- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

From 3D to 2D: Harmonization of Protocols for Two-dimensional Cultures on Cell Culture Inserts of Intestinal Organoids from Various Species
(*contributed equally to this work) Published: Vol 12, Iss 2, Jan 20, 2022 DOI: 10.21769/BioProtoc.4295 Views: 6217
Reviewed by: Giusy TornilloJens PuschhofTakashi Nishina
Original research article
The authors used this protocol in:

Feb 2021
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
In the expanding field of intestinal organoid research, various protocols for three- and two-dimensional organoid-derived cell cultures exist. Two-dimensional organoid-derived monolayers are used to overcome some limitations of three-dimensional organoid cultures. They are increasingly used also in infection research, to study physiological processes and tissue barrier functions, where easy experimental access of pathogens to the luminal and/or basolateral cell surface is required. This has resulted in an increasing number of publications reporting different protocols and media compositions for organoid manipulation, precluding direct comparisons of research outcomes in some cases. With this in mind, here we describe a protocol aimed at the harmonization of seeding conditions for three-dimensional intestinal organoids of four commonly used research species onto cell culture inserts, to create organoid-derived monolayers that form electrophysiologically tight epithelial barriers. We give an in-depth description of media compositions and culture conditions for creating these monolayers, enabling also the less experienced researchers to obtain reproducible results within a short period of time, and which should simplify the comparison of future studies between labs, but also encourage others to consider these systems as alternative cell culture models in their research.
Graphic abstract:
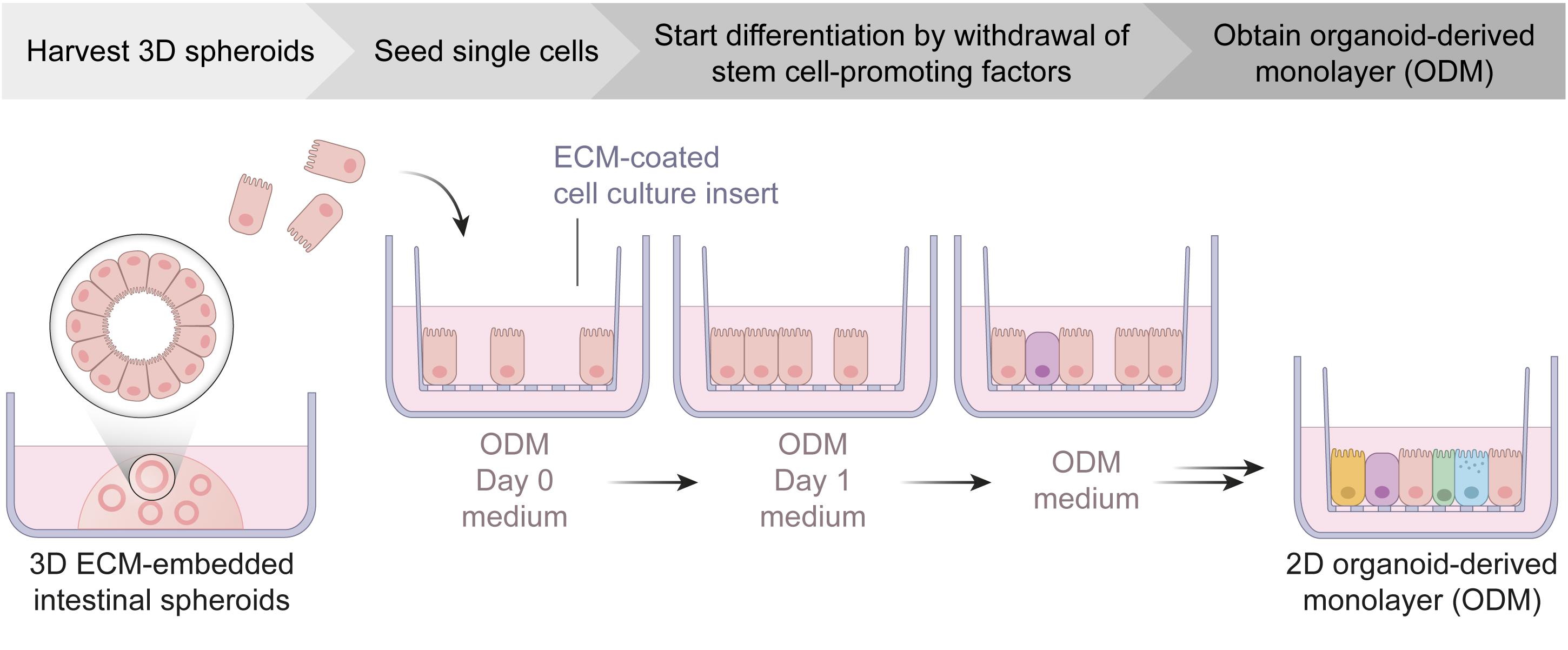
Schematic workflow of organoid-derived monolayer generation from intestinal spheroid cultures. ECM, extracellular matrix; ODM, organoid-derived monolayer.
Background
The small intestinal epithelium is the primary route of infection for many pathogens. In the past, the understanding of host-pathogen interactions has mostly relied on immortalized cell lines or mouse models, which not only lack the complexity of the intestinal epithelium, but also have shortcomings with regard to translatability to the in vivo situation in humans (Klotz et al., 2012; Delgado-Betancourt et al., 2019; Bar-Ephraim et al., 2020; Holthaus et al., 2020). Starting with the development of organoid models by Sato et al. (2009), these concerns have been addressed by many groups, and resulted in the implementation of organoid models for the study of pathogenic bacteria, viruses, and parasites (Bartfeld, 2016; Dutta and Clevers, 2017; Hill and Spence, 2017). Importantly, they are no longer relying solely on the mouse or other animal models, but are moving towards relevant hosts for respective diseases, an important aspect especially in the context of zoonotic pathogens.
There are a multitude of protocols for the establishment of organoid models to study host-pathogen interactions and several labs have developed protocols for the generation of two-dimensional organoid-derived monolayers that provide direct experimental access to the apical and/or basolateral side and allow the study of physiological processes and tissue barrier functions (Moon et al., 2014; VanDussen et al., 2015; Kozuka et al., 2017). However, many protocols focus on only one species, which requires adaptations to suit species-specific needs in case of model switching. Moreover, there is no simple and concise guide to follow, due to the many variations introduced mainly by labs studying a particular host. Here, we provide a protocol using a single common base medium that was tested for four species that play an important role in infectious disease research, namely human, mouse, chicken, and pig.
The starting material for the generation of organoid-derived monolayers are intestinal spheroids, which are stem cell-enriched non-budding three-dimensional structures, obtained by culturing intestinal stem cells in an extracellular matrix hydrogel with Wnt-3a-supplemented media (Miyoshi and Stappenbeck, 2013; VanDussen et al., 2015 and 2019). The term organoid, in a strict sense, refers to more heterogeneous budding structures composed of fewer stem cells and more differentiated epithelial cells, but is also often more broadly used to describe this type of three-dimensional cell culture system.
To achieve intestinal monolayers with a physiological cell composition, differentiation is induced by omitting stem cell-promoting factors from the medium. Previous work by Sato et al. (2009) and Kozuka et al. (2017) provides an excellent overview of the influence of medium conditions on differentiation, and was used as the basis for composing the media in this protocol. We tested the proposed media conditions in our system, and found that the medium we used [withdrawal of Wnt-3a, transforming growth factor beta (TGF-β) receptor signaling inhibitor A83-01, and p38 inhibitor SB202190, but presence of nicotinamide] provided an electrophysiologically thight epithelium, allowed enterocyte-guided differentiation, and enabled prolonged culture of human, mouse, chicken, and pig ODMs for at least six days (Holthaus et al., 2020). With our protocol, one is able to seed and maintain confluent organoid-derived monolayers from human, mouse, chicken, and pig that resemble physiological properties and cell composition, providing an accessible platform for further optimizations and adaptations required for different research approaches. Examples for the application of this protocol can be found in Kraft et al. (2020) and Holthaus et al. (2020).
Materials and Reagents
Materials for organoid culture
12/24-well tissue culture plates (F-base; TPP, catalog numbers: 920 12/24)
15/50 mL conical centrifuge tubes (TPP, catalog number: 910 15/50)
1.5 mL microcentrifuge tubes (BrandTech, catalog number: 780500)
10/200/1,000 μL ART barrier pipette tips (Thermo Scientific, catalog numbers: 10098960/10029040/10313272)
20/100 μL PP filter tips (Nerbe Plus, catalog numbers: 07-662-8300/07-642-8300)
PCF Transwell inserts (0.6 cm2, 0.4 μm pores; Merck, Millicell, catalog number: PIHP01250)
Vacuum filtration system "rapid"-Filtermax (0.22 μm; TPP, catalog number: 99505)
Sartorius MinisartR sterile filters (0.22 μm; Thermo Scientific, catalog number: 10730792)
Disposable hemocytometer (Kisker Biotech, catalog number: M-NZ)
Duran glass beakers (25/600-mL; Schott, catalog numbers: 2110614, 2110648)
Duran glass bottles (500/1,000-mL; Schott, catalog numbers: 4459/5455)
Bemis Parafilm "M" (Thermo Scientific, catalog number: 11762644)
B Braun solo cone Luer syringes (5/10/20-mL; Braun, catalog number: 12752637)
Sterican single-use 21 G syringe needles (Blunt, 25 mm, 0.4 mm; Carl Roth, Braun, catalog number: X134.1)
Single-use 18 G syringe needles (Blunt, 40 mm; VWR, BD Medical, catalog number: BDAM303129)
Note: Examples for providers/manufacturers are given; similar material from others might work as well.
Reagents for organoid culture
Advanced DMEM/F-12 (Thermo Scientific, Gibco, catalog number: 12634028; store at 4°C)
Recombinant Human EGF (Peprotech, catalog number: AF-100-15; store lyophilized at -20°C and aliquots at -80°C)
Recombinant Murine EGF (Peprotech, catalog number: 315-09; store lyophilized at -20°C and aliquots at -80°C)
N-Acetyl-L-cysteine (Sigma-Aldrich, catalog number: A9165; store powder at 4°C and aliquots at -80°C)
Nicotinamide (Sigma-Aldrich, catalog number: N0636; store powder at room temperature and aliquots at -80°C)
A83-01 (Sigma-Aldrich, catalog number: SML0788; store lyophilized at -20°C and aliquots at -80°C)
SB 202190 (Cayman chemicals, catalog number: 10010399-10; store lyophilized at -20°C and aliquots at -80°C)
B-27 Supplement (Thermo Scientific, Gibco, catalog number: 17504044; store at -80°C)
N-2 Supplement (Thermo Scientific, Gibco, catalog number: 17502048; store at -80°C)
HEPES (1 M; Thermo Scientific, Gibco, catalog number: 15630056; store at 4°C)
GlutaMAX Supplement (100×; Thermo Scientific, Gibco, catalog number: 35050038; store at 4°C)
Penicillin/Streptomycin (100×; penicillin [10,000 U/mL] and streptomycin [10,000 µg/mL]; Capricorn scientific, catalog number: PS-B; store at -20°C)
Y-27632 (hydrochloride; Cayman chemicals, catalog number: 10005583; store lyophilized at -20°C and aliquots at -80°C)
TrypLE Express Enzyme (1×; Thermo Scientific, Gibco, catalog number: 12605010; store at 4°C)
ECM Matrigel (Corning, catalog number: 734-0269; store aliquots at -20°C) or Cultrex Reduced Growth Factor Basement Membrane Extract Type 2 (Bio-Techne, R&D Systems, catalog number: 3533-010-02; store aliquots at -80°C)
Note: A protein content of ≥ 8.5 mg/mL is desirable.
Bovine Serum Albumin (BSA, fatty acid free; Sigma-Aldrich, catalog number: A7030-100G; store at 4°C)
Dimethyl sulfoxide (DMSO; Sigma-Aldrich, catalog number: D2650-100ML; store at room temperature)
80% ethanol or other equivalent disinfectant, store at room temperature
MilliQ or other equivalent sterile water, store at room temperature
Note: Found to work as described; similar reagents from other providers/manufacturers might work as well.
Reagents for production of conditioned medium
DMEM High Glucose (4.5 g/L), with Stable Glutamine (Capricorn Scientific, catalog number: DMEM-HPSTA; store at 4°C)
DMEM, high glucose with GlutaMAX Supplement and pyruvate (Thermo Scientific, Gibco, catalog number: 31966047; store at 4°C)
Fetal Bovine Serum (FBS; Capricorn scientific, catalog number: FBS-LE-12A; store aliquots at -20°C)
Note: Test new FBS batches for a minimum of three weeks before use in routine culture. Heat inactivate for 30 min at 56°C and store 50 mL aliquots at -20°C.
Trypsin-EDTA (Capricorn scientific, catalog number: TRY-1B10; store aliquots at -20°C)
Phosphate-buffered saline (PBS; made in-house; store at room temperature or at 4°C)
Penicillin/Streptomycin (100×, penicillin [10,000 U/mL] and streptomycin [10,000 µg/mL]; Capricorn scientific, catalog number: PS-B; store aliquots at -20°C)
Hygromycin B (50 mg/mL; Carl Roth, catalog number: CP12.2; store at 4°C)
Geneticin (G418; 50 mg/mL; Carl Roth, catalog number: CP11.2; store at 4°C)
Zeocin (Invivogen, catalog number: ant-zn-1; store aliquots at -20°C)
Luciferase Assay System (Promega, catalog number: E1500; store aliquots at -20°C)
Note: Found to work as described; similar reagents from other providers/manufacturers might work as well.
Cell lines for conditioned media and its derived media
L-WRN (ATCC CRL-3276) (VanDussen et al., 2019)
HEK 293T HA-noggin-Fc (Heijmans et al., 2013; Bartfeld et al., 2015)
Kindly provided by Hans Clevers, Hubrecht Institute, Utrecht.
HEK 293T R-Spondin1-Fc (Kim et al., 2005)
Kindly provided by Calvin Kuo, Stanford University, USA (subject to MTA).
HEK 293 SuperTopFlash (ATCC CRL-3249)
Luciferase reporter cell line for activity testing of L-WRN and R-Spondin 1 conditioned media.
WERN 3D base proliferation medium for spheroid culture (see Recipes)
Organoid freezing medium (see Recipes)
Dissociation reagent (see Recipes)
ODM Day 0 medium (see Recipes)
ODM Day 1 medium (see Recipes)
ODM medium, day 2 onwards (see Recipes)
Phosphate buffered saline (see Recipes)
L-WRN growth medium (see Recipes)
L-WRN conditioning medium (see Recipes)
R-Spondin 1 and mNoggin growth medium (see Recipes)
R-Spondin 1 and mNoggin conditioning medium (see Recipes)
L-WRN, R-Spondin 1, and mNoggin freezing medium (see Recipes)
Equipment
-20°C freezer
Vacuum Aspirator system, Integra Biosciences Vacusafe Comfort Aspiration system (Fisher Scientific, Integra, catalog number: 10590273)
Adjustable pipettes, Eppendorf Research plus 10/20/200/1,000 pipettes (Eppendorf, catalog numbers: 3123000020, 3123000039, 3123000055, 3123000063)
CO2 cell culture incubator (Humidified atmosphere, 5% CO2, 37°C), CB 170 CO2 incubator (Binder, model number: CB170-230V-O)
Safety cabinet for BSL-2 work, Safe 2020 Class II Biological Safety Cabinet (Thermo Scientific, catalog number: 51026640)
Cooled centrifuge for 15/50 mL conical tubes, Multifuge X1R Multi-Application Centrifuge (Thermo Scientific, catalog number: 15364306)
Cooling containers, Eppendorf IsoRack with IsoPack 0°C (Eppendorf, catalog number: 3880001174)
CoolCell LX Cell Freezing Vial Containers (Fisher Scientific, Corning, catalog number: 15572771)
37°C water bath, WNB 14 (Carl Roth, Memmert, model: WNB 14)
Luminometer, TriStar LB 941 multimode microplate reader (Berthold, model: LB 941)
Chopstick electrode for measurement of transepithelial electrical resistance, Volt-Ohm meter ERS-2 with electrode STX01 (Merck, Millicell, catalog number: MERS00002)
37°C heating block for measurement of transepithelial electrical resistance, digital dry bath incubator (MRC Laboratory Instruments, model: DBD-002N)
Note: Examples for providers/manufacturers are given; similar material from others will probably work as well.
Procedure
For the establishment of 3D organoids from intestinal tissue, standard protocols of Sato et al. (2009) were followed. Passage of all 3D organoids was done once a week (human) or twice a week (all other species), following protocols by Mahe et al. (2015). Medium conditions for 3D organoid cultures are summarized in Holthaus et al. (2020) for each species, and also briefly in the recipes.
Handling of 3D organoids
Thawing 3D organoids
Thaw extracellular matrix (ECM; Matrigel or Cultrex) compounds in a 4°C cooling rack in the refrigerator.
Pre-warm a 24-well plate in a 37°C CO2 incubator.
Pre-warm “WERN 3D base proliferation medium for spheroid culture” in a 37°C water bath.
Get one vial of 3D organoids from liquid nitrogen storage (wear goggles and gloves for protection) and transport it to the bench in a cooling rack.
Thaw the vial in a 37°C water bath under gentle agitation.
Once the content is thawed, remove the vial from the water bath. Disinfect the vial by wiping it with 70% ethanol and move it to the biosafety cabinet.
Note: Always use a BSL-2 biosafety cabinet when handling organoids of human or wildlife origin, unless tested to be free of infectious pathogens.
Transfer the vial content into a 15 mL tube with 9 mL of Advanced DMEM/F-12.
Centrifuge at 500 × g for 5 min at 4°C.
Aspirate the supernatant and resuspend the pellet in 100 µL of Advanced DMEM/F-12. Transfer the cell suspension into a pre-chilled 1.5 mL microcentrifuge tube in a 4°C cooling rack.
Add 100 µL of ECM and mix by pipetting up and down.
Transfer 40-50 µL of ECM/cell mixture per well into a 24-well plate.
Incubate the culture at 37°C for 30 min to solidify the ECM.
Add 500 µL of pre-warmed “WERN 3D base proliferation medium for spheroid culture” to the wells, return the plate to the incubator, and change the medium every other day.
Freezing 3D organoids for cryopreservation
Have a bucket with ice ready.
Fill one or more glass beakers with ice and place them inside the biosafety cabinet.
Place the appropriate number of 15 mL conical tubes on ice.
Note: One 15 mL conical tube is required per 12 wells of the 3D organoid culture.
Coat the plastic surfaces of the 15 mL conical tubes with 0.1% BSA solution.
Note: Prepare the 0.1% BSA solution well in advance in a 50 mL tube and store it at 4°C. The solution may be reused up to 5 times or for two weeks.
Fill each 15 mL conical tube with 6 mL of cold Advanced DMEM/F-12 supplemented with P/S.
Transfer the 24-well plates of ECM-embedded 3D organoid culture from the incubator to the biosafety cabinet.
Collect the ECM-embedded 3D organoids. Scrape the ECM drop off the bottom, with a 1,000 µL pipette tip. Set the pipette to ~600 µL (there should be 500 µL of medium + 40 µL of ECM per well) and transfer the ECM/medium suspension to the 15 mL conical tubes containing 6 mL of Advanced DMEM/F-12 supplemented with P/S. Place the conical tube on ice while harvesting the other wells. Distribute the contents of one 24-well plate between two 15 mL conical tubes on ice.
Note: Scraping for 2-3 s per well should be sufficient to dislodge the ECM droplet.
Centrifuge the collected ECM-embedded 3D organoids at 500 × g for 5 min at 4°C.
Note: The organoids will form a pellet at the bottom of the centrifuge tube, and the ECM will settle on top. If there are still ECM remnants visible in the medium layer, repeat this step.
After centrifugation, carefully aspirate the supernatant and the ECM-containing layer with a vacuum pump.
Add the appropriate amount of organoid-freezing medium to your 15 mL tubes and resuspend.
Note: 4 wells of 3D organoids are normally enough per vial for cryopreservation.
Transfer 1 mL to each cryovial and place it inside a freezing container.
Store the freezing container at -80°C for 24 h and then transfer the vials to liquid nitrogen for final storage.
Creating organoid-derived monolayers
Coat cell culture inserts with ECM
Sterilize the biosafety cabinet.
Note: Always use a BSL-2 biosafety cabinet when handling organoids of human or wildlife origin, unless tested to be free of infectious pathogens.
Thaw an ECM aliquot in a pre-cooled rack in a 4°C fridge (100 µL of ECM is required to coat 12 cell culture inserts).
Sterilize a pair of tweezers with disinfectant. Transfer it to the biosafety cabinet, and wait for the disinfectant to evaporate.
Place the cell culture insert into a 12-well plate using the tweezers.
Note: Be careful not to puncture the permeable support with the tweezers.
Place the 12-well plate with the cell culture inserts in a -20°C freezer for at least 20 min.
In the meantime, transfer a 0°C cold rack (e.g., Eppendorf IsoRack with IsoPack 0°C) from a -20°C freezer to the biosafety cabinet and place the tube containing the thawed ECM aliquot from Step B.1.b in the 0°C cold rack.
Note: ECM starts to solidify above 10°C, but might freeze if the rack is too cold. It may be necessary to wait for a few minutes after taking the 0°C cold rack from the freezer.
Prepare the coating solution. Dilute ECM 1:20 with Advanced DMEM/F-12 supplemented with P/S. Pipette up and down to mix. For a 12-well plate, add 100 µL of ECM to 1,900 µL of pre-cooled Advanced DMEM/F-12 supplemented with P/S.
Pipette 150 µL of the diluted ECM solution into each cell culture insert of the chilled plate.
Note: Carefully pipette directly into the center of the permeable membrane and avoid pipetting against the side walls.
Place the coated plates in the 4°C fridge and leave overnight.
Harvest spheroids and seed cells
Note: To generate ODMs from ECM-embedded organoid cultures, harvest the respective culture when it is at the stem cell-enriched spheroid state (Figure 1). The definition of spheroids and organoids is described in the background section of this protocol. Approximately 2-3 × 106 singularized spheroid cells are required per cell culture insert. For reference, to generate one 12-well plate of ODMs, we use one 24-well plate of ECM-embedded human spheroids. The following steps are also shown in Video 1.
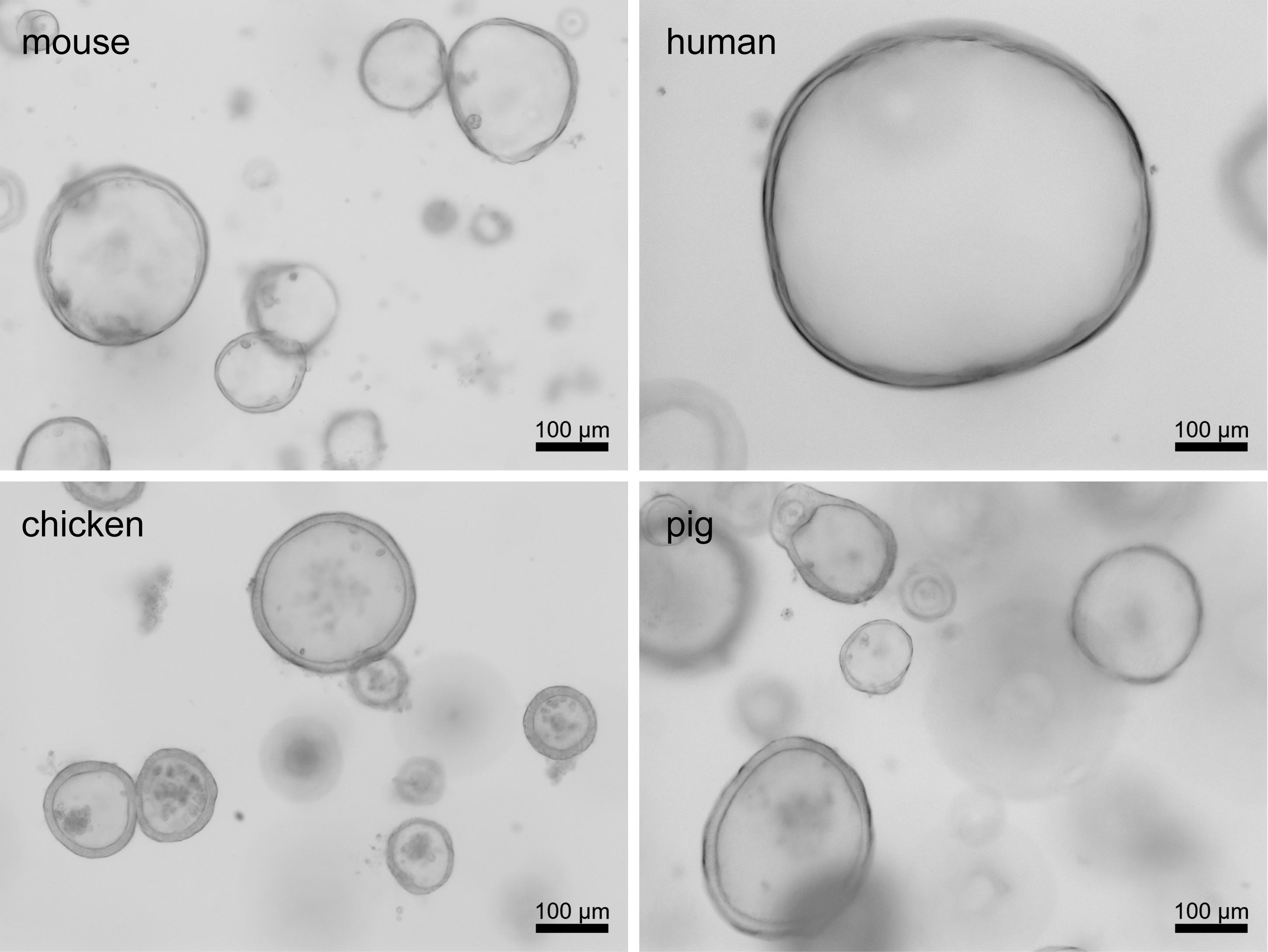
Figure 1. Spheroids before harvesting to make organoid-derived monolayers. Representative brightfield microscopic images of spheroids from mouse, human, chicken, and pig origin are shown. Images were taken on an Axiovert Observer system (Zeiss). Images were adjusted for contrast using Zen Blue 2.6 lite software (Zeiss). Video 1. Experimental procedure for the generation of organoid-derived monolayers and transepithelial-electrical resistance (TEER) measurement.
Video 1. Experimental procedure for the generation of organoid-derived monolayers and transepithelial-electrical resistance (TEER) measurement.Prepare the “ODM Day 0 medium”. For a 12-well plate, 15 mL of this medium are required. To prepare the medium, add the components to a sterile glass beaker. Sterilize the medium using a 0.22 µm syringe filter and transfer it into a 15 mL conical tube.
Optional: Prepare the “ODM Day 1 medium” in parallel and store it in a 4°C fridge until the next day.
Cool down the centrifuge to 4°C.
Prepare a 37°C water bath.
Pre-warm the ODM Day 0 medium in the 37°C water bath.
Sterilize the biosafety cabinet.
Have a bucket with ice ready.
Take the coated plate (from Step B.1) out of the 4°C fridge and carefully aspirate the medium from the cell culture inserts using a vacuum pump (see Video 1).
Place the plate into a 37°C incubator for ~30 min, while continuing with Step B2i.
Fill two or more glass beakers with ice and place them inside the biosafety cabinet.
Put the appropriate number of 15 mL conical tubes on ice.
Note: One 15 mL conical tube is required per 12 wells of spheroid culture.
Assemble a 1 mL syringe and a blunt 18 G needle.
Note: To minimize the risk of injury, use blunt needles and do not recap the needle. Place the assembled syringe in an open 15 mL conical tube that stands in a rack.
Coat the plastic surfaces of the 15 mL conical tubes and the syringe surfaces with 0.1% BSA solution. To coat the conical tube surfaces, pour cold 0.1% BSA solution into each of the 15 mL tubes and aspirate again. To coat the syringe surfaces also fill the syringe with ice-cold 0.1% BSA solution and release (Video 1).
Note: Prepare the 0.1% BSA solution in advance in a 50 mL tube and store it in a 4 °C fridge. The solution may be reused up to 5 times or for two weeks.
Fill each 15 mL conical tube with 6 mL of cold Advanced DMEM/F-12 supplemented with P/S.
Transfer the 24-well plates of ECM-embedded spheroid culture from the incubator to the biosafety cabinet.
Collect the ECM-embedded spheroids. Scrape the ECM drop off the bottom with a 1,000 µL pipette tip. Set the pipette to ~600 µL (there should be 500 µL of medium + 40 µL of ECM per well) and transfer the ECM/medium suspension to the 15 mL conical tubes containing 6 mL of Advanced DMEM/F-12 supplemented with P/S. Place the conical tube on ice while harvesting the other wells. Distribute the contents of one 24-well plate between two 15 mL conical tubes on ice.
Note: Scraping for 2-3 s per well should be sufficient to dislodge the ECM droplet.
Centrifuge the collected spheroids at 500 × g for 5 min at 4°C.
Note: The spheroids will form a pellet at the bottom of the centrifuge tube, and the ECM will settle on top. If there are still ECM remnants visible in the medium layer, repeat this step!
During centrifugation, prepare 1 mL of dissociation reagent (10 μM Y-27632 in TrypLE Express solution) per 15 mL tube of harvested spheroids, e.g., by adding 1 µL of Y-27632 (10 mM stock) to 999 µL of TrypLE Express solution.
After centrifugation, carefully aspirate the supernatant with a vacuum pump.
Resuspend the pellet in 1 mL of dissociation reagent.
Incubate for 2-5 min in a 37°C water bath for enzymatic digestion.
Note: Incubate human spheroids for 5 min; mouse, chicken, and pig for 2 min. Adjustments might be needed for other species.
Mechanically disrupt the spheroids by forcing the suspension through the blunt 18 G needle, seven to ten times (Video 1).
Note: The mechanical disruption is mainly dependent on the experimenter. Normally, 7-10 times is sufficient. Always check your cell suspension under the microscope the first time.
Add Advanced DMEM/F-12 supplemented with P/S to a total of 12 mL.
Centrifuge at 500 × g for 5 min at 4°C.
Carefully aspirate the supernatant.
Resuspend the pellets in ODM Day 0 medium. First, add 1 mL of ODM Day 0 medium to each of the pellets, resuspend the pellets by pipetting, and pool the cell suspensions before adding the remaining medium.
Note: 4.8 mL of ODM Day 0 medium is needed per 12-well plate for the cell suspension and another 9.6 mL for the basolateral side per 12-well plate. We seed around 2-3 × 106 cells per cell culture insert.
Transfer the coated plates from the incubator to the biosafety cabinet.
Carefully pipette 400 µL of the cell suspension into the cell culture insert onto the coated permeable membrane (apical side).
Carefully pipette 800 µL of ODM Day 0 medium to the outer compartment (basolateral side). Return plates to incubator.
On the next day (day 1 after seeding), change the medium from ODM Day 0 medium to ODM Day 1 medium. Aspirate the old medium with a vacuum pump. Start with the outer compartment before aspirating the medium from inside the cell culture insert. Add pre-warmed ODM Day 1 medium in reverse order: first, pipette 400 µL into the cell culture insert and then 800 µL to the outer compartment.
Note: Be careful to follow the sequence when changing the medium! The cell layer could detach from the membrane due to the buoyancy of the surrounding medium if the apical medium is removed first.
On the next day (day 2 after seeding), change medium from ODM Day 1 medium to ODM medium. Aspirate the old medium and add fresh ODM medium as described in Step B2z.iii.
Exchange ODM medium three times a week, as described in Step B2z.iii.
Note: Successful monolayer formation can be assessed by the expression of marker proteins and genes using immunofluorescence microscopy and RT-qPCR, respectively. Epithelial tightness can be confirmed by transepithelial-electrical resistance (TEER) measurements. Figures 2 and 3 show examples of successful monolayer formation. The data are taken from Holthaus et al. (2020), where the methods and interpretation of the data are described in detail. The TEER measurement is also described in Procedure C of this protocol. Figure 4 shows examples of unsuccessful monolayer formation. In this example, the cells failed to reach 100% confluence and did not form an electrophysiologically tight monolayer.
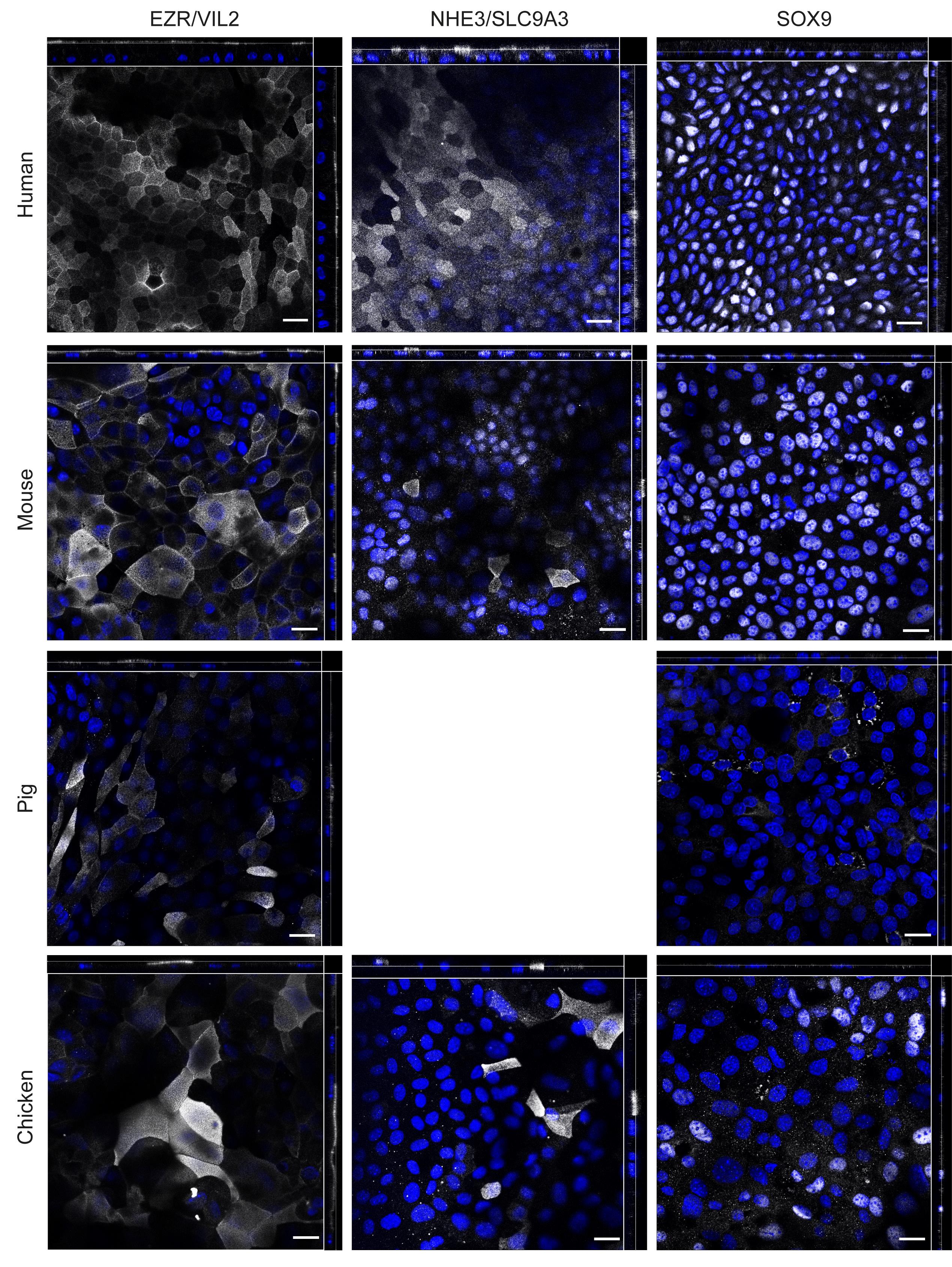
Figure 2. Immunofluorescence analysis of marker protein expression in human, mouse, pig and chicken ODMs. Marker proteins EZR/VIL2 (brush border), NHE3/SLC9A3 (transporter) and SOX9 (proliferative cells) are shown in white, nuclei stained with DAPI are shown in blue. ODMs were fixed after 10 (human), 12 (mouse), 6 (pig), and 15 (chicken) days. NHE3/SLC9A3 staining for pig ODMs was excluded, since the suitability of the antibody was questionable for this species. Representative orthogonal images of z-stacks are shown. Scale bar = 20 µm. Data are from Holthaus et al. (2020), where interpretation of the staining patterns can be found.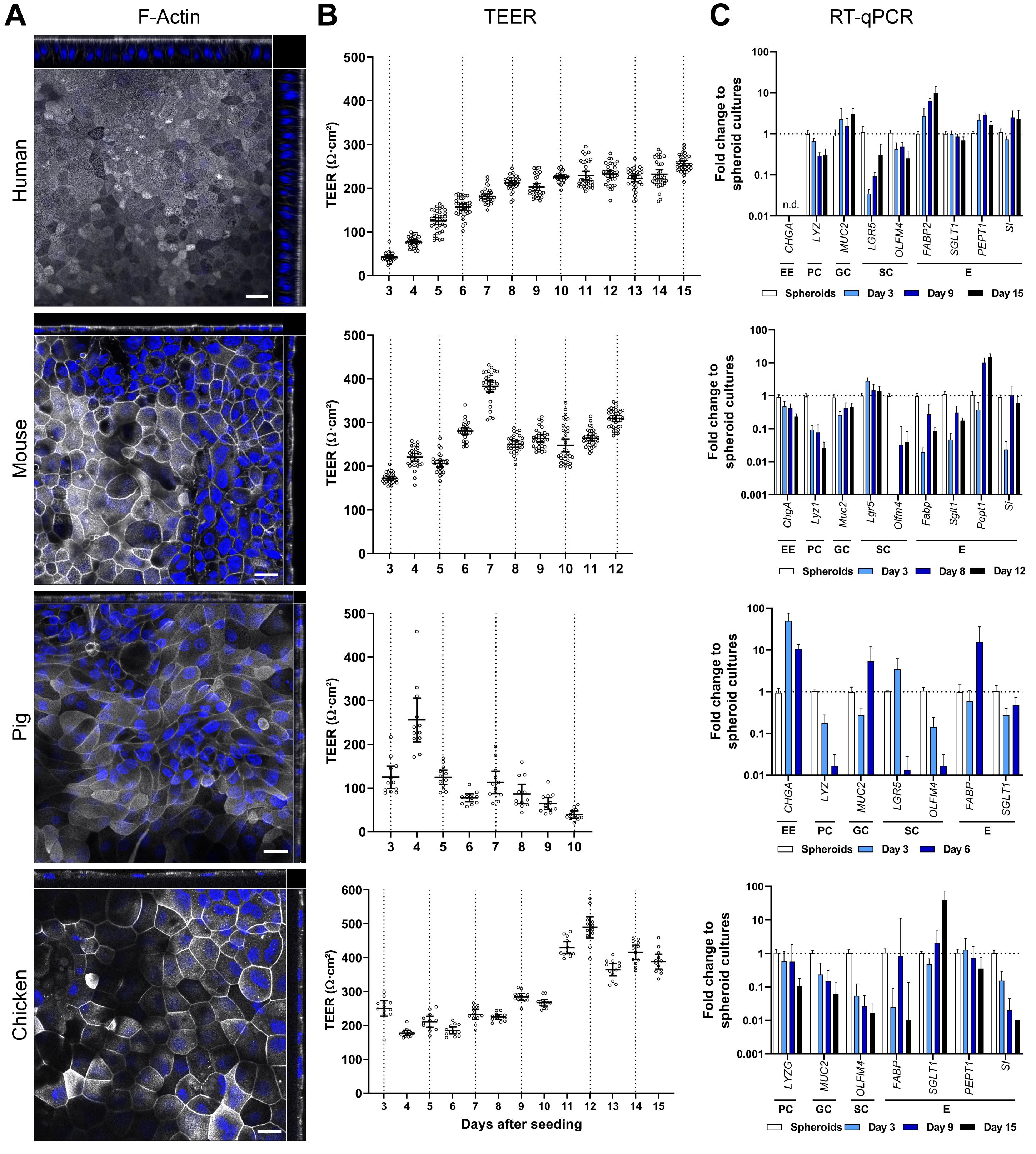
Figure 3. Analysis of brush border orientation, electrophysiological tightness, and transcriptional expression of intestinal marker genes in ODMs. (A) Representative orthogonal images of z-stacks with phalloidin staining for F-actin (white) that accumulated in the apical brush border. ODMs were fixed after 10 (human), 12 (mouse), 6 (pig), and 15 (chicken) days. Nuclei stained with DAPI (blue). Scale bar = 20 µm. (B) Analysis of transepithelial-electrical resistance (TEER) of ODMs after seeding. All ODMs, except for pig, developed an electrophysiologically tight monolayer. Mean (± 95% CI) of 12-36 filter inserts per species is shown. (C) Expression of marker gene transcripts for intestinal cell populations, measured by RT-qPCR. Indicated markers are: EE, enteroendocrine cells; PC, Paneth cells; GC, goblet cells; SC, stem cells; and E, enterocytes. Mean (± 95% CI) of ≥ 4 technical replicas of at least two independent biological replicates are shown. n.d., not detectable. Data are derived from Holthaus et al. (2020), where interpretation of the data can be found.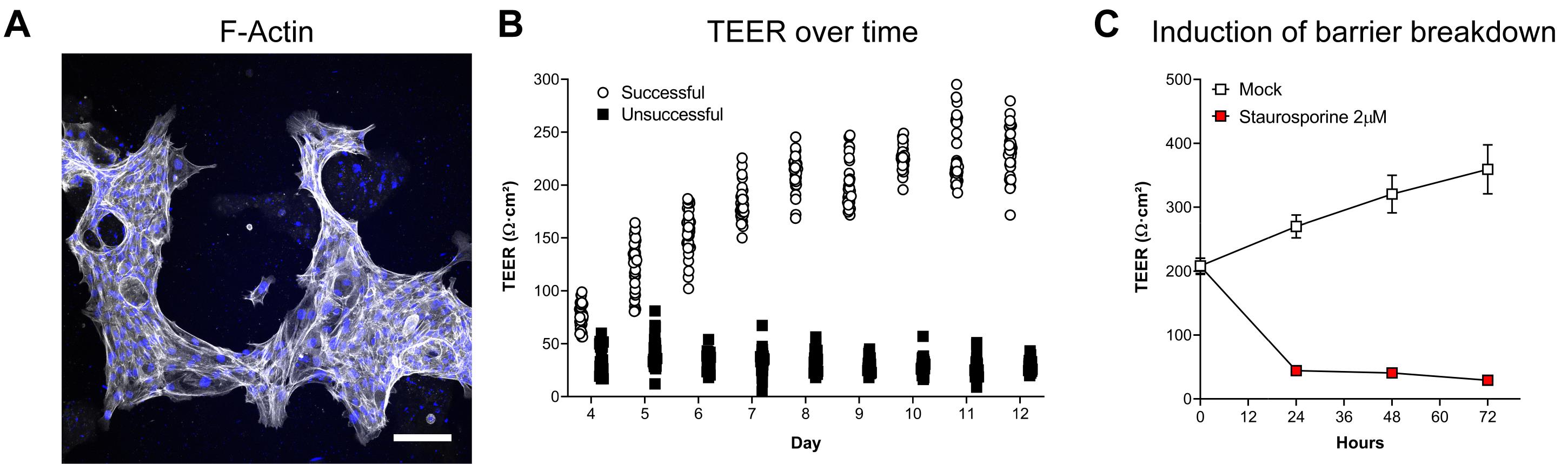
Figure 4. Examples of unsuccessful attempts of ODM establishment and induction of barrier breakdown. (A) Exemplary projection of a z-stack of non-confluent ODMs with phalloidin staining for F-actin (white). Nuclei stained with DAPI (blue). Scale bar = 100 µm. (B) TEER development over time of successful versus unsuccessful establishment of human ODMs, over a time course of 12 days. Means (± 95% CI) of 36 and 28 filter inserts per condition are shown. Successful establishment data correspond to Figure 3B. (C) Induction of barrier breakdown (as a control) with 2 µM of the kinase inhibitor and apoptosis inducer staurosporine (Belmokhtar et al., 2001). Means (± SEM) of three independent experiments per condition are shown. Successful human TEER data is derived from Holthaus et al. (2020).
Measuring transepithelial electrical resistance (TEER) using a chopstick electrode
Pre-warm the heating plate to 37°C.
Note: TEER is temperature-sensitive, therefore using the heating plate is recommended.
Pour 80% ethanol and sterile water in 25 mL sterile glass beakers.
Note: It is important that the water is sterile, to prevent contamination of your sample.
Sterilize the electrode by placing it in 80% ethanol until the heating block is heated up.
Put your ODM plate on the 37°C heating plate.
Turn on the ERS2 voltohmmeter.
Note: Make sure that the voltohmmeter measures Ω and not mV. The values in water and ethanol should be around 12,000-14,000 Ω.
Rinse away the ethanol by placing the electrode in the beaker with water.
Shake off remaining water from the electrode.
Put the electrode into the well you want to measure (Video 1).
Note: The electrode has one shorter and one longer leg. Use the longer leg for the basal compartment and the shorter leg for the apical one. Be cautious not to scratch the cell surface! You can touch the well bottom with the longer leg of the electrode without scratching the cell surface, which gives additional stability. Be cautious not to apply too much weight, as this will bend the electrode. You might want to practice it beforehand.
Wait for the values to stabilize and record them in your notes (raw TEER values range between 500-700 for human and mouse ODMs).
Repeat Steps C.8 and C.9 to measure all your wells. Wash with ethanol and water between different conditions.
Put your plate back into the incubator.
Turn off the voltohmmeter and heating plate, and disinfect the electrode with ethanol.
Put the electrode into a conical 15 mL tube after your measurements and seal it with parafilm.
Production of L-WRN conditioned medium
Note: We use the L-WRN (ATCC CRL-3276) cell line for the production of L-WRN conditioned medium. The following protocol is based on the procedures found on the product sheet, but includes modifications that have proven useful in our hands. In particular, we indicate the days on which each step could be performed, so that harvesting can be done on weekdays.
Thawing of L-WRN cells
Add 10 mL of L-WRN growth medium without geneticin and hygromycin B to one T75 flask, and place it in a 37°C incubator with 5% CO2.
Get a vial of L-WRN cells from the liquid nitrogen storage (wear goggles and gloves for protection) and transport it to the bench in a cooling rack.
Thaw the vial in a 37°C water bath under gentle agitation.
Once the content is thawed, remove the vial from the water bath. Disinfect the vial by wiping it with 70% ethanol and move it to the biosafety cabinet.
Transfer the content of the vial into the T75 flask containing the pre-warmed medium.
Incubate the flask at 37°C and change the medium after 24 h to L-WRN growth medium supplemented with 0.5 mg/mL geneticin and 0.5 mg/mL hygromycin B.
Passaging of L-WRN cells
Note: We usually do this on Mondays as this allows us to harvest on weekdays.
Aspirate and discard the L-WRN growth medium from the T75 flask.
Rinse the cell layer twice with 10 mL of PBS.
Add 2 mL of 0.5% trypsin-EDTA solution to the flask and place it at 37°C for 5-10 min.
Note: You can follow the progression of cell detachment under a microscope.
Add 8 mL of pre-warmed growth medium to stop the trypsinization reaction.
Transfer 1.5 mL (split ratio 1:6.6) of the cell suspension to the appropriate number of new T75 flasks containing 18.5 mL of pre-warmed growth medium.
Note: The number of new T75 flasks depends on the desired volume of L-WRN-conditioned medium you want to produce. Each T75 flask is later expanded to three T150 flasks that are harvested four times, yielding a total of 300 mL of conditioned medium per T75 flask. For reference, we usually expand our L-WRN culture to three to five new T75 flasks per round of conditioned media production.
Incubate the flasks at 37°C in an incubator with 5% CO2.
Preparation and harvest of L-WRN conditioned medium
Note: We usually do this on Thursdays, as this allows us to harvest on weekdays in the following week.
Prepare three T150 flasks per one T75 flask of L-WRN cells.
Add 22 mL of pre-warmed conditioning medium (without geneticin and hygromycin B) to each T150 flask.
Aspirate and discard the L-WRN growth medium from the T75 flask, rinse with PBS, and detach the cells with 2 mL of trypsin-EDTA, as described above for passaging.
Add 8 mL of conditioning medium to the detached cells.
Transfer 3 mL of the cell suspension into each of the prepared T150 flasks.
If you want to continue passaging the L-WRN cells, add 0.5-1 mL of the remaining cell suspension to one additional T75 flask containing 19 mL of L-WRN growth medium (supplemented with geneticin and hygromycin B). Incubate at 37°C for 4 days and repeat Step D.2 to expand the culture for another round of harvesting.
Incubate the T150 flasks at 37°C for 4 days or until the cells become over-confluent.
Note: If you follow our scheme, the cells should be ready on Monday.
Aspirate and discard the medium.
Rinse the cell layer once with 10 mL of conditioning medium and discard the rinse.
Add 25 mL of fresh pre-warmed conditioning medium and incubate for 24 h at 37°C.
On the next day, transfer the conditioned medium to a 50 mL tube. Add 25 mL of fresh conditioning medium to the flasks and return the flasks to the incubator. Centrifuge the conditioned medium at 2,000 × g, 4°C for 5 min and decant the supernatant into a sterile glass bottle. Store the L-WRN-conditioned medium at 4 °C.
Repeat the previous step (Step D3k) every 24 h, harvesting a total of 4 times. Decant the supernatant into the same bottle each time. After the fourth harvest, mix well, and store the batch of conditioned medium at 4°C.
Note: It may be possible to continue harvesting beyond day 4. However, we usually harvest for four consecutive days (Tuesday to Friday) and start with fresh cultures on Monday. Before combining batches, test their activity (see Procedure F).
Sterilize by filtration through a 0.22 µm filter before use. Aliquot and store at -80°C.
Cryopreservation of L-WRN cells
Note: One confluent T150 flask or two T75 flasks should be sufficient to generate 4 cryo-stocks of 5 × 106 cells each.
Pre-warm L-WRN growth medium at 37°C in water bath.
Aspirate and discard the old growth medium.
Rinse the cell layer once with 10 mL of PBS.
Detach the cells with 2 mL of trypsin-EDTA, as described above.
Add 4 mL of pre-warmed growth medium and transfer cell suspension to a 15 mL tube.
Rinse the flask again with an additional 4-5 mL of growth medium, to collect any remaining cells, and add the medium to the 15 mL tube.
Determine the cell number. Fill a Neubauer chamber with 10 µL of the cell suspension and count the cells under the microscope.
Centrifuge the cell suspension for 5 min at 134 × g.
Remove the supernatant and carefully resuspend the cell pellet in the appropriate volume of freezing medium. Each cryovial should contain approximately 5 × 106 cells in 1 mL.
Transfer 1 mL to each cryovial and place it in a freezing container.
Store the freezing container at -80°C for 24 h, and then transfer the vials to liquid nitrogen for final storage.
Production of R-Spondin 1-conditioned medium and mNoggin-conditioned medium
Note: We use the HEK 293T HA-noggin-Fc cell line (Heijmans et al., 2013; Bartfeld et al., 2015), kindly provided by Hans Clevers, Hubrecht Institute, Utrecht, and the HEK 293T R-Spondin1-Fc cell line (Kim et al., 2005), kindly provided by Calvin Kuo, Stanford University, USA, for the production of R-Spondin 1-conditioned medium and mNoggin-conditioned medium. The following protocol is based on the procedures of the respective authors, but includes modifications that have proven useful in our hands. In particular, we indicate the days on which each step could be performed so that harvesting can be done on weekdays.
Thawing of R-Spondin 1 cells and mNoggin cells
Pre-warm R-Spondin 1 and mNoggin growth medium in a 37°C water bath. Add 0.3 mg/mL zeocin for R-Spondin 1 cells and 0.5 mg/mL geneticin for mNoggin cells, respectively. Use media supplemented with the respective antibiotics for all subsequent steps.
Add 19 mL of R-Spondin 1 growth medium and mNoggin growth medium to one T75 flask each, and place them in a 37°C incubator with 5% CO2.
Get one vial each of R-Spondin 1 cells and mNoggin cells from the liquid nitrogen storage (wear goggles and gloves for protection) and transport them to the bench in a cooling rack.
Thaw the vials in a 37°C water bath under gentle agitation.
Once the content is thawed, remove the vials from the water bath. Disinfect the vials by wiping them with 70% ethanol and move them to the biosafety cabinet.
Transfer the content of each vial into a 15 mL tube containing 9 mL of R-Spondin 1 growth medium and mNoggin growth medium, respectively.
Centrifuge at 134 × g for 5 min at 4°C.
Aspirate and discard the supernatant. Resuspend the cell pellets in 1 mL of R-Spondin 1 growth medium and mNoggin growth medium, respectively, and transfer the cell suspensions into one T75 flask, each containing the respective pre-warmed medium.
Incubate the flasks at 37°C and change the medium after 24 h.
Passaging of R-Spondin 1 cells and mNoggin cells
Pre-warm R-Spondin 1 and mNoggin growth medium in a 37°C water bath. Add 0.3 mg/mL zeocin for R-Spondin 1 cells and 0.5 mg/mL geneticin for mNoggin cells, respectively. Use media supplemented with the respective antibiotics for all subsequent steps.
Aspirate and discard the growth medium.
Rinse the cell layer twice with 10 mL of PBS.
Add 2 mL of 0.5% trypsin-EDTA solution to the flask and place it at 37°C for 5-10 min.
Note: You can follow the progression of cell detachment under a microscope.
Add 8 mL of pre-warmed R-Spondin 1 growth medium and mNoggin growth medium, respectively, to stop the trypsinization reaction and transfer each of the cell suspensions to a 15 mL tube.
Centrifuge at 134 × g for 5 min at 4°C.
Aspirate and discard the supernatants.
If you want to start with the preparation of conditioned medium, expand each culture by subculturing it to two T150 flasks. Therefore, resuspend the pellets in 9 mL of pre-warmed R-Spondin 1 growth medium and mNoggin growth medium, respectively. Transfer 3 mL of the cell suspensions into each of the two new T150 flasks containing 37 mL of the respective pre-warmed growth medium.
Note: If you do not want to proceed with harvesting, passage the cells at a sub-cultivation ratio of 1:10.
Incubate the flasks at 37°C in an incubator with 5% CO2 until the cells reach confluency after 3-4 days.
Preparation and harvest of R-Spondin 1-conditioned medium and mNoggin-conditioned medium
Note: We usually do this on Mondays or Thursdays as this allows us to harvest on weekdays.
Pre-warm R-Spondin 1 and mNoggin growth medium (with and without zeocin or geneticin, respectively) in a 37°C water bath. Add 0.3 mg/mL zeocin for R-Spondin 1 cells and 0.5 mg/mL geneticin for mNoggin cells, respectively.
Note: You could also add the antibiotics directly to the flasks in Step E3i.
Check under the microscope that the cells are fully confluent before proceeding.
To expand the cell cultures for subsequent harvesting, prepare 12 new T150 flasks from the two T150 flask of R-Spondin 1 cells and mNoggin cells, respectively.
Add 38.6 mL of pre-warmed growth medium (without zeocin or geneticin) to each of the new T150 flasks.
Aspirate and discard the old growth medium from the T150 flasks, rinse with PBS, and detach the cells with 3 mL of trypsin-EDTA, as described above for passaging.
Add 10 mL of pre-warmed growth medium (without zeocin or geneticin) to stop the trypsinization reaction. Transfer the cell suspension of one flask into one 50 mL tube each.
Centrifuge at 134 x g for 5 min at 4°C.
Aspirate and discard the supernatant. Carefully resuspend the pellets in 10.5 mL of pre-warmed R-Spondin 1 or mNoggin growth medium (without zeocin or geneticin) and transfer 1.4 mL of the cell suspension into each of the new T150 flasks.
If you want to continue passaging, add 1.4 mL of the remaining cell suspensions to two additional T150 flask containing 38.6 mL of R-Spondin 1 growth medium and mNoggin growth medium (with zeocin or geneticin), respectively. Incubate at 37°C for 3-4 days and repeat Step E.3 to start another round of harvesting.
Note: You may add the antibiotics directly to the flasks.
Incubate the 12 T150 flasks at 37°C for 3-4 days.
Aspirate and discard the growth medium and replace it with 50 mL of conditioning medium.
Incubate the flasks at 37°C for 7 days without exchanging the medium.
Harvest the conditioned media and transfer it into 50 mL tubes.
Centrifuge the conditioned medium at 2,000 × g for 5 min and decant the supernatant into a glass bottle.
Sterilize by filtration through a 0.22 µm filter before use. Aliquot and store at -80°C.
Note: Before combining batches, test their activity (see Procedure F).
Cryopreservation of R-Spondin 1 cells and mNoggin cells
For cryopreservation of R-Spondin 1 cells and mNoggin cells, follow the protocol for cryopreservation of L-WRN cells (Step D.4), but use R-Spondin 1 growth medium and mNoggin growth medium, respectively.
Note: For R-Spondin 1 cells and mNoggin cells, we usually prepare cryo-stocks containing 8 × 106 cells.
Activity testing of conditioned media
It is critical to test all conditioned media before use on organoid cultures. Noggin-conditioned media is routinely tested on newly harvested murine crypts using ERN medium (Standard mouse medium – EGF, R-Spondin 1, Noggin), and 1% of a freshly prepared batch has to be sufficient for organoid establishment. Different methods for batch-to-batch quality control have been described and compared (see VanDussen et al., 2019). It is at least recommended to test the Wnt and R-Spondin 1 activity in WRN- and R-Spondin 1-conditioned media with a luciferase reporter cell line that expresses firefly luciferase in response to canonical Wnt signaling, for example the HEK293 SuperTopFlash (STF) cell line (ATCC Number: CRL-3249) in combination with a luciferase assay kit, using a luminometer according to the manufacturer’s instructions (Mahe et al., 2015; Urbischek et al., 2019). Additionally, it is recommended to compare proliferation and spheroid growth between different batches of conditioned media (VanDussen et al., 2019). Batches of all three conditioned media or protein surrogates are commercially available, for example at U-Protein Express B.V. (Puschhof et al., 2021).
Data analysis
For measurements of trans-epithelial electrical resistance (TEER) it is vital that measurements are subsequently normalized to surface area. It is common that measurements are presented in Ω*cm2.
Recipes
WERN 3D base proliferation medium for spheroid culture
50% (v/v) L-WRN-conditioned media (CM) (VanDussen et al., 2019)
20% (v/v) R-Spondin 1-CM (Kim et al., 2005)
10% (v/v) Noggin-CM (Heijmans et al., 2013; Bartfeld et al., 2015)
50 ng/mL EGF
Note: For mouse and human organoids, use mouse EGF and human EGF, respectively. For pig organoids, we use mouse EGF. For chicken organoids, use both human and mouse EGF combined.
1 mM HEPES
2 mM GlutaMax
1× P/S (100 U/mL penicillin and 100 µg/mL streptomycin)
1× N2
1× B27
1 mM N-acetyl-L-cysteine
10 mM nicotinamide
500 nM A83-01 (TGF-β inhibitor)
1 μM SB-202190 (p38 inhibitor)
in Advanced DMEM/F-12.
Use a 0.22 μm filter to sterilize and use within one week. Store at 4°C.
Note: For pig organoids, supplement the 3D base medium with 10 µM Y-27632. For chicken organoids, supplement the 3D base medium with 10 µM Y-27632, 3 µM CHIR99021 (GSK-3 inhibitor), and 10 µM prostaglandin E2.
Organoid freezing medium
10% (v/v) fatty acid-free bovine-serum albumin (BSA)
10% (v/v) DMSO
in Advanced DMEM/F-12.
Use a 0.22 µm filter to sterilize and use within one week. Store at 4°C.
Dissociation reagent
10 µM Y-27632 in TrypLE Express
Prepare fresh immediately before use.
ODM Day 0 medium
50% (v/v) L-WRN-CM (VanDussen et al., 2019)
20% (v/v) R-Spondin 1-CM (Kim et al., 2005)
10% (v/v) Noggin-CM (Heijmans et al., 2013; Bartfeld et al., 2015)
50 ng/mL EGF
1 mM HEPES
2 mM GlutaMax
1× P/S (100 U/mL penicillin and 100 µg/mL streptomycin)
1× N2
1× B27
1 mM N-acetyl-L-cysteine
10 mM nicotinamide
10 μM Y-27632 (ROCK1 inhibitor)
in Advanced DMEM/F-12.
Use a 0.22 μm filter to sterilize and use within one week. Store at 4°C.
ODM Day 1 medium
5% (v/v) L-WRN-CM (VanDussen et al., 2019))
20% (v/v) R-Spondin 1-CM (Kim et al., 2005)
10% (v/v) Noggin-CM (Heijmans et al., 2013; Bartfeld et al., 2015)
50 ng/mL EGF
1 mM HEPES
2 mM GlutaMax
1× P/S (100 U mL−1 penicillin and 100 µg/mL streptomycin)
1× N2
1× B27
1 mM N-acetyl-L-cysteine
10 mM nicotinamide
10 μM Y-27632 (ROCK1 inhibitor)
in Advanced DMEM/F-12.
Use a 0.22 μm filter to sterilize and use within one week. Store at 4°C.
ODM medium day 2 onwards
20% (v/v) R-Spondin 1-CM (Kim et al., 2005)
10% (v/v) Noggin-CM (Heijmans et al., 2013; Bartfeld et al., 2015)
50 ng/mL EGF
1 mM HEPES
2 mM GlutaMax
1× P/S (100 U/mL penicillin and 100 µg/mL streptomycin)
1× N2
1× B27
1 mM N-acetyl-L-cysteine
10 mM nicotinamide
in Advanced DMEM/F-12.
Use a 0.22 μm filter to sterilize and use within one week. Store at 4°C.
Phosphate buffered saline (PBS)
137 mM NaCl
8.0 mM Na2HPO4
2.7 mM KCl
1.5 mM KH2PO4 (pH 7.4)
Use a 0.22 μm filter to sterilize. Store at room temperature or at 4°C.
L-WRN growth medium
10% (v/v) FBS
1% (v/v) P/S (100 U/mL penicillin and 100 µg/mL streptomycin)
0.5 mg/mL hygromycin B
0.5 mg/mL geneticin (G418)
in DMEM High Glucose (4.5 g/L), with Stable Glutamine.
Store at 4°C.
Note: It is recommended to prepare a smaller volume of media to save resources.
L-WRN conditioning medium
10% (v/v) FBS
1% P/S (v/v) (100 U/mL penicillin and 100 µg/mL streptomycin)
in DMEM High Glucose (4.5 g/L), with Stable Glutamine.
Store at 4°C.
R-Spondin 1 and mNoggin growth medium
10% (v/v) FBS
1% (v/v) P/S (100 U/mL penicillin and 100 µg/mL streptomycin)
in DMEM, high glucose with GlutaMAX Supplement and pyruvate.
Store at 4°C.
R-Spondin 1 and mNoggin conditioning medium
1% (v/v) P/S (100 U/mL penicillin and 100 µg/mL streptomycin)
1 mM HEPES
2 mM GlutaMax
in Advanced DMEM/F-12.
Store at 4°C.
L-WRN, R-Spondin 1 and mNoggin freezing medium
20% (v/v) FBS
10% (v/v) DMSO
in DMEM, high glucose with GlutaMAX Supplement and pyruvate.
Use a 0.22 μm filter to sterilize and use within one week. Store at 4°C.
Acknowledgments
For this protocol and the original research paper, financial support was provided by the Deutsche Forschungsgemeinschaft (DFG) via GRK 2046 to TA, CK, and FS. DW, ED-B, DH, and AM are supported by GRK 2046 (Projects B3 & C3). AM is also supported by the Sonnenfeld foundation. The work of SMK and JDS is DFG-funded by TRR 241 (Project B06). Work by GK, TA, CK, and FS is supported by the Robert Koch-Institute. FS is also a member of IRTG 2029 (supported by the DFG), and of TOXOSOURCES (supported by funding from the European Union’s Horizon 2020 Research and Innovation Programme under grant agreement No 773830: One Health European Joint Programme).
This protocol is derived from the original research paper by Holthaus et al. (2020). For this, we thank Luca Bertzbach and Benedikt Kaufer at Freie Universität Berlin, for the provision of intestinal chicken samples and assembly of a library of intestinal marker primer sequences. We also thank Svenja Steinfelder at Freie Universität Berlin, for the provision and extraction of porcine intestinal crypts.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The funding bodies had no role in the design, collection, analysis, and interpretation of data, or in writing of this protocol or the previous study.
Ethics
The isolation and establishment of organoids from the human duodenal specimen were done as described before (Kraft et al., 2020; Holthaus et al., 2020) and were approved by the ethical committee of the Charité, Berlin (#EA4-015-13). The donors of human material provided their written informed consent to participate in scientific studies. For porcine samples, duodenal crypts were isolated from a 10 week-old piglet (Sus scrofa, kindly provided by Svenja Steinfelder, Institute of Immunology, Freie Universitat Berlin, animal license T0002/17). Chicken duodenal crypts were isolated from the intestine of a 14-day-old female chicken (Gallus gallus, kindly provided by Luca Bertzbach & Benedikt Kaufer, Institute of Virology, Freie Universitat Berlin, animal license T0245/14). The mouse duodenal sample was derived from a female C57/Bl6 mouse from an RKI in-house bred colony (animal license T0173/14). The animal studies were reviewed and approved by the Landesamt für Gesundheit und Soziales (LAGeSo) Berlin.
References
- Bar-Ephraim, Y. E., Kretzschmar, K. and Clevers, H. (2020). Organoids in immunological research. Nat Rev Immunol 20(5): 279-293.
- Bartfeld, S. (2016). Modeling infectious diseases and host-microbe interactions in gastrointestinal organoids. Dev Biol 420(2): 262-270.
- Bartfeld, S., Bayram, T., van de Wetering, M., Huch, M., Begthel, H., Kujala, P., Vries, R., Peters, P. J. and Clevers, H. (2015). In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 148(1): 126-136 e126.
- Belmokhtar, C. A., Hillion, J. and Segal-Bendirdjian, E. (2001). Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 20(26): 3354-3362.
- Delgado-Betancourt, E., Hamid, B., Fabian, B. T., Klotz, C., Hartmann, S. and Seeber, F. (2019). From Entry to Early Dissemination-Toxoplasma gondii's Initial Encounter With Its Host. Front Cell Infect Microbiol 9: 46.
- Dutta, D. and Clevers, H. (2017). Organoid culture systems to study host-pathogen interactions. Curr Opin Immunol 48: 15-22.
- Heijmans, J., van Lidth de Jeude, J. F., Koo, B. K., Rosekrans, S. L., Wielenga, M. C., van de Wetering, M., Ferrante, M., Lee, A. S., Onderwater, J. J., Paton, J. C., et al. (2013). ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Rep 3(4): 1128-1139.
- Hill, D. R. and Spence, J. R. (2017). Gastrointestinal Organoids: Understanding the Molecular Basis of the Host-Microbe Interface. Cell Mol Gastroenterol Hepatol 3(2): 138-149.
- Holthaus, D., Delgado-Betancourt, E., Aebischer, T., Seeber, F. and Klotz, C. (2020). Harmonization of Protocols for Multi-Species Organoid Platforms to Study the Intestinal Biology of Toxoplasma gondii and Other Protozoan Infections. Front Cell Infect Microbiol 10: 610368.
- Holthaus D., Kraft M. R., Krug S. M., Wolf S., Müller A., Delgado-Betancourt, E., Schorr M., Holland G., Knauf F., Schulzke J. D., Aebischer T., Klotz C. (2021). Dissection of Barrier Dysfunction in Organoid-Derived Human Intestinal Epithelia Induced by Giardia duodenalis. Gastroenterology S0016-5085(21)03798-7.
- Kim, K. A., Kakitani, M., Zhao, J., Oshima, T., Tang, T., Binnerts, M., Liu, Y., Boyle, B., Park, E., Emtage, P., Funk, W. D. and Tomizuka, K. (2005). Mitogenic influence of human R-spondin1 on the intestinal epithelium. Science 309(5738): 1256-1259.
- Klotz, C., Aebischer, T. and Seeber, F. (2012). Stem cell-derived cell cultures and organoids for protozoan parasite propagation and studying host-parasite interaction. Int J Med Microbiol 302(4-5): 203-209.
- Kozuka, K., He, Y., Koo-McCoy, S., Kumaraswamy, P., Nie, B., Shaw, K., Chan, P., Leadbetter, M., He, L., Lewis, J. G., et al. (2017). Development and Characterization of a Human and Mouse Intestinal Epithelial Cell Monolayer Platform. Stem Cell Reports 9(6): 1976-1990.
- Mahe, M. M., Sundaram, N., Watson, C. L., Shroyer, N. F. and Helmrath, M. A. (2015). Establishment of human epithelial enteroids and colonoids from whole tissue and biopsy. J Vis Exp(97): 52483.
- Miyoshi, H. and Stappenbeck, T. S. (2013). In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc 8(12): 2471-2482.
- Moon, C., VanDussen, K. L., Miyoshi, H. and Stappenbeck, T. S. (2014). Development of a primary mouse intestinal epithelial cell monolayer culture system to evaluate factors that modulate IgA transcytosis. Mucosal Immunol 7(4): 818-828.
- Puschhof, J., Pleguezuelos-Manzano, C., Martinez-Silgado, A., Akkerman, N., Saftien, A., Boot, C., de Waal, A., Beumer, J., Dutta, D., Heo, I. and Clevers, H. (2021). Intestinal organoid cocultures with microbes. Nat Protoc 16(10): 4633-4649.
- Sato, T., Vries, R. G., Snippert, H. J., van de Wetering, M., Barker, N., Stange, D. E., van Es, J. H., Abo, A., Kujala, P., Peters, P. J. and Clevers, H. (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459(7244): 262-265.
- Urbischek, M., Rannikmae, H., Foets, T., Ravn, K., Hyvonen, M. and de la Roche, M. (2019). Organoid culture media formulated with growth factors of defined cellular activity. Sci Rep 9(1): 6193.
- VanDussen, K. L., Marinshaw, J. M., Shaikh, N., Miyoshi, H., Moon, C., Tarr, P. I., Ciorba, M. A. and Stappenbeck, T. S. (2015). Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 64(6): 911-920.
- VanDussen, K. L., Sonnek, N. M. and Stappenbeck, T. S. (2019). L-WRN conditioned medium for gastrointestinal epithelial stem cell culture shows replicable batch-to-batch activity levels across multiple research teams. Stem Cell Res 37: 101430.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Warschkau, D., Delgado-Betancourt, E., Holthaus, D., Müller, A., Kliem, G., Krug, S. M., Schulzke, J. D., Aebischer, T., Klotz, C. and Seeber, F. (2022). From 3D to 2D: Harmonization of Protocols for Two-dimensional Cultures on Cell Culture Inserts of Intestinal Organoids from Various Species . Bio-protocol 12(2): e4295. DOI: 10.21769/BioProtoc.4295.
Category
Stem Cell > Adult stem cell > Intestinal stem cell
Microbiology > Microbe-host interactions > In vitro model
Cell Biology > Cell isolation and culture > 3D cell culture
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.