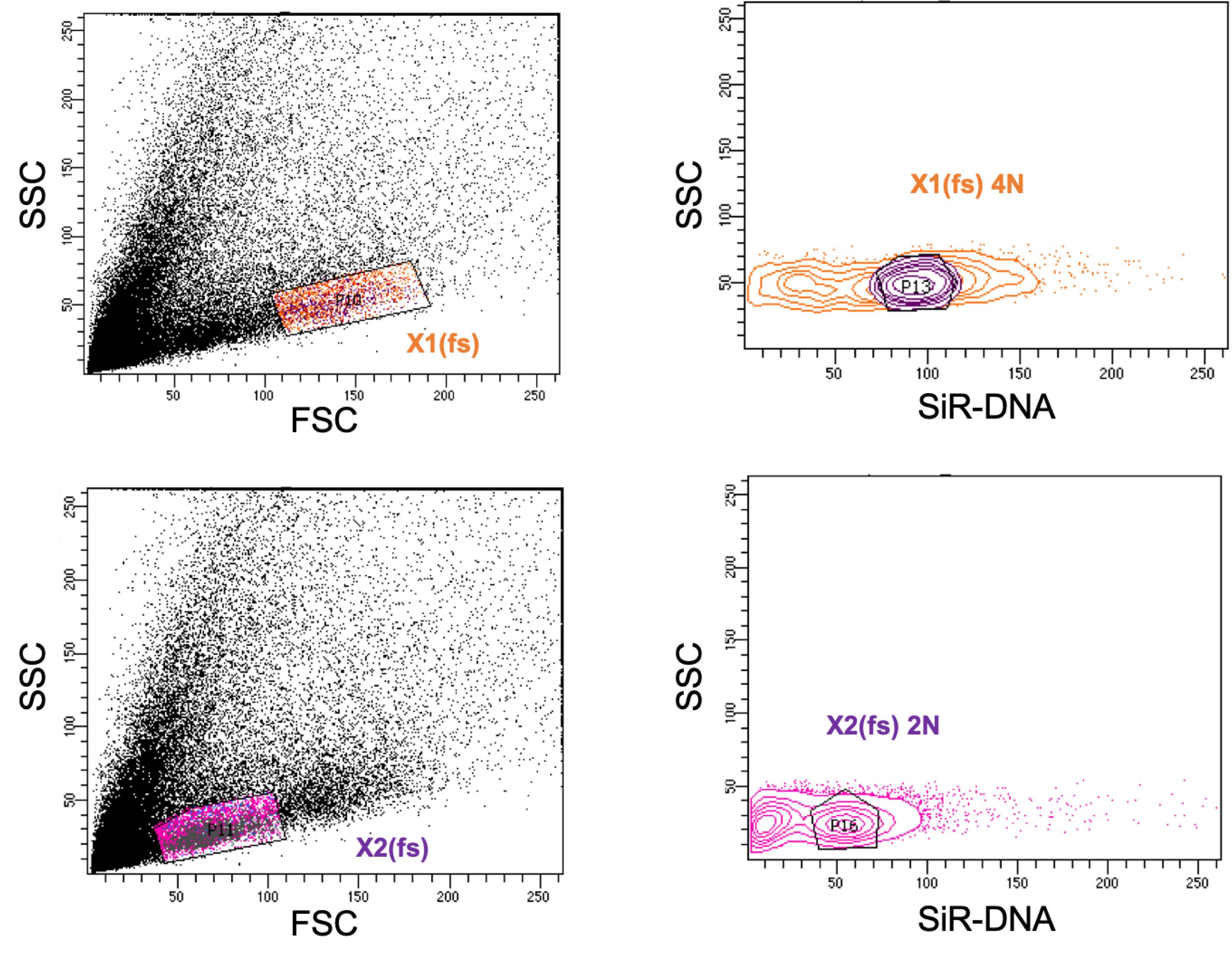
Figure 5. Representative FACS graph for sorting 2N and 4N SiR-DNA population within X2(fs) and X1(fs) gates. This gating is performed to reduce cross-contamination of X1 cells and X2 cells, as the SiR-DNA dye could not resolve the cell cycle stages well, unlike Hoechst.
FACS sorting
Sort the cells in 5 mL FACS tubes containing 2 mL of IPM + 10% FBS media. Keep the collection chamber at 4°C using the appropriate adaptor.
Note: We routinely perform the sorting using the ‘purity’ mode described in BD FACS Aria III and BD FACS Aria Fusion sorters. Such sort precision modes are instrument specific, and the users should refer to the instrument manual for the appropriate settings. The users can run the sorted samples in the FACS machine to ensure the purity of the sort. In addition, fluorescent in situ hybridization or immunostaining for neoblast markers, such as piwi-1, can be performed to check the purity of the sorted samples.
Centrifuge the sorted cells at 300 × g for 10 min at 4°C using a swinging bucket rotor. Discard the supernatant and resuspend the cells in an appropriate volume based on the downstream applications.
Note: The cell pellet after sorting ~100,000 to 150,000 cells will be very small and may not be readily visible.
Data analysis
The flow-cytometry graphs are analyzed using the BD FACS DIVA software. Alternate software such as FlowJo could also be used. The isolated cell populations were characterized by PIWI-1 marker expression and transplantation experiments over a minimum of three independent replicates (Mohamed Haroon et al., 2021). Statistical analysis were performed using an unpaired, two-tailed Student’s t-test.
Recipes
1× Montjuïc salts
NaCl 1.6 mM
CaC3 1 mM
MgSO4 1 mM
MgCl2 0.1 mM
KCl 0.1 mM
NaHCO3 1.2 mM
pH 7.0
Calcium, magnesium free buffer with BSA (CMFB) (For 1,000 mL)
NaH2PO4 400 mg
NaCl 800 mg
KCl 1,200 mg
NaHCO3 800 mg
Note: 10× concentration of the above salts could be prepared (filtered and autoclaved) separately and stored as stock.
Glucose 240 mg
HEPES 15 mM
pH 7.3
BSA 1%
Note: 1% BSA should be added fresh to the buffer.
IPM + 10 % FBS (For 1,000 mL)
HEPES (free acid) 7,208.8 mg
HEPES (sodium salt) 3,514.1 mg
NaCl 985.4 mg
NaHCO3 800.1 mg
KCl 26.4 mg
CaCl2 113.23 mg
MgSO4 44.02 mg
MnCl2 0.19 mg
KH2PO4 68.5 mg
D-biotin 0.3 mg
D-glucose 300 mg
D-trehalose 50 mg
Tricine 2.5 mg
MEM Essential amino acids 2 mL
MEM non-essential amino acids 5 mL
MEM vitamin solution 3 mL
Sodium pyruvate 14 mL
Penicillin/streptomycin 10 mL
L-Glutamine 2 mL
Fetal Bovine Serum 100 mL
pH 7.3
Acknowledgments
The protocol is majorly adapted from the recently published work from our laboratory (Mohamed Haroon et al., 2021). The authors thank Central Imaging and Flow Cytometry Facility (CIFF-BLiSc campus) for the state-of-the-art flow cytometry facility. MMH thanks DBT for funding the graduate study through the DBT-JRF program. DP acknowledges Swarnajayanti fellowship (DST/SJF/LSA-02/2015-2016) from DST. DP and PKV thank the Department of Biotechnology, inStem for core funding.
Competing interests
The authors declare no competing or financial interests.
References
- Lei, K., Mckinney, S. A., Ross, E. J., Lee, H. C. and Alvarado, A. S., (2019). Cultured pluripotent planarian stem cells retain potency and express proteins from exogenously introduced mRNAs. BioRxiv 573725.
- Hayashi, T., Asami, M., Higuchi, S., Shibata, N. and Agata, K. (2006). Isolation of planarian X-ray-sensitive stem cells by fluorescence-activated cell sorting. Dev Growth Differ 48(6): 371-380.
- Higuchi, S., Hayashi, T., Hori, I., Shibata, N., Sakamoto, H. and Agata, K. (2007). Characterization and categorization of fluorescence activated cell sorted planarian stem cells by ultrastructural analysis. Dev Growth Differ 49(7): 571-581.
- Mohamed Haroon, M., Lakshmanan, V., Sarkar, S. R., Lei, K., Vemula, P. K. and Palakodeti, D. (2021). Mitochondrial state determines functionally divergent stem cell population in planaria. Stem Cell Reports 16(5): 1302-1316.
- Niu, K., Xu, H., Xiong, Y. Z., Zhao, Y., Gao, C., Seidel, C. W., Pan, X., Ying, Y. and Lei, K. (2021). Canonical and early lineage-specific stem cell types identified in planarian SirNeoblasts. Cell Regen 10(1): 15.
- Wagner, D. E., Wang, I. E. and Reddien, P. W. (2011). Clonogenic neoblasts are pluripotent adult stem cells that underlie planarian regeneration. Science 332(6031): 811-816.
- Wang, I. E., Wagner, D. E. and Reddien, P. W. (2018). Chapter 20: Clonal analysis of planarian stem cells by subtotal irradiation and single-cell transplantation. Methods Mol Biol 1774: 475-495.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Mohamed Haroon, M., Vemula, P. K. and Palakodeti, D. (2022). Flow Cytometry Analysis of Planarian Stem Cells Using DNA and Mitochondrial Dyes . Bio-protocol 12(2): e4299. DOI: 10.21769/BioProtoc.4299.
Category
Stem Cell > Pluripotent stem cell > Regenerative medicine
Cell Biology > Cell-based analysis > Flow cytometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.