- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

High-throughput Growth Measurements of Yeast Exposed to Visible Light
Published: Vol 12, Iss 2, Jan 20, 2022 DOI: 10.21769/BioProtoc.4292 Views: 3217
Reviewed by: Gal HaimovichIndranil MalikAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Light is a double-edged sword: it is essential for life on the planet but also causes cellular damage and death. Consequently, organisms have evolved systems not only for harvesting and converting light energy into chemical energy but also for countering its toxic effects. Despite the omnipresence and importance of such light-dependent effects, there are very few unbiased genetic screens, if any, investigating the mechanistic consequences that visible light has on cells. Baker’s yeast, Saccharomyces cerevisiae, is one of the best annotated organisms thanks to several easily available mutant collections and its amenability to high-throughput genetic screening. However, until recently this yeast was thought to lack receptors for visible light, therefore its response to visible light was poorly understood. Nevertheless, a couple of years ago it was discovered that yeast senses light via a novel and unconventional pathway involving a peroxisomal oxidase, hydrogen peroxide, and a particular type of antioxidant protein, called peroxiredoxin. Here, we describe in detail a protocol for scoring yeast genes involved in the resistance to visible light (400-700 nm) on a genome-wide scale. Because cells in dense cultures shield each other from light exposure, resulting in apparent light resistance, our method involves adaptations to reduce inoculum size under conditions amenable to high-throughput screens, to properly be able to identify light-sensitive mutants. We also describe how to measure growth in the presence of light, including two follow-up validation tests. In this way, this method makes it possible to score light-sensitivity on a genome-wide scale with high confidence.
Graphic abstract:
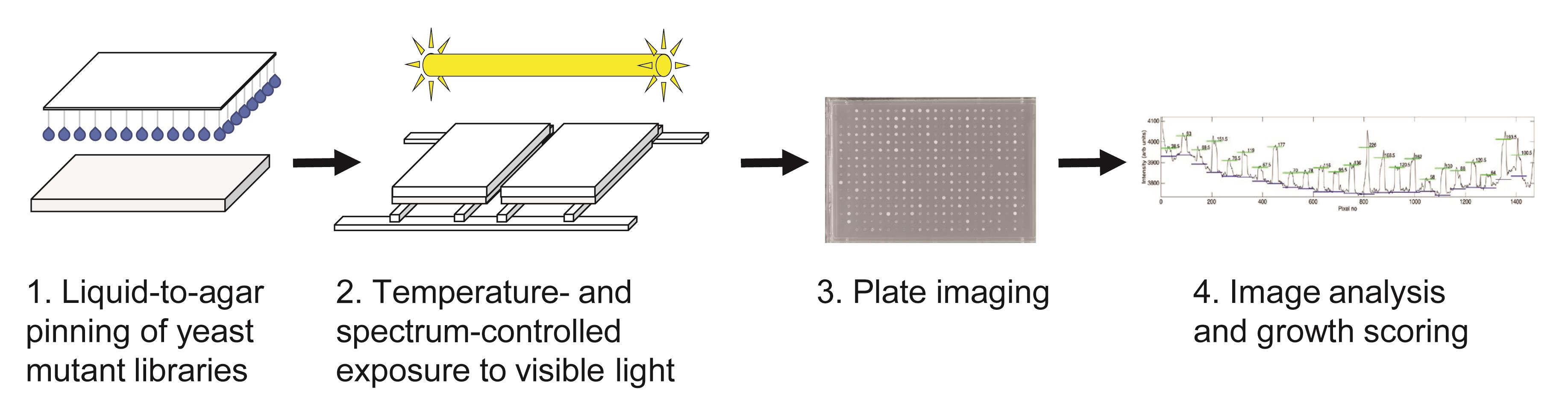
Overview of strategy for high-throughput determination of yeast growth upon visible light stress.
Background
Light is, on the one hand, essential for life on the planet as a source of energy but, on the other hand, it can also be involved in regulatory processes, as well as cause substantial cellular damage and demise. Studies in later years have suggested that cells from the three domains of life, including most human cells, keep stimulus-independent rhythms of activity to adapt to the prevailing recurring alterations of sunlight and darkness (i.e., circadian rhythms). A key stimulus entraining circadian rhythms is visible light. Characterized clock mechanisms, including those in filamentous fungi, involve cryptochrome, rhodopsin, and melanopsin photoreceptors (Yu and Fischer, 2019; Patke et al., 2020). However, circadian rhythms persist in red blood cells in the absence of canonical clock components (O'Neill and Reddy, 2011; O'Neill et al., 2011), indicating that alternative clock mechanisms must exist. In red blood cells, the unusual anti-oxidants and signaling regulators peroxiredoxins were proposed to constitute conserved and transcription-independent regulators and markers of circadian rhythms (Edgar et al., 2012). On a similar note, baker’s yeast has long been thought to lack light receptors (Idnurm et al., 2010). Recently, however, it was discovered that yeast can sense light via a peroxisomal oxidase, hydrogen peroxide, and the peroxiredoxin Tsa1 (Bodvard et al., 2017). Thus, yeast is a model for understanding responses to light in cells and organisms lacking dedicated light receptors. In the yeast Saccharomyces cerevisiae, high-throughput genetic screening by ordered arrays of mutant libraries has become a central tool in understanding gene function on a genome-wide scale (Giaever et al., 2002).
Several filamentous fungi are able to sense light of different wavelengths through photoreceptors homologous to those in mammals and plants. In fungi, light controls circadian clocks and morphological responses, as well as growth/stress response decisions. Investigations of their responses to light have mainly focused on identifying photoreceptors, circadian clock components, and regulators of morphological responses. In these organisms, large-scale investigations of light responses have so far been limited to transcriptomics (Lewis et al., 2002; Dong et al., 2008; Chen et al., 2009; Wu et al., 2014), forward genetics screens using light-dependent reporters (Yu et al., 2016), and reverse genetic screens of limited gene sets (e.g., genes encoding transcription factors) affecting circadian clock reporters (Munoz-Guzman et al., 2021). Here, we outline a protocol for assessing baker’s yeast functions relevant in responses to visible light (400-700 nm) on a genome-wide scale, by scoring light-dependent growth of mutants. Potentially, by using suitable reporters (Kainth et al., 2009; D'Orazio et al., 2019) and with proper modifications to the setup, this protocol could be used to study other types of responses to light or, depending on the availability of genome-wide mutant collections, also understand light-resistance in other microorganisms.
The procedure we describe here, to score genes involved in the resistance of yeast to visible light, starts at a genome-wide scale, and subsequently narrows down to smaller scale validation tests for selected light-sensitive strains. In our case, the haploid BY4741 deletion collection was examined, in an arrayed format used in the Boone lab for synthetic genetic array analysis (Tong et al., 2001), in which all plate edges are lined by a row of wild-type-like (his3∆) control strains. We describe a detailed protocol for achieving the initial genome-wide screen, despite a strong dependence of yeast light-dependent growth on cell density that complicated analyses. Furthermore, to be able to reliably score light-sensitivity, we next describe appropriate methods for image analysis and growth scoring in detail. Following this, we describe a medium-throughput assay suitable for retesting a hundred or so strains, which should preferably be used on multiple deletion mutant collections, such as those containing the haploid (BY4741) and homozygous diploid (BY4743) gene knockouts. Finally, we outline a serial dilution drop test assay suitable for a smaller subset of genes, which in our case was used to retest gene deletions from the homozygous diploid BY4743 collection.
Materials and Reagents
96 well plates: TPP® Tissue culture plate 96 well, flat-bottom (Merck, catalog number: Z707902)
Experimental agar plates: PlusPlates (Singer Instruments, catalog number: PLU-003)
Pinning pads: RePads 96 long pin (Singer Instruments, catalog number: REP-001)
14 mL Falcon® tubes round-bottom with snap-cap (VWR, catalog number: 734-0985)
Yeast haploid deletion mutant collection (BY4741 (Mat a) transOMIC technologies, catalog number: TKY3502)
Yeast homozygous diploid deletion mutant collection (BY4743, transOMIC technologies, catalog number: TKY3500)
Glucose (Sigma-Aldrich, catalog number: G8270)
Peptone (VWR, catalog number: ICNA0210480805)
Yeast extract (VWR, catalog number: J850)
Yeast Nitrogen Base without Amino Acids and without Ammonium Sulphate (Formedium, catalog number: CYN0510)
Agar (VWR, catalog number: J637)
Ammonium sulfate (Sigma-Aldrich, catalog number: A4418)
Complete Supplement Mixture with all amino acids (Formedium, catalog number: DCS0019)
Succinic acid (Sigma Aldrich, catalog number: 398055)
NaOH (Sigma Aldrich, catalog number: S5881)
40% (w/w) glucose (see Recipes)
YPD liquid medium (see Recipes)
10× stock of amino acid mixture (see Recipes)
10× stock of succinate buffered Yeast Nitrogen Base (YNB) (see Recipes)
10× stock of amino acid mixture (see Recipes)
Ultrapure (Milli-Q) water (see Recipes)
Agar mixture (see Recipes)
Equipment
For shaking plates (Vibrax VXR basic, IKA, Germany)
Robot for pinning (RoToR HDA; Singer Instruments, UK)
Open industrial light tube fitting for 3 × 58 W light tubes (Mir, Elektroskandia, product number E72 106 41)
UV radiation-filtered fluorescent light tubes (Osram L 58 W/940)
Sample holders: for each plate two ribs holding the plate above ground to allow for air circulation and specific distance from lamp
Spectrometer (QE65000-FL, Oceans Optics)
Imaging of plates (Digital camera D5000 Nikon and Epson Perfection V700)
Software
MATLAB® (for high- and medium-throughput assays)
QuantityOne (Bio-Rad, for serial dilution drop test)
Procedure
Cell plate preparation
It is important that agar plates are made correctly and consistently, to achieve a standardized and even transfer of cells upon robotic pinning. When casting the agar plates, it is important to always use the same volume (precisely 50 mL/plate) of well-mixed media per plate, and let the plates solidify on a completely level surface. If the solidification surface is not level, this will produce plates with a non-level agar surface, where the pinning robot applies the cells unevenly across the plate during pinning. If the plates are made with agar medium volumes different than 50 mL, uneven hydration and variability due to altered agar surface shapes and properties between plates may result. All agar plates should be inspected before screening, to ensure plate quality and hydration levels are consistent among the set of plates. Because some plate imperfections are only detected at the pinning stage, make sure to cast a few extra plates than are strictly needed. A related source of variability is the specific firmness of the agar medium, which can affect the pinning and transfer of cells. The optimal pinning pressure to use with the Rotor HDA robot will vary depending on the firmness of the specific agar plates used (and is usually related to the agar concentration).
The procedure of cell transfer onto agar plates is a crucial step in assessing the ability of yeast cells to grow in visible light (Molin et al., 2020). If too many cells are transferred to the same spot, the cells on top will shield and protect the cells below from the light (which makes them grow), resulting in the scoring of growth as light-resistant, i.e., at higher initial cell density all cells/mutants will appear light-resistant (Figure 1A). Figure 1B shows how the wild-type (wt) control and the positive control strain (lacking the antioxidant transcription factor Yap1, and therefore being light-sensitive) that were used to set up the screening conditions respond differently to illumination, depending on the cell density spotted. Agar to agar pinning that results in larger chunks of cells being transferred to the same spot, results in all cells growing, including the positive control (Molin et al., 2020). Thus, for high-throughput and medium throughput screens for which pinning machines are used, liquid-to-agar transfer should be the mode of cell-transfer used to better be able to control the initial cell density on the experimental plates. Care should be taken to ensure that an even number of cells per strain is being transferred.
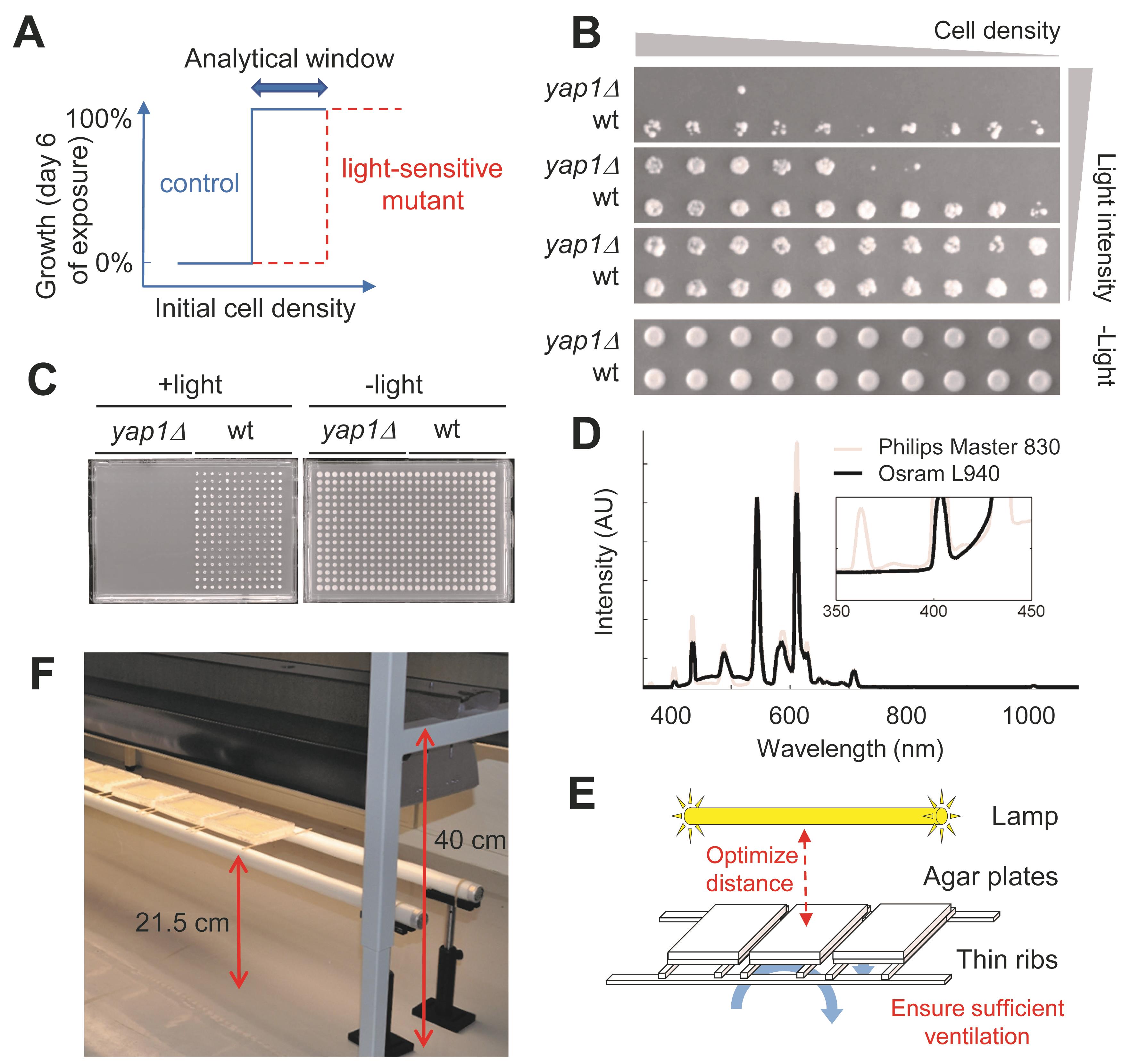
Figure 1. Because of a strong dependence on the initial cell density, and the impact of illumination on temperature, high-throughput screening of yeast mutant libraries for light-sensitivity necessitates several special adaptations. A. Schematic representation of the growth of light-sensitive mutants compared to the control strain, taking into account the intital cell density. The screen is delicate to perform, since the analytical window where light-sensitivity can be scored is rather narrow. B. Both the light intensity and the initial cell density affect the light sensitivity of yeast strains, as observed here on both the wild-type and the yap1∆ mutant strains. C. Images show the contrast in growth between the wild-type and the positive control yap1∆, using the same experimental procedure and format (384 colonies/plate) as used in the genome-wide screening experiments. D. The spectrum of the UV-filtered fluorescent lamp (Osram L 940) in comparison to the spectrum of a normal fluorescent lamp in the lab (Philips Master 830). The inset shows an enlargement of the 350-450 nm range. Note that the peak in the UV range at ~360 nm in the normal lamp is absent for the UV filtered Osram L 940. E-F. Overview (E) and picture (F) of the experimental setup used.Using such a setup, it is possible to generate growth conditions amenable to high-throughput screening, during which the positive control strain yap1∆ does not grow upon illumination, while the control strain grows fine (Figure 1C). Due to the difficulty in achieving an optimal cell density for all strains in a high-throughput screen, it is recommended that each strain is serially pinned onto a number of positions, thus obtaining a range of cell-densities. In our study, serial pinning was done in our first validation test (Molin et al., 2020). However, this procedure could be applied to the primary genome-wide screen as well, but at the cost of handling a much larger number of plates in representing the whole yeast mutant collection. For verification studies with a selected number of mutants, serial drop tests should be used to even better control for comparable initial cell densities during light exposure.
Screening collection preparation step 1 – liquid-to-agar pinning of mutant collection
The yeast deletion collection is stored long-term at -80°C and the 96 well plates need to be thawed at room temperature (RT) before pinning.
Pin from thawed stock source plates, using 96-format long-pin pads onto solid YPD agar experimental plates, in a 384-array format using the pinning robot. The YPD medium in this step ensures good survival and recovery of the frozen cells.
For the source plates, apply 2D target mixing for 10 repetitions at a distance of 0.1 mm from the bottom of the wells, with a mixing speed of 25 mm/s and a mixing diameter of 1 mm. For pinning onto the agar, the pinning pressure is 7%, with an agar overshoot of 2 mm and a pinning speed of 19 mm/s.
Note: The deletion collection contains multiple mutants which are sensitive to being picked up directly onto liquid media. Thus, it is strongly recommended that the collection is taken up onto solid YPD agar first. This also allows condensation of the collection down to a 384 array format, which will use ¼ as many plates. In addition, any missing colonies can often be picked up and inoculated-in manually at a later stage.
Screening collection preparation step 2 – agar-to-liquid pinning of mutant collection
Pin from a 384-array format collection YPD agar stock plates, using 96-format long-pin pads, into flat bottom 96 well plates (manually pre-loaded with 300 µl of YPD per well) using the pinning robot.
Notes:
For efficient stirring, round bottom plates should not be used, since in them cells will collect in the bottom-center of the wells, irrespective of the stirring.
Although not tested here, it should be pointed out that it may be possible to transfer liquid mutant cultures to pinning plates using a liquid handling robot. As a rough estimate of the amounts of liquid culture suitable, we measured the amount of liquid transferred by each pin on the Singer RePads to involve volumes in the sub-μL range (~0.1 μL).
For pinning from the agar source plate, the pinning pressure is 7%, with an agar overshoot of 2 mm, a pinning speed of 19 mm/s, and mixing on the source plate using default values. For the target liquid-medium plates, 2D source mixing for 10 repetitions at a distance of 0.1 mm from the bottom of the wells, with a mixing speed of 25 mm/s, and a mixing diameter of 1 mm is applied.
Note: Applying mixing on the agar source plates allows for more consistent pinning of small colonies, as the pins of the pinning pad effectively cover larger areas.
Primary genome-wide screen – liquid-to-agar pinning onto plates
Preculture cells in 96 well microtitre plates for 48 h at 30°C in 300 μL of liquid YPD medium without agitation.
Note: Although we grew liquid precultures in rich undefined YPD medium (Molin et al., 2020), we see no reason for why it should not be possible to preculture cells in synthetic complete liquid medium instead (similar to the medium we used in the experimental plates but lacking agar). In relation to this, we don't think the YPD medium would be appropriate for the experimental plates because of their coloration, which would influence the exposure to light (in particular the reflected light that travels through the agar).
Before pinning, shake plates at 500 rpm (using the Vibrax VXR basic shaker) for 10 min to ensure proper mixing and optimized/consistent cell transfer. Yeast cells are dense and easily sediment when plates are standing still for too long, which is why we shake plates in batches of 4-5 plates before pinning (the pinning takes about 3-5 min per plate, including manual mounting of plates and refilling of pinning pads). The whole collection of mutants is represented on 61 plates in the 96-well format (including five where slow growing mutants have been gathered) or 16 plates in the 384 format (including two with slow growers). Thus, for the whole mutant collection, the pinning procedure will take a couple of hours.
Pin from liquid preculture source plates (four different 96 well plates per agar experimental plate) using 96-format long-pin pads onto solid agar experimental plates in a 384-array (Figure 2), using the pinning robot and the 1:4 array setup (see the Singer Rotor HDA User manual, v1.2, p. 24, available at SINGER INSTRUMENTS ROTOR HDA USER MANUAL Pdf Download | ManualsLib). Agar target plates for assessing cell growth during light exposure contained synthetic complete medium with 2% agar.
We used the default version of the pinning program. For good pick-up of cells from the liquid culture source plate, it is optimal to use 2D source mixing for 10 repetitions at a distance of 0.1 mm from the bottom of the wells, with a mixing speed of 25 mm/s, and a mixing diameter of 1 mm. For pinning onto the agar target plates, the pinning pressure is 7%, with an agar overshoot of 2 mm, and a pinning speed of 19 mm/s. Each colony is pinned once, since repeated pinning on the same spot will result in a higher number of cells inoculated, which can be detrimental for assessing light-sensitivity.

Figure 2. An example plate in the primary screen for yeast light-sensitivity imaged at different time points of light-exposure along with the corresponding dark control. To identify light sensitive mutants, an automated image analysis algorithm was used to analyze photos of plates exposed to light taken at day 0 and 6, and control plates incubated in the dark at day 0 and 1. Green and orange rings indicate mutant colonies scored as light-sensitive, with a high and moderate confidence, respectively. The red ring denotes a mutant colony growing neither on plates incubated in the dark nor light, illustrating the need to take into account growth under both conditions in scoring light sensitivity. The white rectangles at the border of the plates enclose the wild type controls. High-resolution images of plates that may be used for testing out the scoring algorithm are available in Figures S1-S4.Note: We pinned strains at only one position on the experimental plates, to minimize the number of plates to be handled in the light-exposure set-up. However, a number of technical replicates per plate may be alternatively included, to obtain a better precision in the measurements, at the cost of having to set up a more extensive light-exposure stand. Our strategy was to apply a two-tiered subsequent validation of the positive hits in the initial screen instead (Molin et al., 2020).
It is important to incorporate a high number of wild-type or wild-type-like controls in the mutant collection used, which will later serve as controls for setting the scoring thresholds, and to experimentally estimate the number of false-positives, as explained in the data analysis part of this protocol. This is not provided in the deletion collections listed in Materials and Reagents. We used 76 (in our case his3∆ mutant strain) controls per plate, as realized in the Boone lab setup of the haploid Mat a deletion collection (Tong et al., 2001), adding up to 1,064 controls in total for the whole experiment.
First validation test, medium-throughput assay – liquid-to-agar pinning onto plates using repeated pinning with variable cell densities.
To achieve an improved precision in the final scoring, selected light-sensitive strains from the primary screen may be serially pinned to a number of plate positions, to obtain a concentration gradient of cell-densities (Figure 3A-3B). Using the same pinning procedure as described for the primary screen, but where only one 96 well plate is used to inoculate one 384 format, results in four colonies of each strain on the plate. However, by introducing a time-delay between each of the four pinnings (and in this case, no shaking of plates in-between), cell sedimentation will result in lower amounts of cells being transferred for each pinning round. The default program options should be used for the liquid-to-agar transfer, which means no 2D source mixing. Example images of plates from a medium-throughput follow-up screen performed this way are seen in Figure 3C.
Note: The use of deletion mutants from a different collection than in the primary screen (e.g., the homozygous diploid deletion mutant collection) during the validation tests reduces the risk of cross-contamination influencing the final scoring. In addition, the homozygous diploid deletion mutant collection is expected to be buffered against recessive suppressor mutations that may have accumulated, since any such mutations would most likely be heterozygotic.
Second validation test, low-throughput assay - serial dilution drop test to more precisely control for a lower number of light-sensitive strains
A second confirmation test based on quantitative serial dilutions can be performed (Figure 3D-3E). This method enables a quantitative measure of light sensitivity that ranks the mutants’ rate of growth during illumination (growth during illumination relative to their growth in the dark control). We suggest preparing five tenfold serial dilutions, with a starting dilution of OD610 ~1 (24 h of growth in liquid medium resulted in an OD ~4) and spot 7 μL of each dilution onto SC agar plates prepared as above, which are then either subjected to light-exposure or incubated in the dark.
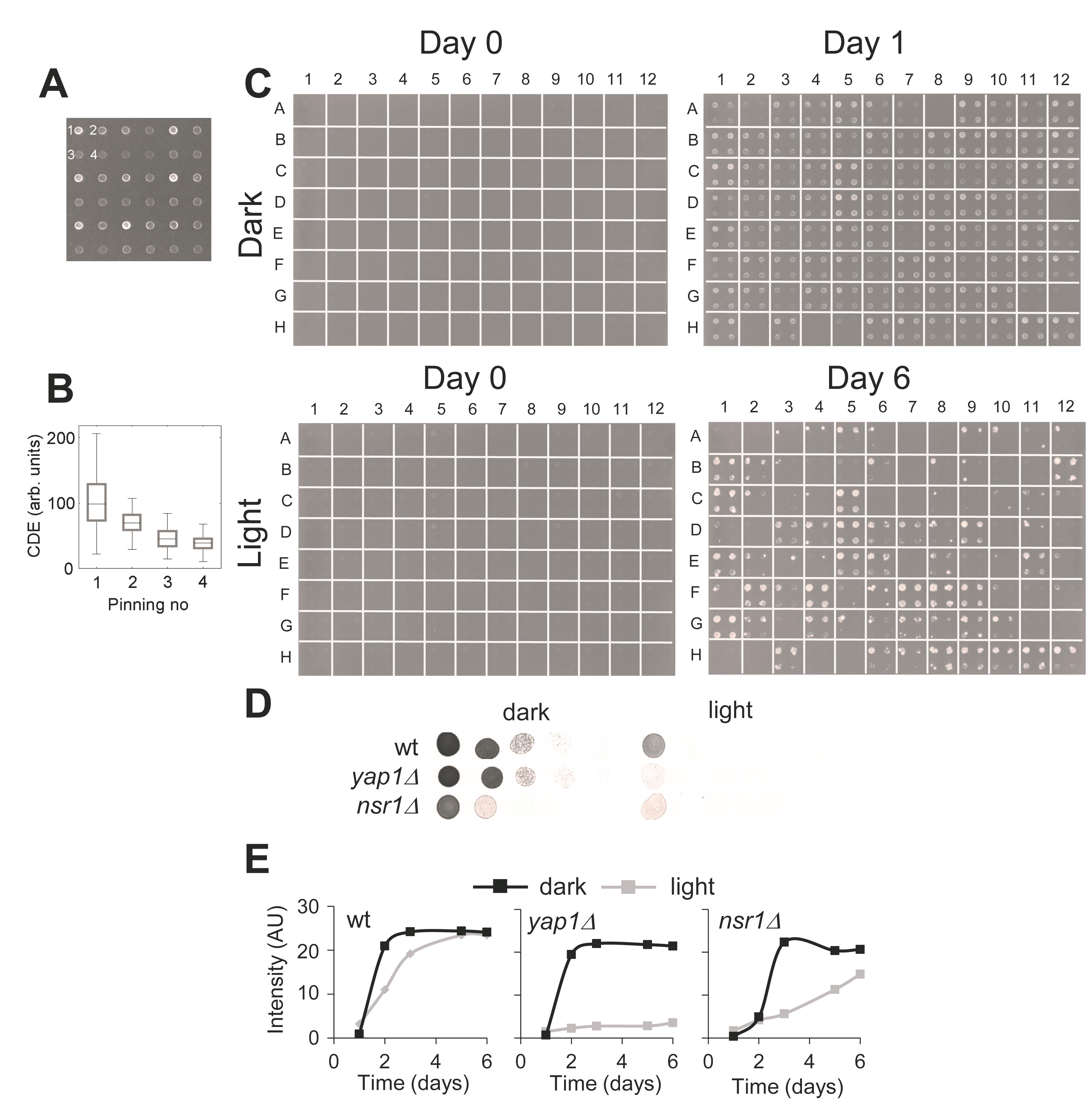
Figure 3. Medium-throughput and low-throughput verification assays suitable for scoring yeast light-sensitivity. A. In the medium-throughput assay, each strain is pinned into four different positions on the plate (like a quadrant). The pinning order is indicated for one of the strains in the upper left corner. A slight delay between the four pinnings was imposed, resulting in a gradient of cells being transferred like in a dilution series. B. Quantification of the average dilutions imposed by the repeated pinnings among all the 96 strains tested. The line in the box indicates the median and box boundaries the 25th and 75th percentiles. C. Example images of plates from medium-throughput verification assay of 96 strains at different time-points. D. Quantitative serial dilution drop test validation assay. Strains are first diluted to the same cell density (OD610 ~1), further diluted in five sequential 10-fold dilutions, and spotted onto agar plates. Plates are either exposed to light or kept in the dark and imaged daily. Images of wild type, yap1∆, and nsr1∆ strains grown on dark control and light exposed plates after 2 days. E. Growth curves of the strains shown in D, where the cell-spot intensity has been estimated for images taken over 6 days.
Experimental setup for light-exposure
In order to minimize UV light induced DNA damage in cells, i.e., to ensure that exclusively growth defects caused by visible light (400-700 nm) are measured, use a light source consisting of a UV radiation-filtered fluorescent lamp, e.g., Osram L 940. We used three light tubes in parallell, 58 W each, fitted in an open standard light tube fitting, equipped with a reflector, but without a deflecting grid. Figure 1D shows the spectrum of this lamp in comparison to a commonly used non-UV filtered fluorescent lamp (Philips Master 830). Spectra from the lamps can be measured directly using a fiber-coupled spectrometer (e.g., QE65000-FL, Oceans Optics). Make sure to measure the spectra through the lid of the agar plate, since the plates will be covered with the lid during the experiment to minimize evaporation. To prevent dehydration of the agar, the plate edges between the lid and the base should be closed with parafilm. To increase the light-intensity, and to get an even distribution of the light irradiation over the plate, a white paper should be fitted to the outside bottom of each plate.
Place the plates below the light source with the top (lid-side) up (Figure 1E-1F). Make sure to measure the light intensities at different distances from the edge of the reflector. In our case, light intensities dropped towards the edges and, hence, we made sure to not put any experimental plates closer than roughly 30 cm from the edge of the lamp (Figure 1F). The distance between the lamp and the plates should be optimized in such a way that a substantial contrast in growth between the wild-type and the positive control yap1∆ is obtained. The on-site intensity should be ~35 W/m2 (in our case with the three 58W lamps, the distance used was 20 cm). As the temperature in the plate is dependent on the distance from the light source and the temperature in the laboratory, the environment around the plates must be controlled to achieve the desired temperature in the plates. In our case, putting the plates on thin ribs (Figure 1E-1F), allowing good ventilation, in a room with a temperature of about 21°C resulted in a temperature of ~30°C in the plates (note that the temperature has to be measured inside the plates), compared to ~34°C if the plates were placed directly on a bench.
The control plates (no illumination) should be kept in the dark at the same temperature as the plates incubated in light (30°C).
Image or scan the plates regularly. For the high- and medium-throughput screens, we imaged the plates after 0, 1, 2, and 6 days, using a 12.3-megapixel digital camera (D5000, Nikon, Japan). Example images taken at different time points during a genome-wide screen are seen in Figure 2. For the low-throughput validation serial drop test, we scanned the plates after 1, 2, 3, 5, and 6 days with a photo scanner (Epson Perfection V700) set in transmission mode (Figure 3D-3E).
Note: To achieve a higher precision in cell number estimations, plates used in the high- and medium-throughput screen may be scanned in transmission mode and cell numbers quantified, like performed in the plate-based Scan-o-matic high-throughput phenotyping method (Zackrisson et al., 2016).
Data analysis
Primary genome-wide screen
Due to the difficulty in transferring an equal number of cells per strain, and because of the cell density dependence of growth, the light-sensitivity of the mutants in the genome-wide screen should be evaluated with care. In order to objectively evaluate the light-sensitive growth, an automated image analysis tool should be used. In our study, we used a set of image analysis algorithms developed in house that were implemented in MATLAB® (Figure 4). The overall design of our image analysis method is based on calculating a cell density estimate, not only measuring the colony area, to obtain the best quantitative precision (Zackrisson et al., 2016). The main reasons for this are two-fold: 1) to give a good estimation of the cell density at day 0, where all cell patches after pinning are of about the same size but might contain different amounts of cells, and 2) to circumvent the drastic spatial effects encountered during yeast growth on plates where colonies on the edges cover much larger areas (Baryshnikova et al., 2010). Our image analysis algorithms were formulated in the following way and can be adopted after proper scaling pixel sizes and intensity values. A cell density estimate (CDE) for each of the 384 colonies per plate was calculated based on the pixel values in a photo (reflective image) of each plate (Figure 4). A CDE corresponds approximately to the sum of the pixel-intensities in a colony after subtracting the local background. The MATLAB® code was designed such that each row of 24 colonies (in our case, each row is 30 pixels high and 1440 pixels wide) was initially analyzed individually. An intensity profile over each row was computed by summing the top 25 pixel values for each column (the 30 pixel-high column, Figure 4A-4C). The rationale behind extracting the sum of each column is to enhance the signal-to-background relation for faint colonies (see e.g. the left-most of the colonies in Figure 4A-4B). Next, the intensity profile for each colony was extracted by stepping over the profile (Figure 4B), with a step of 60 pixels (corresponding to the center-to-center distance between the colonies), with the colony pixel values centered in each of the steps. For each colony profile, the colony intensity was then calculated by taking the median of the top 10 pixel values of the central 20 pixel values (green lines in Figure 4B). The background intensity (blue lines in Figure 4B) was calculated by taking the mean of the minimum summed pixel values on the left and the right side of the colony (corresponding to pixel 5-20 and pixel 40-55) in the profile. The CDE was then calculated by subtracting the background intensity (blue lines in Figure 4B) from the colony intensity (green lines in Figure 4B), using the MATLAB code outlined in Figure 4D.
As the cell concentration at day 0 influences the cells’ growth response during illumination, and because the cell concentration at day 0 varies between the pinned colonies, both the CDE at days 0 and 6 are used in the scoring of light sensitivity. The settings of thresholds for identifying light-sensitive mutants are based on the experimental distribution of the wild-type controls (in our case the 1066 his3∆; treated as wild-type controls). The light-dependent growth of the controls in relation to the initial cell density distributes in a binary fashion as seen in Figure 5A.
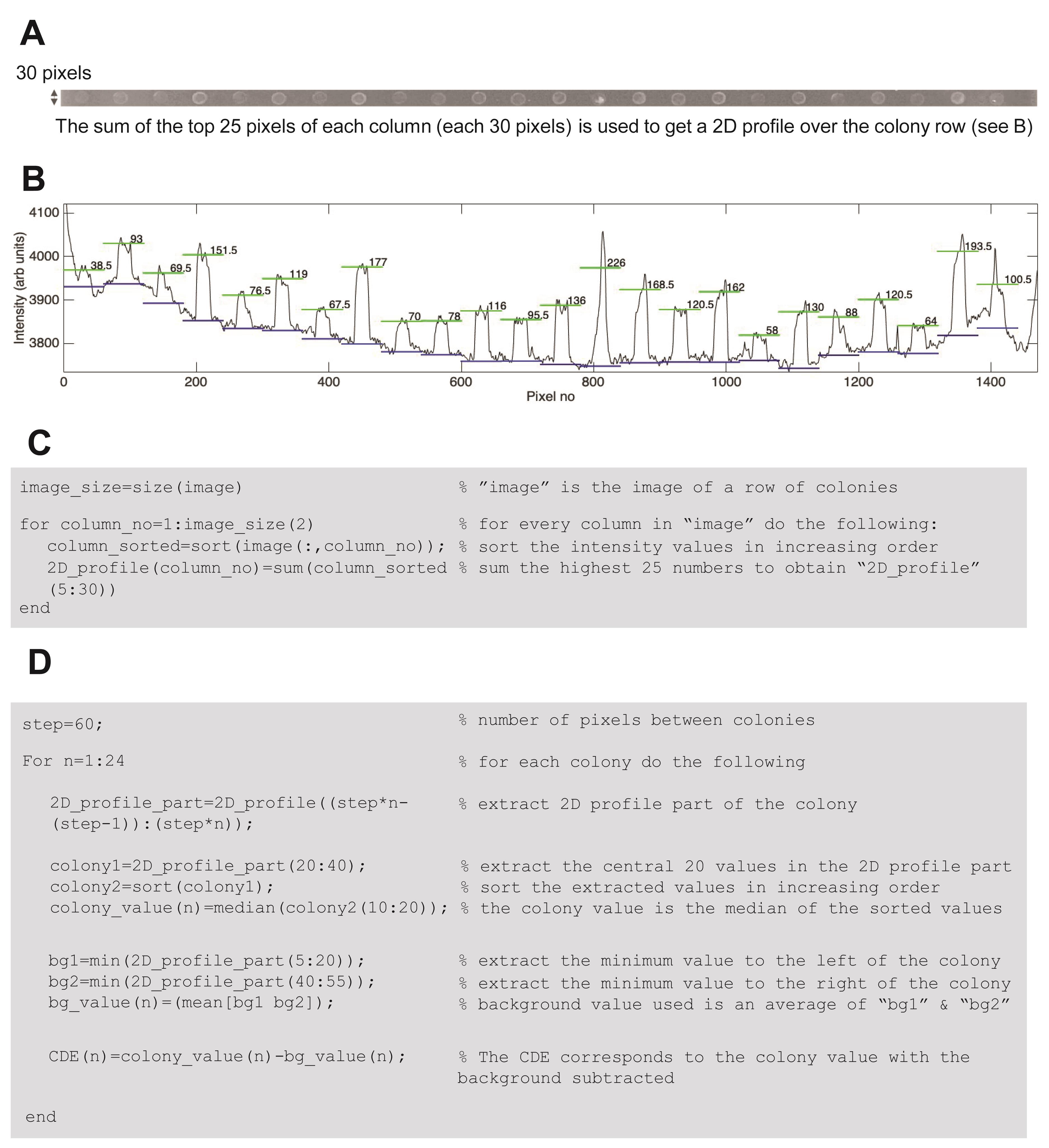
Figure 4. Image analysis algorithm used to estimate yeast growth in light. A. Each plate is divided into 16 rows, each 30 pixels high. B-D. A two-dimensional intensity profile of the 24 colonies in the row is generated, by summing the 25 highest pixels in each column. The median of the top 10 pixels of each colony is used as the colony intensity (green line). Background values for each colony are calculated on each side, and these values should be averaged (blue lines). The CDE used in scoring growth is calculated by subtracting the background from the colony intensity. C-D. The central MATLAB® code used in image analysis.This binary distribution is used to identify what we call “confident sets” for identification of light sensitive mutants. In our case, control colonies with an initial CDEday0 ≥ 121 CDE showed good growth (being light-resistant) under illumination, while colonies with a CDEday0 of < 121 exhibited variable scores, with control colonies exhibiting CDEday0 ≤ 60 all being light-sensitive (Figure 5A). Thus, two different criteria were used to select for light-sensitive mutant strains, based on the response profile of the control cells: (i) a high-confidence set: CDEday0 ≥ 121, and CDEday6 < 900 (Figure 5A-5B, light grey box), and (ii) a moderate-confidence set: 61 ≤ CDEday0 ≤ 120, and CDEday6 < 400 (Figure 5A-5B, dark grey box). The estimated rate of false positives, based on the distribution of the 1066 controls used in our assay, was 0.1%, and approximately 5% for the high- and moderate-confidence sets, respectively (Molin et al., 2020).
To avoid erroneously scoring generally slow-growing deletion strains as light-sensitive, the growth of each mutant under light exposure needs to be compared to its corresponding growth in the dark (Figure 2, Figure 5C). Because growth is much slower under light exposure, it is advisable to compare the growth up until day 6 during light exposure to growth up until day 1 without light exposure. Finally, the light-sensitivity of each deletion strain should be normalized to the light sensitivity of the median of the internal controls (his3∆) present on the same plate, to estimate a double-normalized light growth (normalized to dark growth and to control strain light/dark growth). Therefore, LGnormj for each deletion strain j is:
LGnormj = (LGj/DGj)/(LGctr/DGctr)
where LGj and LGctr are the CDE for deletion strain j and the CDE for controls (n = 384 in our case), respectively, after 6 days of light growth, and DGj and DGctr are the CDE for deletion strain j and CDE for controls, respectively, after 1 day of dark growth. The median of the light/dark growth of the controls on each plate is used, and this is done independently for the high-confidence range and the medium-confidence range of initial cell densities (due to slightly different median values for the two ranges; in case of missing values for any of the ranges, median values from the neighboring plates may be used). Light-sensitive deletion strains in both the primary genome-wide screen and the confirmation assay are called as those with LGnormj < 0.75. It is recommended that very slow growing deletion strains with less than a 1.5-fold increase in colony size during the first day of dark growth are excluded from the analysis, because of insecure data from overall poor growth throughout the measurement period that is independent of light.
For detecting resistant mutants with better growth than the control during illumination, mutants with a CDEday6 > 1642 may be considered resistant [52 mutants in our screen (Molin et al., 2020)] using the same threshold criteria for cell density after pinning, 61 ≤ CDEday0, as for the light-sensitive mutants. However, the analytical window is much tighter for light-resistance and, based on the growth of the controls, the estimated rate of false positives in this case is 20%.
Notes:
Different genome-wide collections of yeast mutants come with specific spatial designs over the plate. We used a design in our screen for light-sensitivity (Molin et al., 2020) that distributed the control strain to the outer regions of plates, as a “shield" for the mutants on the plate against the confounding effects of extra nutrients on the outer sides of plates (Tong and Boone, 2006; Tong et al., 2004). However, one can equally well, and maybe even preferably, use a design where the control strain is evenly distributed over the whole surface of the plate (Zackrisson et al., 2016) to get a spatially distributed response of the controls.
Because the exact values for the threshold-CDEs obtained are going to vary with the setup used, below we describe in short a rationale for how to setup the scoring criteria.
Firstly, evaluate the cell density estimates of the wild-type controls on the plates by plotting their values at day 6 against values at day 0. The logic behind this is that the cell density at the start of the experiment will strongly influence the extent of light-sensitivity observed. Therefore, the cell density inoculated has to be compensated for in the scoring regime applied.
Secondly, decide scoring thresholds for light-sensitivity based on the distribution of wild-type CDEs after light exposure. High confidence scoring thresholds can, for example, be assigned in a manner that the two CDE value thresholds both become low enough that only 0.1% wild-type inoculates grow [light grey box in Figure 5A (Molin et al., 2020)].
Thirdly, because of the distribution of CDE values at day 0 and day 6 in the wild-type controls, this high-confidence set will invariably result in a relatively high number of false negatives i.e. light sensitive mutants being missed. Therefore, it may be advisable to design a second set of slightly more relaxed scoring criteria where CDE day 6 values are indeed very low, but so are day 0 values, in total allowing ~5% of wild-type colonies to grow [dark grey box in Figure 5A (Molin et al., 2020)].
First validation test, medium-throughput assay
In the follow-up medium-throughout study of selected light-sensitive mutants, strains should be scored as for the primary screen, using the experimental distribution of the control strains to define scoring thresholds (as described above). The following thresholds from our screen may serve as a guide line – haploid set: high confidence CDEday0 ≥ 13 and CDEday6 < 900 (no moderate set defined, since all control strains, even at low initial cell densities, grew well during illumination); diploid set: high confidence CDEday0 ≥ 105 and CDEday6 < 900, and moderate confidence 45 ≤ CDEday0 ≤ 104 and CDEday6 < 500. The control plates (no illumination) that were wrapped up in aluminum foil and kept in the dark at 30°C were used to construct dark-normalized light-sensitivities of all mutants as above, using the same 75% threshold in relation to the control as for the primary screen. Examples of differential scoring of light-sensitivity of mutants, dependent on the cell density spotted in the medium-throughput confirmation assay, can be seen in Figure 5D (diploid set).
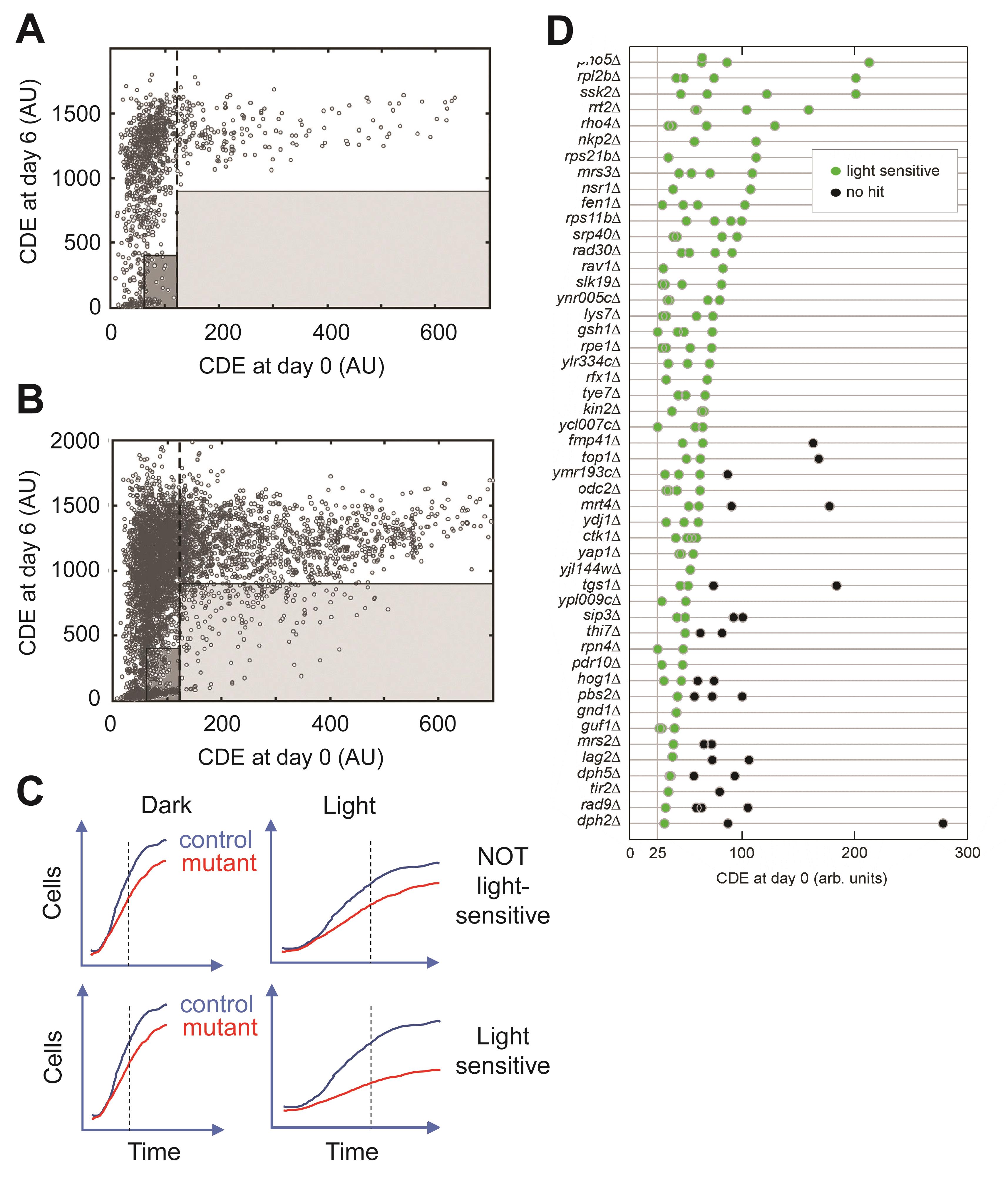
Figure 5. The strong cell density-dependence of yeast light-sensitivity and the slow growth of some mutant strains requires careful set up of scoring criteria. A. Extracted CDEs at day 6 versus day 0 for the control strains (in total 1,064 colonies). High confidence set (light shaded box) and moderate confidence set (dark shaded box) are indicated (see main text for explanation of the scoring – scoring criterion I). B. The light sensitivity distribution of the mutants. High confidence set (light shaded box) and moderate confidence set (dark shaded box). C. Schematic growth curves of light-sensitive and not light-sensitive mutants, according to the scoring procedure used taking both slow growth in light as well as in the dark into account (scoring criterion II). D. The light-sensitivity of 96 mutants selected from the initial genome-wide screen was also confirmed in the medium througput confirmation assay, as a function of the starting cell density. Only the hits based on criterion I are shown. Green dots indicate light-sensitive hits and black dots indicate no light-sensitivity (no hits). The x-axis shows the various initial cell densities (CDE) at day 0. The mutants are sorted according to their light-sensitivity, where a sensitive hit with a high CDE value at day 0 is regarded as a stronger hit than a strain with a lower CDE intensity.Second validation test, low-throughput assay
The plate scans from day 3, of plates incubated in the light, are suitable to be used for quantitation using QuantityOne (Analysis Software version 4.6.8, Figure 3D-3E). To optimize the growth resolution, and to get the best quantitative data, compare the second dilution of the dark control (OD610 ~0.1) to the first dilution (OD610 ~1) of the light-stressed cells, since growth rates differ substantially under light and dark conditions. To compensate for slower growth of the deletion strains in the dark compared to wild type, a relative cell-spot intensity per area unit after background subtraction (INT/mm2) can be applied to calculate the strain-specific light/dark ratio. For a number of slow-growing strains, the light value may be extracted at day 5 instead (at the same dilutions), for a more reliable quantification.
As discussed above (Procedure, step A3d), we chose not to replicate mutant spots on experimental plates, but instead to perform a three-tiered validation analysis. Regarding assay reproducibility, it can be mentioned that we were able to confirm 19 out of the 22 mutants (86%) scored as light-sensitive at high-confidence in the primary screen, i.e., we were able to score them as light-sensitive also in the medium-throughput verification assay (Molin et al., 2020). Meanwhile, 36 of the 52 (69%) scored as light-sensitive at moderate confidence in the primary screen displayed light-sensitivity also in the medium-throughput verification assay. It should also be mentioned that no less than 6 out of the 16 negative calls from the primary screen, displayed light-sensitivity when reassessed in the medium-throughput verification assay, testifying to the large number of false negatives in the high-throughput screening assay setup used (Molin et al., 2020). Thus, in future screens for light-sensitivity, it may be advisable to increase the number of mutant colony replicates scored, as well as to carefully design scoring criteria to reduce the large number of false negative calls.
Using the low-throughput verification assay (spot-tests), we reassessed light-sensitivity for 34 of the mutants from the haploid deletion collection scored as sensitive in the primary screen (Molin et al., 2020). Here, statistical significance was assessed for each strain in relation to the control by two-sided Student’s t-tests, assuming equal variance (in our case n = 2). Out of the 34 mutants from the haploid deletion mutant collection scored as positive in the primary screen, 24 (71%) scored as positive when light-sensitivity of the corresponding homozygous diploid gene knockouts was assessed. As mentioned in the original publication, the low overlap between the primary screen performed in haploid deletion mutants and the follow-up in a different mutant collection (homozygous diploids) may be related to differences in gene-environment effects on growth in haploids vs diploids (Molin et al., 2020). Supporting this idea, only 46 out of 76 mutants (60%) scored as light-sensitive in the primary screen of haploid deletion mutants, when retested using the medium-throughput verification assay in the homozygous diploid deletion collection, as compared to 55 out of 74 (74%) in total, when the medium-throughput verification assay was used on the haploid deletion mutants.
Notes
Notes about the experimental setup including cautionary points and suggestions for improvements have been added to the relevant sections above.
Recipes
For preparing inoculum liquid medium (YPD) for pre-cultures, prepare the following using sterile technique.
40% (w/w) glucose
Dissolve 20 g of glucose in 30 g of Milli-Q filtered water under stirring.
Add ultrapure water up to 50 g total weight.
Autoclave glucose mixture and store at room temperature.
YPD liquid medium
Add 5 g of yeast extract and 10 g of peptone to a suitable flask.
Fill up to 475 mL with Milli-Q filtered water under stirring.
Autoclave YP liquid and allow to cool down.
Add 25 mL of sterile 40% (w/w) glucose solution
10× stock of amino acid mixture
Mix 10 times the suggested amount of Complete Supplement Mixture (CSM) containing all amino acids into 0.1 L of Milli-Q filtered water.
Stir and let it dissolve completely. This can take several hours.
Sterile filter the amino acid mixture into a sterile bottle, for storage at RT.
For casting experimental agar medium plates (20 synthetic complete medium plates), prepare the following using sterile technique.
10× stock of succinate buffered Yeast Nitrogen Base (YNB)
Mix 1.7 g of YNB without Amino Acids and without Ammonium Sulphate, 5 g of ammonium sulfate, 10 g of succinic acid, and Milli-Q filtered water into a total volume of 95 mL.
Let the components dissolve under stirring.
Set pH to 5.8 using NaOH.
Fill up to 0.1 L with Milli-Q filtered water.
Sterile filter 10× succinate buffered YNB into a sterile bottle, for storage at RT.
10× stock of amino acid mixture
Mix 10 times the suggested amount of Complete Supplement Mixture (CSM) containing all amino acids into 0.1 L of Milli-Q filtered water.
Stirr and let it dissolve completely. This can take several hours.
Sterile filter the amino acid mixture into a sterile bottle, for storage at RT.
Sterilize ultrapure water and store at RT.
Agar mixture (should be prepared immediately before plate casting)
Mix 0.75 L of Milli-Q filtered water with 20 g of agar.
Autoclave and immediately proceed to step 8.
Add stock solutions to agar mixture
To the warm freshly autoclaved agar mixture, add while stirring: 100 mL of 10× succinate buffered YNB, 100 mL of 10× CSM mixture, and 50 g of 40% glucose. Add sterile water up to 1,000 mL, and allow complete mixing of the ingredients.
Pour plates using a volume of 50 mL per plate.
Bubbles forming during plate pouring can be removed directly, using a red hot platinum inoculation loop.
Make sure plates are left to solidify on a completely level surface.
To assure a level surface, do not stack plates higher than stacks of four.
As the YPD plates are not used for quantification, they can be prepared by simply adding 2% (wt/vol) agar to the YPD medium prior to autoclaving. Directly after autoclaving and with thorough mixing, the YPD plates should be cast in the same careful manner as described in point 8(b) and 9(c).
Acknowledgments
This study was supported by grants from the Swedish Research Council (VR, #2011-5170; 2020-05422), Cancerfonden (#2017-778), the foundation Olle Engkvist byggmästare, and the Carl Trygger Foundation to MM and the Swedish Research Council (VR) to AB (#621-2007-5421). This protocol is derived from the original research article Molin et al. (2020); doi: 10.1186/s12915-020-00867-4.
Competing interests
The authors declare no competing interests.
References
- Baryshnikova, A., Costanzo, M., Kim, Y., Ding, H., Koh, J., Toufighi, K., Youn, J. Y., Ou, J., San Luis, B. J., Bandyopadhyay, S., et al. (2010). Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat Methods 7(12): 1017-1024.
- Bodvard, K., Peeters, K., Roger, F., Romanov, N., Igbaria, A., Welkenhuysen, N., Palais, G., Reiter, W., Toledano, M. B., Kall, M. and Molin, M. (2017). Light-sensing via hydrogen peroxide and a peroxiredoxin. Nat Commun 8: 14791.
- Chen, C. H., Ringelberg, C. S., Gross, R. H., Dunlap, J. C. and Loros, J. J. (2009). Genome-wide analysis of light-inducible responses reveals hierarchical light signalling in Neurospora. EMBO J 28(8): 1029-1042.
- D'Orazio, K. N., Wu, C. C., Sinha, N., Loll-Krippleber, R., Brown, G. W. and Green, R. (2019). The endonuclease Cue2 cleaves mRNAs at stalled ribosomes during No Go Decay. eLife 8: e49117.
- Dong, W., Tang, X., Yu, Y., Nilsen, R., Kim, R., Griffith, J., Arnold, J. and Schuttler, H. B. (2008). Systems biology of the clock in Neurospora crassa. PLoS One 3(8): e3105.
- Edgar, R. S., Green, E. W., Zhao, Y., van Ooijen, G., Olmedo, M., Qin, X., Xu, Y., Pan, M., Valekunja, U. K., Feeney, K. A., et al. (2012). Peroxiredoxins are conserved markers of circadian rhythms. Nature 485(7399): 459-464.
- Giaever, G., Chu, A. M., Ni, L., Connelly, C., Riles, L., Veronneau, S., Dow, S., Lucau-Danila, A., Anderson, K., Andre, B., et al. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature 418(6896): 387-391.
- Idnurm, A., Verma, S. and Corrochano, L. M. (2010). A glimpse into the basis of vision in the kingdom Mycota. Fungal Genet Biol 47(11): 881-892.
- Kainth, P., Sassi, H. E., Pena-Castillo, L., Chua, G., Hughes, T. R. and Andrews, B. (2009). Comprehensive genetic analysis of transcription factor pathways using a dual reporter gene system in budding yeast. Methods 48(3): 258-264.
- Lewis, Z. A., Correa, A., Schwerdtfeger, C., Link, K. L., Xie, X., Gomer, R. H., Thomas, T., Ebbole, D. J. and Bell-Pedersen, D. (2002). Overexpression of White Collar-1 (WC-1) activates circadian clock-associated genes, but is not sufficient to induce most light-regulated gene expression in Neurospora crassa. Mol Microbiol 45(4): 917-931.
- Molin, M., Logg, K., Bodvard, K., Peeters, K., Forsmark, A., Roger, F., Jörhov, A., Mishra, N., Billod, J.-M., Amir, S., et al. (2020). Protein kinase A controls yeast growth in visible light.BMC Biology 18(1): 168.
- Munoz-Guzman, F., Caballero, V. and Larrondo, L. F. (2021). A Global Search for novel Transcription Factors impacting the Neurospora crassa Circadian Clock. G3 (Bethesda).
- O'Neill, J. S. and Reddy, A. B. (2011). Circadian clocks in human red blood cells. Nature 469(7331): 498-503.
- O'Neill, J. S., van Ooijen, G., Dixon, L. E., Troein, C., Corellou, F., Bouget, F. Y., Reddy, A. B. and Millar, A. J. (2011). Circadian rhythms persist without transcription in a eukaryote. Nature 469(7331): 554-558.
- Patke, A., Young, M. W. and Axelrod, S. (2020). Molecular mechanisms and physiological importance of circadian rhythms. Nat Rev Mol Cell Biol 21(2): 67-84.
- Tong, A. H. and Boone, C. (2006). Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol Biol 313: 171-192.
- Tong, A. H., Evangelista, M., Parsons, A. B., Xu, H., Bader, G. D., Page, N., Robinson, M., Raghibizadeh, S., Hogue, C. W., Bussey, H., et al. (2001). Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294(5550): 2364-2368.
- Tong, A. H., Lesage, G., Bader, G. D., Ding, H., Xu, H., Xin, X., Young, J., Berriz, G. F., Brost, R. L., Chang, M., et al. (2004). Global mapping of the yeast genetic interaction network. Science 303(5659): 808-813.
- Wu, C., Yang, F., Smith, K. M., Peterson, M., Dekhang, R., Zhang, Y., Zucker, J., Bredeweg, E. L., Mallappa, C., Zhou, X., et al. (2014). Genome-wide characterization of light-regulated genes in Neurospora crassa. G3 (Bethesda) 4(9): 1731-1745.
- Yu, Z., Armant, O. and Fischer, R. (2016). Fungi use the SakA (HogA) pathway for phytochrome-dependent light signalling. Nat Microbiol 1: 16019.
- Yu, Z. and Fischer, R. (2019). Light sensing and responses in fungi. Nature Reviews Microbiology 17(1): 25-36.
- Zackrisson, M., Hallin, J., Ottosson, L. G., Dahl, P., Fernandez-Parada, E., Landstrom, E., Fernandez-Ricaud, L., Kaferle, P., Skyman, A., Stenberg, S., et al. (2016). Scan-o-matic: High-Resolution Microbial Phenomics at a Massive Scale. G3 (Bethesda) 6(9): 3003-3014.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Logg, K., Andersson, M., Blomberg, A. and Molin, M. (2022). High-throughput Growth Measurements of Yeast Exposed to Visible Light . Bio-protocol 12(2): e4292. DOI: 10.21769/BioProtoc.4292.
Category
Microbiology > Microbial physiology > Stress response
Microbiology > Microbial signaling > Sensory receptor
Cell Biology > Cell-based analysis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.