- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Real-Time Analysis of Mitochondrial Electron Transport Chain Function in Toxoplasma gondii Parasites Using a Seahorse XFe96 Extracellular Flux Analyzer
Published: Vol 12, Iss 1, Jan 5, 2022 DOI: 10.21769/BioProtoc.4288 Views: 4207
Reviewed by: Alexandros AlexandratosAndrew MacLeanSebastien Besteiro
Original research article
The authors used this protocol in:

Feb 2021
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


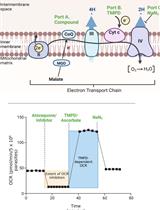
Abstract
The mitochondrial electron transport chain (ETC) performs several critical biological functions, including maintaining mitochondrial membrane potential, serving as an electron sink for important metabolic pathways, and contributing to the generation of ATP via oxidative phosphorylation. The ETC is important for the survival of many eukaryotic organisms, including intracellular parasites such as the apicomplexan Toxoplasma gondii. The ETC of T. gondii and related parasites differs in several ways from the ETC of the mammalian host cells they infect, and can be targeted by anti-parasitic drugs, including the clinically used compound atovaquone. To characterize the function of novel ETC proteins found in the parasite and to identify new ETC inhibitors, a scalable assay that assesses both ETC function and non-mitochondrial parasite metabolism (e.g., glycolysis) is desirable. Here, we describe methods to measure the oxygen consumption rate (OCR) of intact T. gondii parasites and thereby assess ETC function, while simultaneously measuring the extracellular acidification rate (ECAR) as a measure of general parasite metabolism, using a Seahorse XFe96 extracellular flux analyzer. We also describe a method to pinpoint the location of ETC defects and/or the targets of inhibitors, using permeabilized T. gondii parasites. We have successfully used these methods to investigate the function of T. gondii proteins, including the apicomplexan parasite-specific protein subunit TgQCR11 of the coenzyme Q:cytochrome c oxidoreductase complex (ETC Complex III). We note that these methods are also amenable to screening compound libraries to identify candidate ETC inhibitors.
Background
Parasites in the phylum Apicomplexa cause a number of serious diseases in humans and other animals, including toxoplasmosis (Toxoplasma gondii) and malaria (Plasmodium species). Up to a third of the global human population is infected with T. gondii, making it one of the most successful parasites (Pappas et al., 2009). Several anti-parasitic compounds act by targeting the parasite mitochondrion (Alday and Doggett, 2017; Smith et al., 2021); for instance, the clinically used drug atovaquone inhibits the mitochondrial electron transport chain (ETC) (Fry and Pudney, 1992; Pfefferkorn et al., 1993; McFadden et al., 2000).
The ETC is composed of a series of multi-protein complexes (I-V) embedded in the inner mitochondrial membrane. The ETC functions to transport electrons derived from cellular metabolism through ETC Complexes I-IV to the final electron acceptor, oxygen. In mammalian cells, numerous dehydrogenases, including a large multimeric NADH dehydrogenase complex (Complex I) and a succinate dehydrogenase complex (Complex II), oxidize their substrates and reduce coenzyme Q (CoQ), a molecule that functions as an electron shuttle in the inner mitochondrial membrane (Hayward and van Dooren, 2019). CoQ binds to the so called Qo site of Complex III of the ETC, donating two electrons to iron-based co-factors in this complex. In a process known as the Q-cycle, one electron is passed via the iron-sulfur cluster of the Rieske protein and the heme group of cytochrome c1 to the soluble intermembrane space protein cytochrome c, while the other is passed via heme groups of cytochrome b to the Qi site, where CoQ docks and accepts the electron (Mitchell, 1975). Reduced cytochrome c transports electrons to Complex IV, which ultimately consumes oxygen as the final electron acceptor. In many organisms, electron transport through Complexes I, III, and IV is coupled to translocation of protons from the mitochondrial matrix into the intermembrane space, generating a proton motive force across the inner mitochondrial membrane. This proton gradient is harvested by ATP synthase (Complex V) to generate ATP, as well as being important for mitochondrial processes such as protein import (Schmidt et al., 2010).
The ETC of apicomplexan parasites differs in numerous ways from that of mammalian hosts such as humans (Figure 1) (Hayward and van Dooren, 2019). For instance, the large multimeric ETC Complex I found in mammalian cells is missing in T. gondii, and is replaced instead by single subunit NADH dehydrogenases (termed NDH2-I and NDH2-II), which are able to feed electrons from NADH to CoQ but do not contribute to the proton gradient (Saleh et al., 2007; Lin et al., 2008). The genome of T. gondii encodes additional dehydrogenases that oxidize substrates such as malate, glycerol 3-phosphate, dihydroorotate, and succinate, in each instance donating the electrons directly to CoQ (Figure 1). As in mammals, electron transport through Complexes III and IV in the parasite is coupled to proton translocation across the mitochondrial inner membrane, and oxygen is consumed as the terminal electron acceptor at Complex IV. However, the ETC complexes III, IV and V of T. gondii and related parasites contain many additional subunits that are not found in their mammalian counterparts (Huet et al., 2018; Seidi et al., 2018; Salunke et al., 2018; Evers et al., 2021; Hayward et al., 2021; Maclean et al., 2021; Mühleip et al., 2021). Furthermore, the CoQ binding sites of Complex III are sufficiently different from the equivalent sites in the mammalian complex to serve as the targets for anti-parasitic drugs, such as atovaquone and endochin-like quinolones (Fry and Pudney, 1992; Doggett et al., 2012; Fisher et al., 2020). There are currently few options available to assess ETC function in apicomplexan parasites. Those previously used include determination of O2 consumption using a Clark electrode (Vercesi et al., 1998), measurements of mitochondrial membrane potential using dyes such as rhodamine 123 (Divo et al., 1985), 3,3′dihexyloxacarbocyanine iodide (Srivastava et al., 1997), safranin O (Vercesi et al., 1998), and JC-1 (Brooks et al., 2010), and spectrophotometric assays to measure cytochrome c reduction (Fry and Beesley, 1991). Therefore, developing new methods to assess parasite ETC function are of interest both to study novel aspects of parasite biology/metabolism and to identify new anti-parasitic compounds.

Figure 1. The mitochondrial electron transport chain of T. gondii parasites. Dehydrogenases (pink) of the inner mitochondrial membrane (IMM) – namely, glycerol 3-phosphate dehydrogenase (G3PDH), dihydrooratate dehydrogenase (DHODH), succinate dehydrogenase (SDH; Complex II), malate:quinone oxidoreductase (MQO) and type 2 NADH dehydrogenases (NDH2) – donate electrons to coenzyme Q (CoQ; yellow) via oxidation of their substrates. CoQ interacts with ubiquinone:cytochrome c oxidoreductase (Complex III; orange) at the Qo and Qi sites in a process called the Q cycle. These interactions transfer electrons from CoQ to a soluble protein of the intermembrane space (IMS) called cytochrome c (CytC, dark red), as well as translocating protons (H+) from the mitochondrial matrix (MM) into the IMS. CytC donates electrons to cytochrome c oxidase (Complex IV; green), which consumes oxygen as the terminal electron acceptor and also contributes to the proton gradient across the IMM. The proton motive force generated by Complexes III and IV is harnessed by adenosine triphosphate (ATP) synthase (Complex V; purple), which couples the rotation of the F0 domain (caused by movement of protons from the IMS back into the MM) to ATP synthesis at the F1 catalytic domain. Modified from Hayward and van Dooren (2019).
Given that the primary cellular process that utilizes oxygen is the mitochondrial ETC, many methods used to assess ETC function measure changes in the concentration of dissolved oxygen in the assay medium, thereby calculating the oxygen consumption rate (OCR) of cells or isolated mitochondria. A traditional way to measure OCR is by polarography using a Clark electrode (Clark et al., 1953), which uses an electrochemical oxygen electrode to measure changes in the dissolved oxygen concentration in a suspension of cells/mitochondria, one sample at a time, in a closed chamber. A more recent technology, the Seahorse XFe96 analyzer, uses two solid state optical sensor probes to simultaneously measure the dissolved oxygen and proton concentration (i.e., the pH) in the assay medium surrounding cells in a high-throughput 96 well plate format, to derive the OCR and the extracellular acidification rate (ECAR), respectively. The sensor cartridge contains four ports above each well that enable the injection of compounds (e.g., substrates or inhibitors) at predetermined time points during the assay. ECAR is typically interpreted as a measure of glycolytic activity, since the extrusion of lactate (an end product of glycolysis) from cells is coupled with the extrusion of a proton, thus acidifying the culture medium (Zeng et al., 2021). However, as other metabolic pathways that produce and extrude H+ into the medium also contribute to ECAR (Moreno et al., 1998; Mookerjee et al., 2015), the interpretation that ECAR only assesses glycolytic activity is probably an oversimplification. The Seahorse XFe96 analyzer has been used successfully to assess mitochondrial function in several protozoan parasites, including Plasmodium falciparum (Sakata-Kato and Wirth, 2016) and Trypanosoma cruzi (Shah-Simpson et al., 2016).
Recent studies in our lab have used the Seahorse XFe96 system to measure the OCR and ECAR of intact extracellular T. gondii parasites, and thereby characterized the function of novel parasite-specific subunits of ETC Complexes III (Hayward et al., 2021), IV (Seidi et al., 2018), and V (Huet et al., 2018) (Figure 2). We have also utilized these assays to measure the effects of the loss of biosynthetic pathways for co-factors and prosthetic groups that play key roles in the ETC, such as heme (Tjhin et al., 2020) and iron-sulfur clusters (Aw et al., 2021). Additionally, we have developed methods to pinpoint the site of ETC dysfunction by assessing the OCR of permeabilized parasites supplied with different ETC substrates (Hayward et al., 2021). We note that these methods are also amenable to screening compound libraries for identifying inhibitors of the ETC, and for determining the targets of hit compounds. Here, we describe these experimental methods in detail as useful tools to investigate and dissect mitochondrial ETC function in T. gondii parasites.
Figure 2. Overview of the Seahorse XFe96 assay procedure (modelled after Plitzko and Loesgen, 2018).
Materials and Reagents
25 cm2 tissue culture flasks (Greiner Bio-One, catalog number: 690175)
Sterile filter tips (20, 200 and 1,250 μL) (Interpath, catalog numbers: 24500, 24700, 24900)
Sterile serological pipettes (10, 25 mL) (Sigma, catalog numbers: CLS4489-200EA, CLS4488-200EA)
Sterile 2 mL aspiration pipettes (Greiner Bio-One, catalog number: 710183)
Cell scrapers (Sarstedt, catalog number: 83.3951)
Needle hypodermic 26 G × 13 mm (Terumo, catalog number: NN.2613R)
10 mL Luer Lock syringes (Terumo, catalog number: SS10L)
Millex-SV syringe filter unit 5 μM PVDF (Millipore, catalog number: SLSV025LS) or 25 mm Whatman Nuclepore 3 µm polycarbonate membrane filters (Cytiva, catalog number: 10418306). The 3 µm polycarbonate filters must be assembled into reusable 25 mm Whatman polypropylene Swin-Lok filters (Cytiva, catalog number: 420200).
Polypropylene centrifuge tubes (15, 50 mL) (Greiner Bio-One, catalog numbers: 188271, 227261)
Sterile Eppendorf tubes (1.5 mL) (Axygen, catalog number: MCT-150-C)
Reagent reservoirs (Sigma, catalog number: R1400)
Seahorse XF96 FluxPak (culture plates, sensor cartridge, calibrant; Agilent, catalog number: 102416-100)
Seahorse XF base medium (Agilent, catalog number: 102353-100)
Sodium bicarbonate (Sigma, catalog number: S6297-1KG)
Cell-Tak adhesive (Corning, catalog number: 354241)
L-glutamine (Sigma, catalog number: G8540-100G)
D-glucose (BDH AnalaR, catalog number: 101174Y)
Potassium hydroxide (AJAX-Finechem Univar, catalog number: AJA405-500G)
Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (Sigma, catalog number: C2920-10MG)
Dimethyl sulfoxide (DMSO) (Sigma, catalog number: D8418-250G)
Digitonin (Sigma, catalog number: D141-500MG)
L-malic acid (Sigma, catalog number: M1000-100G)
sn-glycerol 3-phosphate bis(cyclohexylammonium) salt (Sigma, catalog number: G7886-1G)
Atovaquone (Sigma, catalog number: A7986-10MG)
N,N,N,N’-tetramethyl-p-phenylenediamine (TMPD) (Sigma, catalog number: T7394-5G)
L-ascorbic acid (Sigma, catalog number: A5960-25G)
Sodium azide (AJAX-Finechem Univar, catalog number: AJA1222-100G)
Ethylene glycol-bis(2-amino-ethylether)-N,N,N,N’-tetraacetic acid (EGTA) (Sigma, catalog number: E3889-25G)
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (Sigma, catalog number: H3375-250G)
Potassium phosphate monobasic (KH2PO4) (ChemSupply, catalog number: PA009-500G)
Magnesium chloride hexahydrate (MgCl2·6H2O) (AJAX-Finechem Univar, catalog number: AJA296-500G)
D-mannitol (Sigma, catalog number: M9546-250G)
Sucrose (Sigma, catalog number: S9378-5KG)
Essentially fatty acid free bovine serum albumin (FAF-BSA) (Sigma, catalog number: A3803-10G)
MilliQ H2O
Sodium bicarbonate (0.1 M) (see Recipes)
Glutamine stock (200 mM) (see Recipes)
Glucose stock (1 M) (see Recipes)
Base medium with glucose (5 mM) and glutamine (1 mM) (BM+Glc+Gln) (see Recipes)
FCCP stock (50 mM) (see Recipes)
Atovaquone stock (10 mM) (see Recipes)
Digitonin stock (5% w/v) (see Recipes)
Mitochondrial Assay Solution (MAS) buffer (1×, 500 mL) (see Recipes)
Malate stock (320 mM) (see Recipes)
Glycerol 3-phosphate stock (200 mM) (see Recipes)
TMPD stock (500 mM) (see Recipes)
Ascorbate stock (200 mM) (see Recipes)
Sodium azide stock (110 mM) (see Recipes)
Equipment
Pipettes 0.1-2.5 μL, 2-20 μL, 20-200 μL and 100-1,000 μL (Eppendorf, catalog numbers: 3120000011, 3120000038, 3120000054, 3120000062)
Multichannel pipettes 10-100 μL and 30-300 μL (Eppendorf, catalog numbers: 3122000035, 3122000051)
Seahorse XFe96 Analyzer (Agilent)
Humidified, 37°C, non-CO2 incubator
Humidified, 37°C, 5% CO2 incubator
Hemocytometer, Improved Neubauer bright line cell counting chamber (Thermo Fisher Scientific, 81002.04)
Standard inverted light microscope (Olympus CKX41), fitted with 4×, 10×, 20× and 40× objective lenses
Biological Safety Cabinet Class II
Aspirator vacuum pump
Water bath, 37°C
4°C fridge and -20°C freezer
pH meter
Software
Wave Software (Agilent)
Excel (Microsoft)
Python (version 3.7 or higher)
R (version 4.0.5 or higher)
GraphPad Prism (GraphPad)
Procedure
See Figure 2 for an overview of the various procedures.
Preparation before the day of the assay
Adjust the Seahorse XF base medium to pH 7.4.
Prepare stock solutions of all drugs and reagents listed in the recipes section.
Note: Even though the stock solutions can be prepared in advance, it is important to check the pH of all reagents is 7.4 again before use in the assay.
Culture T. gondii parasites so that they egress from the host cells (or are present in large vacuoles inside the host cell) on the day of the assay. Some points to consider:
Parasites should not be extracellular for too long before the assay. This is to ensure parasites are assayed when they are at their most healthy/viable.
Note: Though we have not done this routinely in our experiments, the infected host-cell flask could be gently washed the evening before the assay to remove any extracellular parasites that have egressed prematurely.
We frequently use conditional knockdown parasite lines [e.g., where the gene encoding the protein of interest is under the control of an anhydrotetracycline (ATc) regulatable promoter]; in such instances, pre-culture parasites for the required amount of time in ATc (e.g., 0-3 days) before the assay.
We inoculate wells of the Seahorse XFe96 culture plate with 1.5 × 106 parasites/well, and test each condition in triplicate (so 4.5 × 106 parasite cells are required per condition). One 25 cm2 tissue culture flask containing a monolayer of human foreskin fibroblasts yields ~2.5-10 × 107 parasites, and so is usually sufficient for an experiment. If more parasites are required, inoculate multiple 25 cm2 tissue culture flasks, or inoculate larger (e.g., 175 cm2) tissue culture flasks.
Note: The number of parasites inoculated per well may need to be optimized depending on the metabolism of the T. gondii strain being tested. To do this, we recommend titrating different number of parasites in a preliminary experiment.
All the parasite strains you would like to compare should be included on the same plate. This is because we apply a linear mixed effects model in our data analysis pipeline (discussed in the Data analysis section), which compares conditions within a plate to recognize the variation between replicates on different days, and thereby measures the treatment effect as precisely as possible.
Coat a 96 well Seahorse XFe96 culture plate with Cell-Tak. Perform this procedure in a Biological Safety Cabinet to keep the plate sterile.
Combine 750 μL of sterile sodium bicarbonate (0.1 M) with 15 μL of Cell-Tak in a 1.5 mL Eppendorf tube and mix thoroughly. This activates the Cell-Tak and allows it to polymerize and adhere to plastic.
Aliquot 15 μL of this mixture into half the wells of the 96 well plate (48 wells).
Make a second batch of Cell-Tak solution, as in step a, and aliquot into the second half of the plate, as in b. Two batches are recommended to ensure the solution does not polymerize too quickly.
Gently tap the plate to remove any air bubbles from the bottom of the wells, then incubate for 20-30 min at room temperature (RT) to allow the Cell-Tak to adhere to the bottom of each well.
Remove the Cell-Tak solution and wash each well 3 times with 100 μL of sterile MilliQ water to remove residual sodium bicarbonate from the wells.
Remove the final wash and allow the plate to dry.
In our experience, Cell-Tak-coated 96 well Seahorse XFe96 culture plates can be stored in the fridge in a zip-lock bag for up to a week before the assay.
The evening before the assay, hydrate the Seahorse XFe96 sensor cartridge according to the manufacturer’s instructions.
The current manufacturer’s instructions are to add 200 μL of sterile water per well into the calibrant plate and lower the sensor cartridge into this (Figure 3).
To properly hydrate each of the 96 sensor probes, you must ensure that no air bubbles are trapped beneath the sensor probes.
To dislodge air bubbles, gently raise and lower the sensor cartridge into the water in the calibrant plate several times.
Inspect for remaining trapped bubbles and, if necessary, repeat Step 5c.
The plate and cartridge should then be incubated overnight in a humidified incubator set at 37°C and at atmospheric CO2. A 50 mL aliquot of XF calibrant solution should also be kept in the 37°C incubator overnight to degas.
Pre-calculate appropriate concentrations of drugs.
The Seahorse XFe96 system is set up so that 25 μL volumes are injected into each of the 96 wells in the Seahorse XFe96 culture plate from ports A-D of the sensor cartridge during the assay (Figure 3).
The volume in each of the 96 wells in the Seahorse XFe96 culture plate at the start of the assay is 175 μL. Therefore, solutions injected from port A must be prepared at 8 times the desired final concentration in the well, to account for volume change (25 μL in 175 μL = 1:8 dilution). Similarly, solutions injected from port B must be 9 ×, port C 10 ×, and port D 11 × the desired final concentration to account for the sequential volume change.
The drugs should be prepared in the medium being used in the assay [i.e., either Seahorse XF base medium or mitochondrial assay solution (MAS) buffer]. A volume of 25 μL is loaded into each injection port, so calculate the amount of drug needed for the number of wells required (e.g., 48 wells × 25 μL = 1.2 mL). Make excess if economical to allow loading the ports with a multichannel pipette.
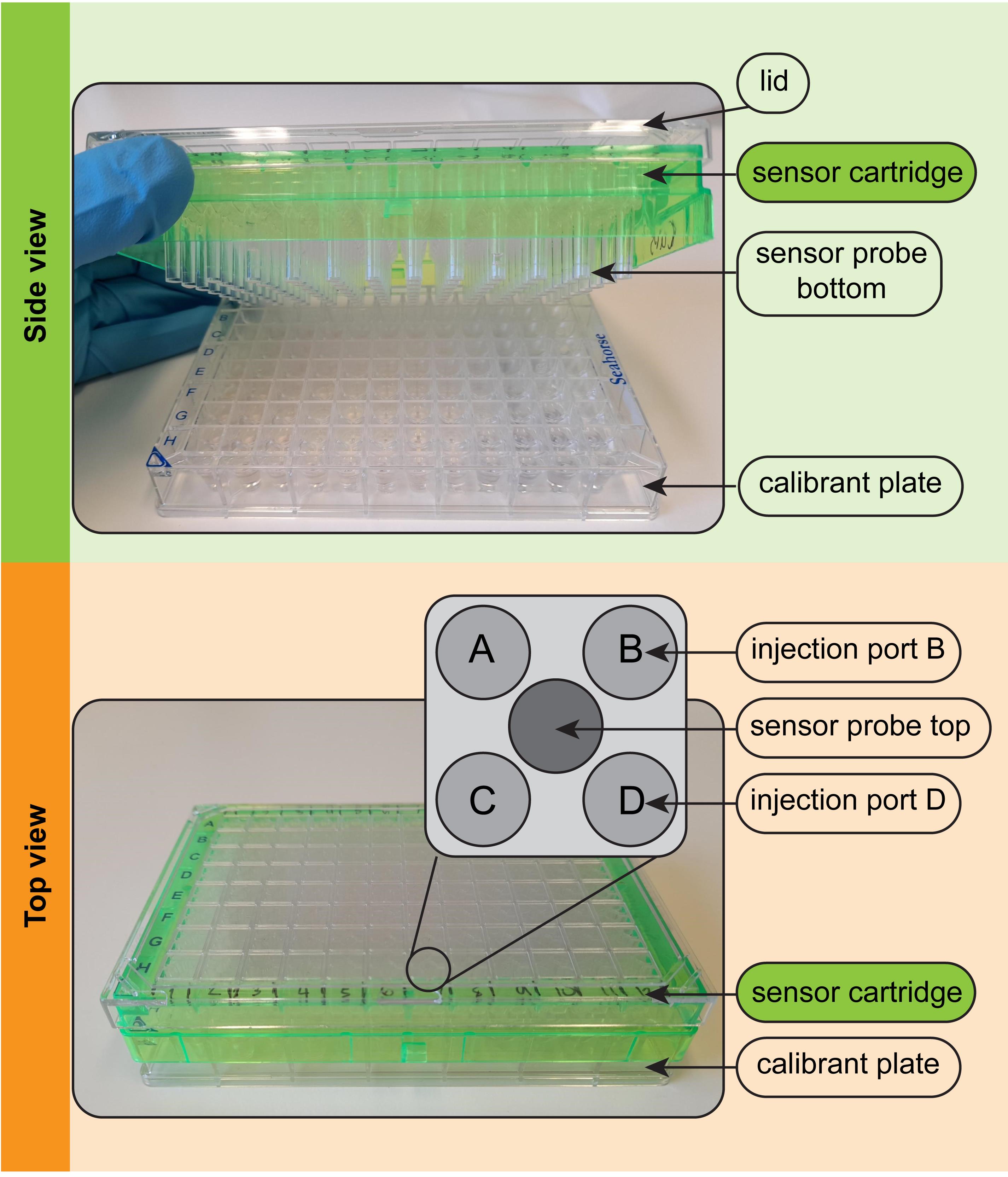
Figure 3. Parts of the Seahorse XFe96 sensor cartridge. (Top) In side view, the sensor probes attached to the green sensor cartridge can be seen. The probes contain solid state optical sensors that must be hydrated overnight before use in the assay. (Bottom) In top view, the four injection ports above each of the 96 wells can be seen. The inset depicts the positions of ports A-D and the central sensor probe. Compounds are injected via ports A-D into the well below at user-defined times during the assay.
Measurement of OCR and ECAR of intact parasites
This method describes the measurement of OCR and ECAR of intact extracellular T. gondii parasites that are supplied glucose and glutamine as substrates (Figure 4A). This is based on the typical mitochondrial stress test (MitoStress) assay used to assess mitochondrial function in mammalian cells (Divakaruni et al., 2014). Basal OCR and ECAR are initially measured to determine baseline ETC activity and general metabolic activity of the parasite, respectively (Figure 4B and 4C). The protonophore FCCP is then injected (via port A) to uncouple electron transport from ATP synthesis, allowing the maximal OCR and spare capacity to be calculated (Figure 4A and 4B). Finally, the ETC Complex III inhibitor atovaquone is injected (via port B) to assess non-mitochondrial OCR (Figure 4A and 4B).
Our assay differs from a typical MitoStress assay in two main ways. Firstly, oligomycin is typically injected before FCCP to inhibit ATP synthase (Complex V) by blocking the proton channel formed by the F0 domain (Symersky et al., 2012). The addition of oligomycin therefore assesses proton leak and ATP-linked respiration (Divakaruni et al., 2014). However, as oligomycin has limited effect on T. gondii parasite OCR in our hands, we do not include it in our assay. Why oligomycin has limited effect in our assays is unclear. The ATP synthase complex of T. gondii differs considerably in protein composition (Huet et al., 2018; Salunke et al., 2018) and structure (Mühleip et al., 2021) from that of many other eukaryotes, and it is conceivable that oligomycin does not bind effectively to the protein complex. Alternatively, it is conceivable that the ETC of extracellular T. gondii tachyzoites is largely uncoupled from ATP synthase activity, in which case inhibiting ATP synthase would have minimal effect on OCR [although, we note that the addition of FCCP causes an increase in parasite OCR (Figure 4D), implying that there is some coupling between ATP synthesis and electron transport in T. gondii]. Secondly, the ETC Complex I inhibitor rotenone is commonly combined with the ETC Complex III inhibitor antimycin A to assess non-mitochondrial OCR (Divakaruni et al., 2014). As Complex I is absent from T. gondii parasites (Hayward and van Dooren, 2019), rotenone is also not included in our assay.
We frequently use this method when characterizing the function of novel T. gondii proteins, to determine whether mutant parasites that lack the protein-of-interest have a defect in ETC function (Seidi et al., 2018; Hayward et al., 2021). For instance, we generated a parasite strain in which we could knockdown expression of the ETC Complex III protein TgQCR11 through the addition of ATc (Hayward et al., 2021). We observed that TgQCR11 knockdown led to a decrease in basal OCR (Figure 4D). We have also observed that knocking down proteins that function selectively in the ETC [e.g., TgQCR11 (Hayward et al., 2021), or the ETC Complex IV subunit TgApiCox25 (Seidi et al., 2018)] does not result in a concomitant impairment of ECAR, and in some cases results in a slight but significant increase in ECAR (Figure 4E). We interpret the observed decrease in OCR but not ECAR as a signature for a selective ETC defect. By contrast, knocking down proteins that are important for general parasite survival leads to a decrease in both OCR and ECAR, and we interpret this as a signature of defects beyond the ETC. For example, knockdown of the heme biosynthesis enzyme TgUroD, or treatment with the protein synthesis inhibitor cycloheximide, led to a simultaneous decrease in OCR and ECAR (Seidi et al., 2018; Tjhin et al., 2020). The assay therefore differentiates defects in ETC function from defects in broader parasite metabolism, or effects on parasite survival. We note that by substituting atovaquone in Port 2 for a test compound, this assay can also determine whether a compound of interest inhibits the ETC.
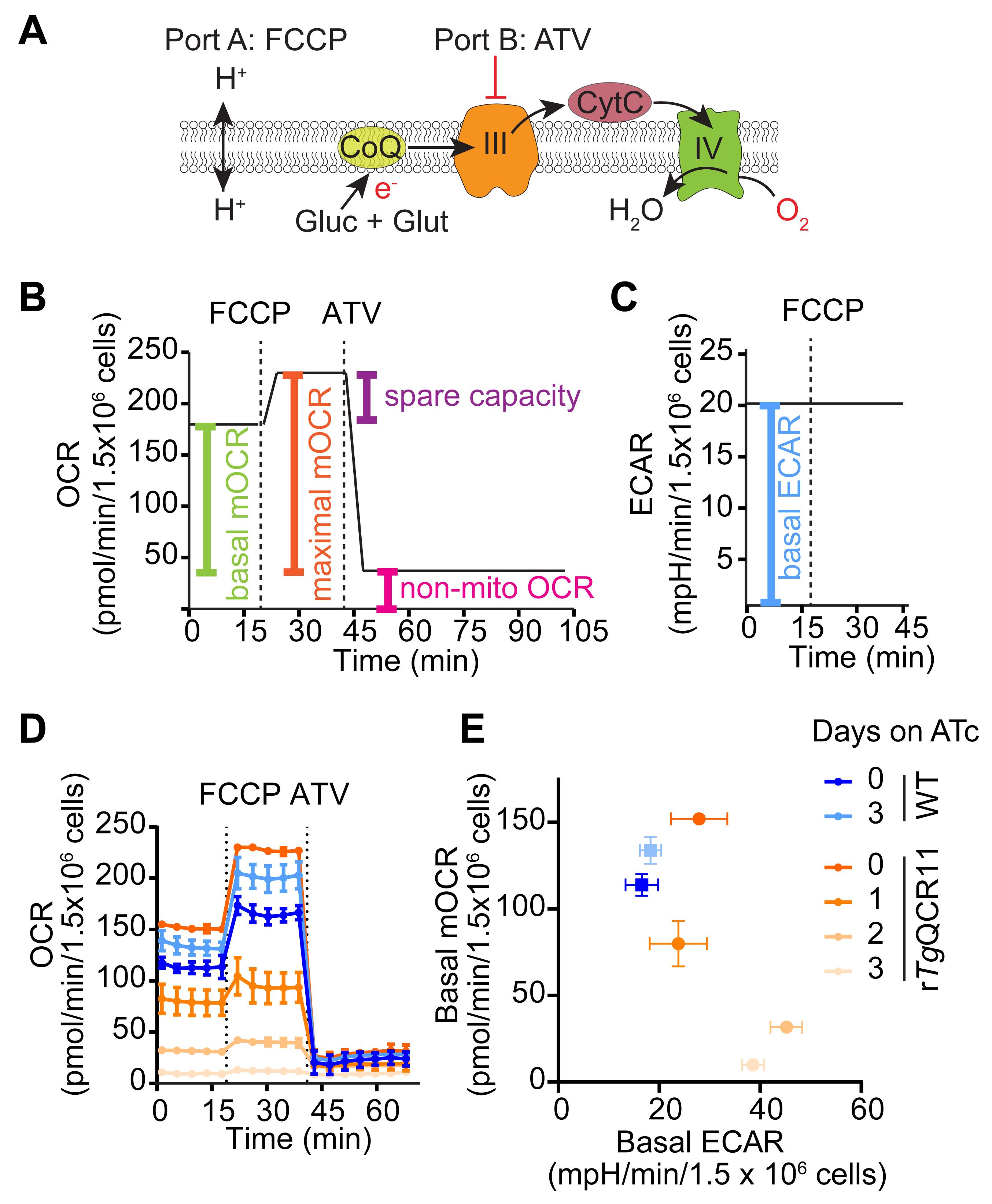
Figure 4. Measurement of OCR and ECAR of intact T. gondii parasites in a MitoStress assay. A. Schematic of the assay. Intact T. gondii parasites are supplied glucose (Glc) and glutamine (Gln) as energy/electron sources in the Seahorse XF base medium. FCCP is injected via port A to dissipate the proton gradient across the inner mitochondrial membrane and thereby uncouple electron transport from ATP synthesis. Atovaquone (ATV) is injected via port B to inhibit ETC Complex III. B. Mock traces highlighting the typical OCR response to FCCP and ATV injection, and the calculations that can be performed on the resulting data: namely, the non-mitochondrial OCR (pink) is obtained by averaging the OCR measurements after injection of atovaquone (ATV). The non-mitochondrial OCR can then be subtracted from the measurements before injection of FCCP to calculate the basal mitochondrial OCR (basal mOCR, green). The non-mitochondrial OCR can also be subtracted from the measurements after injection of FCCP to calculate the maximal mOCR (orange). Finally, the spare capacity is the difference between the maximal and basal mOCR (purple). C. ECAR measurements before injection of FCCP represent the basal ECAR. D. Real traces depicting the OCR of WT (blue) and rTgQCR11 (orange) parasites that were cultured in the absence of ATc or in the presence of ATc for 1-3 days prior to the assay. Data represent the mean ± SD of three technical replicates and are representative of three independent experiments (derived from Hayward et al., 2021). E. Basal mOCR versus basal ECAR of WT (blue) and rTgQCR11 (orange) parasites that were cultured as described in (D). Data represent the mean ± SD of three technical replicates from a single experiment. Data were exported from the Wave software into GraphPad Prism to generate the graphs. Colors match the legend in (D).Seahorse XFe96 assay template preparation for an intact MitoStress assay in Wave software:
Download the Wave software from the Agilent website.
Under ‘Group Definitions’ tab (Figure 5A), fill in the injection strategies as follows:
Port A: FCCP.
Port B: Atovaquone.
Port C: Base medium with glucose and glutamine (BM+Glc+Gln).
Port D: BM+Glc+Gln.
Under ‘Group Definitions’ tab (Figure 5A), add ‘groups’, naming them using the conventions outlined below (must be strictly followed for the Python and R scripts used to analyze the data to work):
X day (where x is the number of days parasites were grown on ATc).
The name of the parasite line being used (e.g., WT, rTgQCR11).
Intact (indicates the assay is being performed on intact parasites, rather than on permeabilized parasites, as occurs in the experiments described in Procedure Section C).
Under the ‘Plate Map’ tab (Figure 5B), configure the plate map ensuring appropriate background wells (wells without cells, blanks) are included, at a minimum of four per plate.
Note: Including and subtracting background wells from the experimental wells is important to account for any background (parasite-independent) OCR and ECAR.
Under the ‘Protocol’ tab (Figure 5C), specify which ports are to be injected by adding injections after the baseline reading.
Under the ‘Protocol’ tab (Figure 5C), specify the timing and number of mixing and measuring cycles:
3 cycles of measurements (in the test data set provided, we did 5 cycles).
30 s mixing.
0 s waiting.
3 min measuring.
Save this template. When opened at the Seahorse XFe96 machine, open the ‘Run Assay’ tab at the top to start the assay.
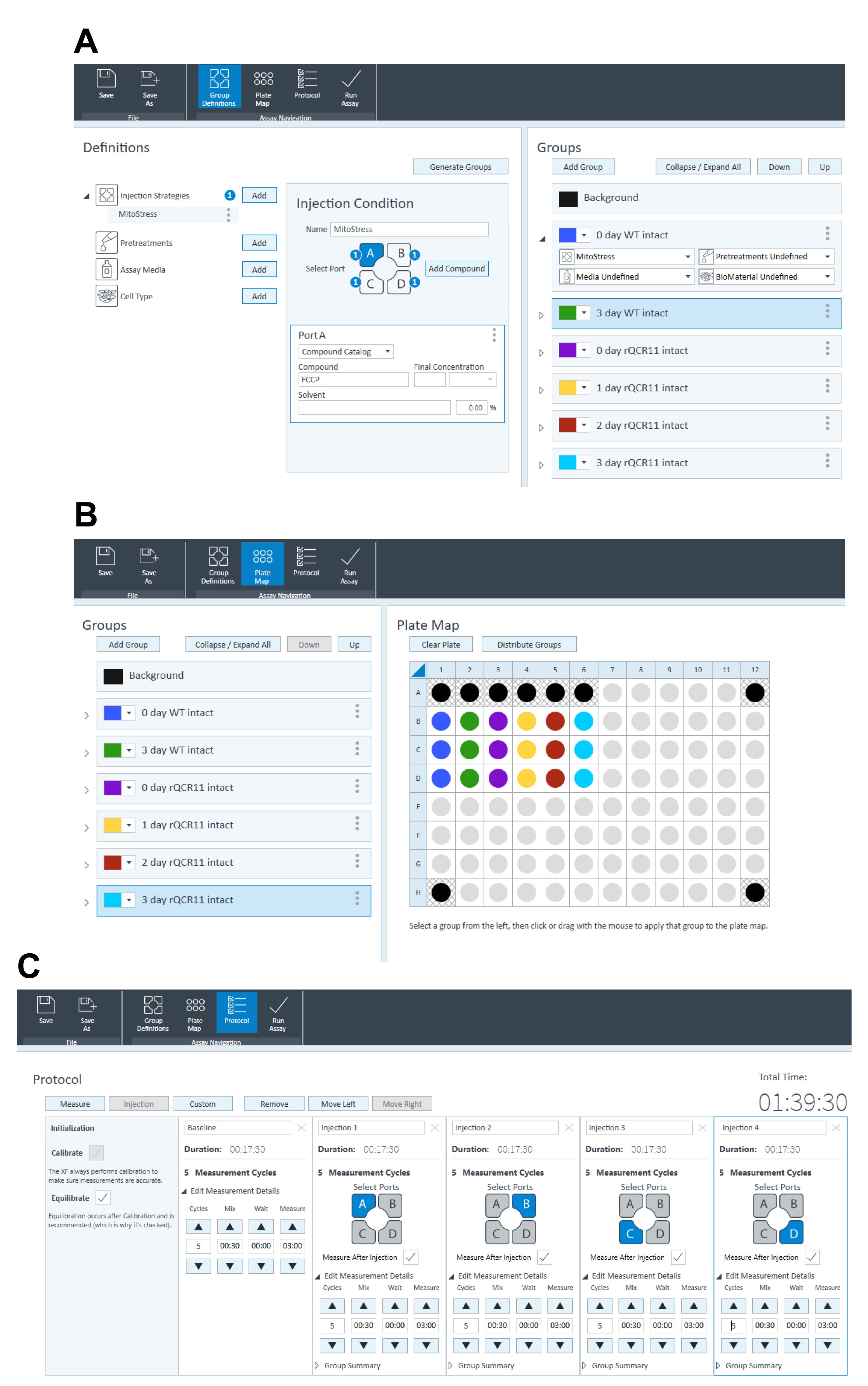
Figure 5. Designing a Seahorse XFe96 experimental template for an intact MitoStress assay in the Wave software. A. Under ‘Group Definitions’ tab, fill in the injection strategies as follows: Port A: FCCP; Port B: Atovaquone; Port C: Base medium with glucose and glutamine (BM+Glc+Gln); Port D: BM+Glc+Gln. Add ‘groups’, and ensure to name them using the conventions x day (where x is the number of days parasites were grown on ATc), the name of the parasite line being used (e.g., WT, rTgQCR11), and intact (indicates the assay is being performed on intact parasites, rather than on permeabilized parasites as described in Procedure Section C). B. Under the ‘Plate Map’ tab, configure the plate map ensuring appropriate background wells (wells without cells, blanks) are included. C. Under the ‘Protocol’ tab, specify which ports are to be injected by adding injections after the baseline reading. Specify the timing and number of mixing and measuring cycles as follows: 3 cycles (in the test data set provided we did 5 cycles); 30 s mixing; 0 s waiting; 3 min measuring. Save the template. When opened at the Seahorse XFe96 machine, open the ‘Run Assay’ tab at the top to start the assay.
Experimental procedure for intact MitoStress assay:
Prepare Seahorse XF base medium supplemented with glucose (5 mM) and glutamine (1 mM) (BM+Glc+Gln – see Recipes section). Check the pH and, if necessary, adjust to 7.4. Warm to 37°C in a water bath.
Prepare the following in BM+Glc+Gln (5 mL of each is enough for an entire 96 well plate with excess):
FCCP (prepare 8 μM for 1 μM final concentration). Add 0.8 μL of FCCP stock (50 mM) to 5 mL of BM+Glc+Gln.
Atovaquone (prepare 9 μM for 1 μM final concentration). Add 4.5 μL of atovaquone stock (10 mM) to 5 mL of BM+Glc+Gln.
Note: In previous studies, we also included a second Complex III inhibitor, Antimycin A (prepared 90 μM for 10 μM final), in combination with atovaquone (Hayward et al., 2021); however, this is unnecessary as atovaquone yields sufficient inhibition of OCR to allow calculation of non-mitochondrial OCR. While we suspect that antimycin A would also yield sufficient inhibition of OCR when used alone, we would recommend testing this in a pilot experiment.
Note: The above concentrations were determined by titrating each compound and selecting the lowest concentration that gave an optimal response. Additional optimization may be required depending on the strain of T. gondii parasites used in the experiments.
Inspect parasite-infected host cell flasks. Ideally, parasites should be recently egressed from host cells or still intracellular on the day of the assay, to ensure optimal parasite viability. For safety and sterility, conduct steps involving parasites in a Class II Biological Safety Cabinet.
If parasites are mostly still intracellular, release parasites from host cells by first removing host cells from the bottom of the flask using a cell scraper, then passaging infected host cells through a 26 gauge needle to mechanically egress parasites.
If extracellular, skip the scraping and needle-passing step.
Pass parasite-containing culture medium through a 5 μm polyvinylidene fluoride (PVDF) filter into a 15 mL tube to remove host cell debris (note that a 3 μm polycarbonate filter can also be used).
Note: This is step is important to ensure that intact host cells (which contain active mitochondria) do not contribute to OCR measurements.
Count parasites using a hemocytometer. Calculate total number of parasites and the volume of base medium required to give 1.5 × 107 parasites/mL.
Note: It is critical to have accurate parasite counts to ensure similar number of parasites are used for each condition and between replicates, so we recommend a single person counts all samples for a particular assay to ensure internal consistency.
Pellet the parasites by centrifugation at 1,500 × g for 10 min at RT.
Aspirate media.
Wash parasite pellet in ~1-2 mL of BM+Glc+Gln. This wash step is important to ensure that NaHCO3-buffered culture medium, which will interfere with ECAR measurements, is sufficiently removed from the samples.
Either keep the parasite solution in the 15 mL tube and centrifuge as above, or transfer to a sterile 1.5/2 mL tube and centrifuge at 12,000 × g for 1 min at RT. Whether you use the 15 mL or the 1.5/2 mL tube will depend on the volume of base medium the pellet needs to be resuspended in to be at a concentration of 1.5 × 107 parasites/ mL.
Aspirate the media from the tubes.
Resuspend the parasite pellet to 1.5 × 107 parasites/mL in BM+Glc+Gln.
Add 100 μL of the parasite suspension (containing 1.5 × 106 parasites) to designated wells of a Cell-Tak-coated Seahorse XFe96 culture plate.
Fill the blank wells (containing no parasites) with 100 μL of BM+Glc+Gln.
Centrifuge the plate at 800 × g for 3 min at RT on a low brake centrifuge setting.
Note: We recommend using a low brake setting to enable an even monolayer of parasites to form on the bottom of each well.
Inspect the wells using a standard inverted light microscope under the lowest magnification setting, to ensure that an even monolayer of parasites has formed on the bottom of the wells.
Note: This is also a good opportunity to visually check whether there are a similar number of parasites in each condition.
Top up each well with 75 μL of BM+Glc+Gln for a total volume of 175 μL.
Remove the green sensor cartridge from the calibrant plate and, in a Class II Biological Safety Cabinet, place it to the side so that the sensor probes are facing up. Replace the water in the wells of the calibrant plate with 200 μL of XF calibrant solution. Place the sensor cartridge back into the calibrant plate and ensure no air bubbles are trapped beneath the sensor probes.
Take the Seahorse XFe96 culture plate containing the parasites, the sensor cartridge in the calibrant plate, and the reagents prepared in step 2 to the Seahorse XFe96 machine. Also bring a multichannel pipette that can dispense 25 μL solutions, tips, and reagent reservoirs.
Incubate the Seahorse XFe96 culture plate containing the parasites at 37°C in a humidified CO2 free incubator while setting up the Seahorse XFe96 software and loading the cartridge ports.
Open the assay template prepared earlier using the Wave software.
Using a multichannel pipette, load 25 μL of solutions into appropriate ports of the sensor cartridge following the injection strategy outlined below.
Port A: FCCP.
Port B: Atovaquone.
Port C: BM+Glc+Gln.
Port D: BM+Glc+Gln.
Notes:
Care is needed to ensure proper loading of solutions into the port and the cartridge must not be roughly bumped or tapped once the ports are loaded. In our experience, the best practice is to:
Use reverse pipetting to prevent air bubbles.
Insert the pipette tip right to the bottom of the port, raise the tip slightly so it is no longer touching the bottom, angle the tip so that it is touching the side of the port, and then gently release the solution into the port. Solution should be visible in each port after correct loading.
Common loading mistakes include:
Positioning the pipette tip right at the bottom of the port and releasing the solution too forcefully, resulting in injection through the port into the bottom plate. The port will look empty if this mistake is made.
Positioning the tip too close to the top of the port, such that an air bubble forms between the bottom of the port and the solution. The port will appear overly full if this mistake is made. This can cause injection failure during the assay.
Once ports of the sensor cartridge are loaded, click “Run Assay” in the Wave software. This will open the tray in the side of the Seahorse XFe96 machine.
Ensuring that the lid has been removed, gently place the loaded sensor cartridge sitting in the calibrant plate onto the tray of the Seahorse XFe96 machine and click the prompt to close the tray.
Note: The lid must be removed to prevent damage to the machine.
The Seahorse XFe96 machine then undergoes a calibration/verification process for approximately 30 min to ensure that the sensor probes are functioning correctly.
Note: If a sensor cartridge fails these calibration/verification steps, we recommend re-starting the Seahorse XFe96 machine and trying a second time. If it fails a second time, the sensor cartridge may be faulty and we would not recommend including the data obtained in the subsequent assay in publications.
When prompted, remove the calibrant plate from the tray (the sensor cartridge is retained inside the Seahorse XFe96 machine).
Carefully remove the Seahorse XFe96 culture plate (containing parasites) from the 37°C incubator, remove the lid, place it on the tray, and click the prompt to close the tray.
The assay starts at this point and, depending on how many cycles of mixing and measuring were selected, takes between 1-2 h to run.
Analyses of the resulting data are described in Data analysis Section A.
Measurement of substrate-elicited OCR in permeabilized parasites
The Seahorse XFe96 measurements described in the previous section measure OCR and ECAR in intact extracellular parasite fed glucose and/or glutamine as carbon sources. These experiments rely on the uptake and active metabolism of these substrate in parasites. Seahorse XFe96 assays can also be adapted for measuring OCR that is elicited by direct substrates of the ETC. This involves permeabilizing the parasite plasma membrane with a low concentration of digitonin to allow substrates that would otherwise not enter intact parasites to reach the mitochondrion. Parasites are then incubated with selected substrates that can be directly oxidized by the various dehydrogenases that feed electrons into the ETC (Figure 1). This approach offers information that can be difficult to obtain with intact cells, and has the added benefit of not needing to purify mitochondrial organelles from the parasite to directly assay mitochondrial activities. Utilizing these approaches, we are able to more deeply investigate ETC function in T. gondii. For example, these approaches allow us to determine the specific step of the ETC that is impaired upon the knockdown of genes encoding proteins that function in the ETC (Hayward et al., 2021), or to elucidate the molecular target of ETC inhibitors. This section describes a method to assess the functionality of different ETC complexes by measuring the OCR of permeabilized T. gondii parasites that have been supplied specific substrates of the ETC (Figure 6A and 6B).
In this assay, T. gondii parasites are starved to deplete endogenous energy sources, then the plasma membrane of the parasite is permeabilized with digitonin. Next, a substrate is injected via injection port A to stimulate O2 consumption, together with FCCP to ensure that OCR is occurring at maximum capacity. We have utilized two different substrates for these experiments, both of which independently feed electrons to CoQ: i) malate, which feeds electrons into the ETC via the TCA cycle enzyme malate:quinone oxidoreductase; and ii) glycerol 3-phosphate (G3P), which feeds electrons into the ETC via the mitochondrial G3P dehydrogenase independently of the TCA cycle (Figure 6A; Hayward et al., 2021). If OCR elicited by both substrates is compromised in a parasite mutant, this provides evidence that the defect is occurring downstream of coenzyme Q (i.e., on ETC Complex III or IV). For instance, knockdown of the ETC Complex III subunit TgQCR11 led to a decrease in both malate-dependent (Figure 6C) and G3P-dependent (Figure 6D) OCR (Hayward et al., 2021). If OCR elicited by only one of the substrates is compromised in a parasite mutant, then likely the effect on the ETC is specific to that dehydrogenase.
The next step of the assay involves injecting the Complex III inhibitor atovaquone (ATV) via port B. This will inhibit the ETC at Complex III and enable determination of the non-mitochondrial OCR. Next, to differentiate between a defect in Complex III and Complex IV, reduced N,N,N,N’-tetramethyl-p-phenylenediamine (TMPD), a substrate that donates electrons directly to CytC (and thereby bypasses Complex III; Figure 6A), is injected via port C. If OCR can be rescued by addition of TMPD, this provides evidence that the defect is occurring upstream of CytC (e.g., at ETC Complex III), whereas if OCR cannot be rescued, this points to a defect downstream of CytC (i.e., in Complex IV). As validation of this approach, we demonstrated that OCR can be rescued by the addition of reduced TMPD in mutant parasites lacking the ETC Complex III subunit TgQCR11 but not the Complex IV subunit TgApiCox25 (Figure 6C; Hayward et al., 2021). Finally, the Complex IV inhibitor sodium azide (NaN3) is injected via port D as validation that the TMPD-dependent OCR is mediated by the ETC upstream of Complex IV (Figure 6B-D).
We note that this assay can be easily modified to supply different substrates via port A. A titration of the new substrate should be performed to determine the lowest concentration that gives the maximal OCR response. This assay can also be used to pinpoint the target of ETC inhibitory compounds by substituting atovaquone with the compound of interest in port B. In this case, we would recommend starting with a high concentration of the test compound to yield maximal inhibition, then titrating the concentration of the candidate inhibitor to determine its potency in targeting the ETC.
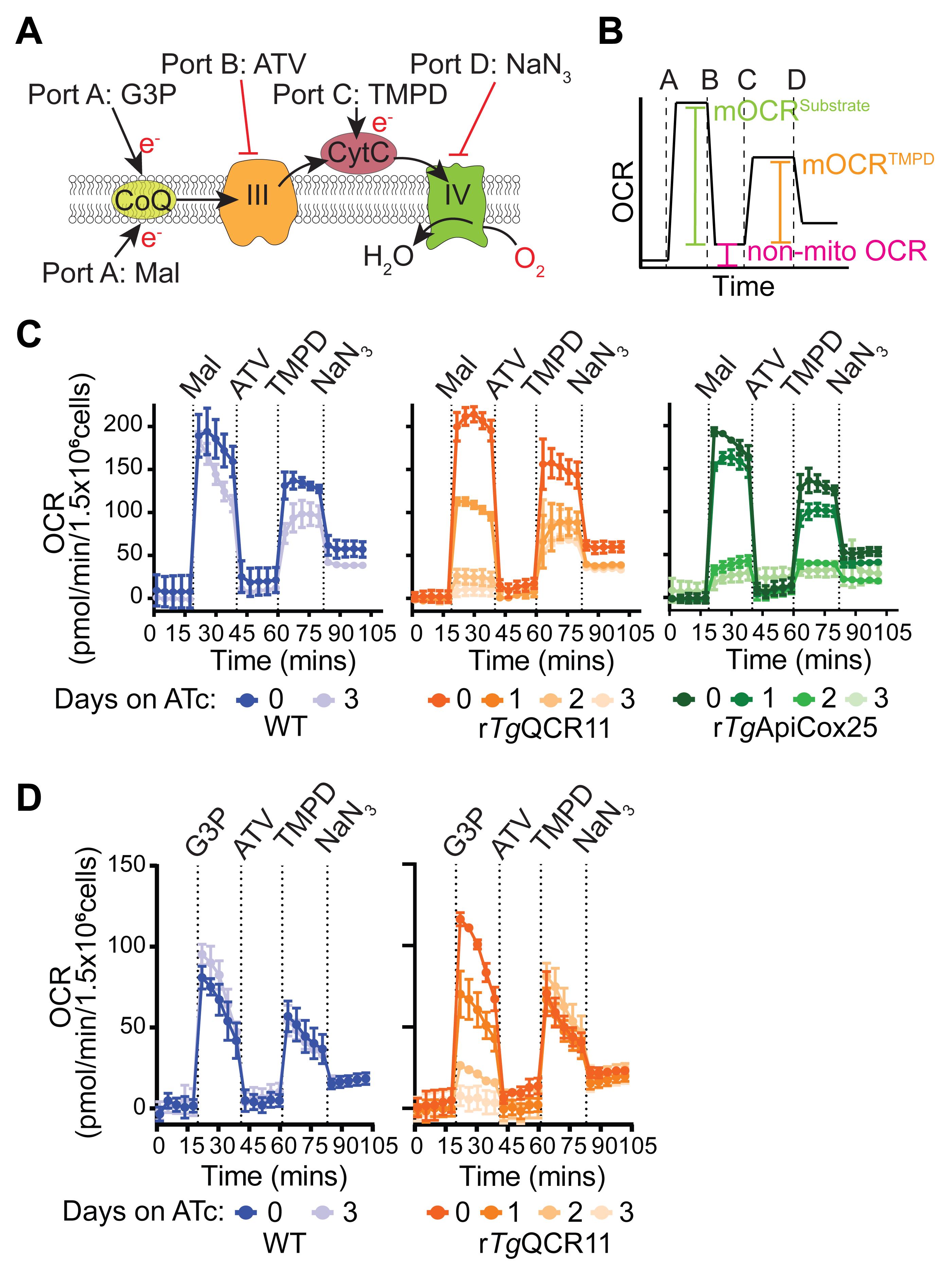
Figure 6. Measurement of substrate-elicited OCR in permeabilized parasites (Hayward et al., 2021). A. Schematic of the assay. T. gondii parasites are starved for 1 hour in base media to deplete endogenous energy sources, then permeabilized with 0.002% (w/v) digitonin before being subjected to the following injections of substrates or inhibitors: Port A, FCCP plus the substrates malate (Mal) or glycerol 3-phosphate (G3P); Port B, the ETC Complex III inhibitor atovaquone (ATV); Port C, the cytochrome c (CytC) substrate TMPD (TMPD); Port D, the ETC Complex IV inhibitor sodium azide (NaN3). B. Mock trace highlighting the typical OCR response of WT parasites, and the calculations that can be performed on the resulting data: namely, the non-mitochondrial OCR (pink) is obtained by averaging the OCR measurements after injection of atovaquone (ATV) via port B. The non-mitochondrial OCR can then be subtracted from the measurements after the injection of Mal or G3P via port A to calculate the substrate-elicited mitochondrial OCR (mOCRSubstrate, green). The non-mitochondrial OCR can also be subtracted from the measurements after injection of TMPD via port C to calculate the TMPD-elicited mOCR (mOCRTMPD, orange). Finally, as a measure for how effectively TMPD stimulates OCR relative to the substrate, the ratio of mOCRTMPD/mOCRSubstrate can also be calculated. C. Real traces depicting the OCR of WT (blue), rTgQCR11 (orange), and rTgApiCox25 parasites (green) that were cultured in the absence of ATc for 3 days, or in the presence of ATc for 1-3 days prior to the assay, and were supplied Mal as a substrate via port A. Data represent the mean ± SD of three technical replicates and are representative of three independent experiments. D. Real traces depicting the OCR of WT (blue) and rTgQCR11 (orange) that were cultured as described in (C), but supplied G3P as a substrate via port A. Data represent the mean ± SD of three technical replicates and are representative of three independent experiments. Data were exported from the Wave software into GraphPad Prism to generate the graphs. Data were described previously in Hayward et al. (2021).Seahorse XFe96 assay template preparation for a permeabilized assay in Wave software:
Under ‘Group Definitions’ tab (Figure 5A), fill in the injection strategies as follows:
Port A: FCCP + Malate OR FCCP + Glycerol 3-phosphate.
Port B: Atovaquone.
Port C: TMPD + Ascorbate.
Port D: Sodium azide.
Under ‘Group Definitions’ tab (Figure 5A), add ‘groups’, naming them using the conventions outlined below (must be strictly followed for the Python and R scripts used to analyze the data to work):
X day (where x is the number of days parasites were grown on ATc).
The name of the parasite line being used (e.g., WT, rTgQCR11).
Perm (indicates the assay is being performed on permeabilized parasites, rather than on intact parasites as occurs in Procedure Section B).
Under the ‘Plate Map’ tab (Figure 5B), configure the plate map ensuring appropriate background wells (wells without cells, blanks) are included.
Under the ‘Protocol’ tab (Figure 5C), specify which ports are to be injected by adding injections after the baseline reading.
Under the ‘Protocol’ tab (Figure 5C), specify the timing and number of mixing and measuring cycles:
3 cycles of measurements (in the test data set provided, we did 5 cycles).
30 s mixing.
0 s waiting.
3 min measuring.
Save this template. When opened at the Seahorse XFe96 machine, open the ‘Run Assay’ tab at the top to start the assay.
Experimental procedure for an assay with permeabilized parasites:
Warm non-supplemented Seahorse XF base medium (BM) to 37°C in the water bath.
Prepare 0.002% (w/v) digitonin in mitochondrial assay solution (MAS) buffer: add 8 μL of digitonin stock (5% w/v) to 20 mL of MAS buffer. This is sufficient for an entire plate with excess.
Note: The 5% w/v digitonin stock may need to be heated to ~90°C for ~5 min to fully dissolve the digitonin prior to use.
Notes:
The 0.002% (w/v) digitonin concentration was optimized by titrating the digitonin and identifying the lowest concentration that dissipated ECAR (indicating the plasma membrane had been permeabilized) but retained OCR response to substrates such as malate and TMPD. Optimizing the concentration of digitonin is critical, as too low concentrations may not permeabilize the plasma membrane and limit compound entry, while too high concentrations can disrupt the mitochondrial membranes.
MAS buffer is a high [K+], low [Na+] buffer designed to mimic intracellular ion concentrations. It is critical that MAS buffer, and not BM, is used in these assays on permeabilized parasites.
Prepare substrates/drugs in MAS buffer; 5 ml is enough to cover a plate with excess:
8× FCCP (8 μM for 1 μM final), together with 8× of the substrates malate (prepare 80 mM for 10 mM final) or glycerol 3-phosphate (prepare 200 mM for 25 mM final).
Note: In previous studies, we used a combination of malate and glutamate (Hayward et al., 2021); however, subsequent testing showed that it is the malate that contributes to OCR as glutamate alone yields no OCR response in these assays.
Malate (5 mL): Add 1.25 mL of malate stock (320 mM) and 0.8 μL of FCCP stock (50 mM) to 3.75 mL of MAS buffer.
Glycerol 3-phosphate (5 mL): Add 0.8 μL of FCCP stock (50 mM) to 5 mL of glycerol 3-phosphate stock (200 mM).
Note: As G3P is a more expensive substrate, this can be prepared on a per-well basis (make 25 μL per well) rather than making 5 mL.
9× Atovaquone (prepare 9 μM for 1 μM final). Add 4.5 μL of atovaquone stock (10 mM) to 5 mL of MAS buffer.
Note: In previous studies, we also included a second Complex III inhibitor, Antimycin A (prepared 90 μM for 10 μM final), in combination with atovaquone (Hayward et al., 2021); however, this is unnecessary as atovaquone yields sufficient inhibition of OCR.
10× TMPD (prepare 2 mM for 0.2 mM final) and ascorbate (prepare 33 mM for 3.3 mM final). Add 835 μL of ascorbate stock (200 mM) to 4.155 mL MAS buffer and mix. Then, add 20 μL of TMPD stock (500 mM) and mix again. The solution should be light brown in color.
Note: It is important to add the ascorbate to the MAS buffer first and then add the TMPD. The purpose of the ascorbate is that it keeps the TMPD in a reduced state. If the solution appears green/blue in color rather than light brown, the TMPD is in an oxidized state. In this instance, we recommend re-making the ascorbate solution to ensure the TMPD is reduced prior to the assay.
Sodium azide (NaN3, prepare 110 mM for 10 mM final). The sodium azide stock (110 mM) can be made in advance and is ready to use as is.
Note: The above concentrations of substrates and inhibitors were determined by titrating each compound and selecting the lowest concentration that gave an optimal response. Additional optimization may be required depending on the strain of T. gondii parasites.
Inspect parasite-infected host cell flasks. Ideally, parasites should be freshly egressed from host cells or exist in large intracellular vacuoles within host cells on the day of the assay to ensure optimal parasite viability.
If the bulk of the parasites are intracellular, mechanically egress parasites from host cells by scraping infected host cells from the flask using a cell scraper and passing infected host cells through a 26 gauge needle.
If parasites are extracellular, skip the scraping and needle-passing step.
Remove host cell debris by passing egressed parasite cultures through a 3 µm polycarbonate or a 5 μm PVDF filter into a 15 mL tube. This step is important because contaminating host cells can contribute to OCR.
Count parasites using a hemocytometer. Calculate total number of parasites. It is critical to have accurate parasite counts to ensure that a similar number of parasites are used for each condition, so we recommend a single person counts all samples for a particular assay to ensure internal consistency.
Centrifuge the 15 mL tubes containing the parasites at 1,500 × g for 10 min at RT to pellet parasites.
Aspirate media from the tubes.
Wash parasite pellet in ~1-2 mL of non-supplemented BM.
Either keep the parasite solution in the 15 mL tube and centrifuge as above, or transfer to a sterile 1.5/2 mL tube and centrifuge at 12,000 × g for 1 min at RT.
Aspirate the media from the tubes.
Gently resuspend the pellet to 1.5 × 107 parasites per mL in non-supplemented BM.
Incubate the parasites for 1 h in a 37°C incubator to starve them of endogenous substrates. Note: It is important to optimize the length of starvation for each parasite strain. In our hands, over-starving the parasite can lead to an apparent loss of viability, culminating in lower-than-expected OCR readings. Under-starving parasites can result in higher than expected preliminary OCR readings, which can complicate subsequent data analysis. Our preliminary experiments suggest that different lengths of time may be required to optimally starve different parasite strains.
Add 100 μL of the parasite suspension (containing 1.5 × 106 parasites) to designated wells of a Cell-Tak-coated Seahorse XFe96 culture plate.
Fill the blank wells (containing no parasites) with 100 μL of BM.
Centrifuge the plate on low brake at 800 × g for 3 min at RT.
Using a microscope, check that a parasite monolayer has formed at the bottom of the well. This is also a good opportunity to visually check whether there are a similar number of parasites in each condition.
Remove the sensor cartridge from the calibrant plate and place it to the side so that the sensor probes are facing up. Replace the water in the wells of the calibrant plate with 200 μL of XF calibrant solution. Place the sensor cartridge back into the calibrant plate and ensure no air bubbles are trapped beneath the sensor probes.
Take the Seahorse XFe96 culture plate containing the parasites, the sensor cartridge in the calibrant plate, the digitonin in MAS buffer prepared in step 2, and the reagents prepared in step 3 to the Seahorse Xfe96 machine. Also bring multichannel pipettes that can dispense 25 μL and 200 μL volumes, tips, and reagent reservoirs.
Incubate the Seahorse XFe96 culture plate containing the parasites at 37°C in a humidified incubator at atmospheric CO2.
Open the assay template prepared earlier using the Wave software.
Load 25 μL of drug solutions into appropriate ports of the sensor cartridge following the injection strategy outlined below.
Port A: FCCP + Malate OR FCCP + Glycerol 3-phosphate.
Port B: Atovaquone.
Port C: TMPD + Ascorbate.
Port D: Sodium azide.
Note: Care is needed to ensure proper loading of solutions into the port. See the tips provided in the Procedure Section B notes.
Once loaded, click “Run Assay” in the Wave software.
When prompted, and ensuring that the lid has been removed, place the loaded sensor cartridge sitting in the calibrant plate into the Seahorse XFe96 machine.
The Seahorse XFe96 machine then undertakes a calibration/verification process for approximately 30 min to ensure the sensor probes are functioning correctly.
Toward the end of the calibration/verification process, remove the Seahorse XFe96 culture plate containing the parasites from the 37°C incubator.
Using the P200 multichannel pipette, gently remove the BM from the well. Note: care needs to be taken not to disturb the parasite monolayer during this step.
Carefully add 175 μL of 0.002% (w/v) digitonin in MAS buffer to each well using the P200 multichannel pipette. This solution permeabilizes the plasma membrane of the parasites.
Notes:
We recommend angling the pipette tips against the wall of the wells and slowly releasing the solution so that the parasite monolayer is not disturbed during this step.
For best results, we recommend limiting the amount of time the parasites are permeabilized for prior to the start of the assay, and keeping this time consistent between independent replicates of an assay. We find that response to substrates decreases with increasing time since permeabilization.
When prompted, remove the calibrant plate from the machine and replace with the culture plate containing permeabilized parasites, ensuring the lid has been removed.
The assay starts at this point and, depending on how many cycles of mixing and measuring were selected, takes between 1-2 h to run.
Analyses of the resulting data are described in Data analysis Section B.
Data analysis
Data analysis of intact MitoStress assay
The assay performed on intact parasites described in Procedure Section B is based on a typical MitoStress assay: basal OCR is measured first, then FCCP is injected (port A) to assess maximal OCR and spare capacity, then finally atovaquone is injected (port B) to assess non-mitochondrial OCR (Figure 4A and 4B). The output from the Seahorse XFe96 assay is a trace of OCR or ECAR over time for each condition tested, providing an informative visual representation of how parasites responded to injections of the various compounds (Figure 4C). Another way to depict the data from this experiment is to calculate the basal mitochondrial OCR (mOCR), maximal mOCR, spare capacity in mOCR, and basal ECAR from the raw data (Figure 4B), and present these data as column graphs. This representation enables clearer statistical comparisons between parasite strains and conditions. The first step in these calculations is to average the values obtained after injection of atovaquone to obtain the non-mitochondrial OCR (Figure 4B). The non-mitochondrial OCR can then be subtracted from each of the basal OCR points and each of the points after injection of FCCP, to yield the basal and the maximal mOCR, respectively (Figure 4B). The spare capacity is the difference between the maximal and the basal mOCR (Figure 4B). The basal ECAR is the ECAR measurements before injection of FCCP.
While these calculations are quite simple and can be done manually in Excel, we have provided a Python script “pre_R_script.py” that cleans up the raw Excel data files, performs these calculations and outputs four csv files containing basal mOCR, maximal mOCR, spare capacity mOCR and basal ECAR. The Python script then uses these csv files as inputs for subsequent analysis in the R software environment using the R script “treat_seahorse_data.r”. First, we use a linear mixed effects model to analyze the data. In the linear mixed effects model, plates (between experiment variation) and wells (within experiment, between measurement variation) are set as random effects, and the cell lines and days on drug (e.g., ATc) are set as fixed effects. In doing so, the linear mixed effects model compares conditions within plates to allow for greater precision in the treatment effect estimates (see Data analysis Section C for further considerations of the statistical analyses of the data). Analysis of the estimated marginal means (previously referred to as least square means; Hayward et al., 2021) of the linear mixed effects model is also performed in R and statistical differences are assessed using p-values adjusted using Tukey’s method. The Python script outputs the results of these analyses into four more csv output files, and these analyzed data can then be used to create graphs in GraphPad Prism. We have provided three test data files (rep1_intact, rep2_intact and rep3_intact) for the rTgQCR11 intact MitoStress experiment described in Hayward et al. (2021). These can be downloaded from the “test_data” subfolder in a Bitbucket repository (https://bitbucket.org/ngMiaNG/seahorse-scripts/src/master/).
After the assay is finished, save the Wave results file and make this file accessible on the computer on which you will perform the subsequent analyses (e.g., save onto a USB flash drive or email it to yourself).
Open the file using the Wave program.
Click on “Add - Overview” to view the plate map and results simultaneously.
Ensure that the “Background correction” box has been ticked – this subtracts the oxygen consumption rate of the blank wells from the experimental wells.
Note: If the OCR within the background wells is higher than the OCR within sample wells, negative values will result. This does sometimes happen in our experiments, particularly when knocking down an ETC protein. Our current analysis strategy includes these negative values in the analysis; see Data analysis Section C for further discussion of this point.
Occasionally, compounds do not inject properly from one or more of the ports, or inject at the wrong time in the assay, leading to erroneous values in the data. Inspect each well of the data for any large outliers. If you have strong reason to believe an incorrect injection has occurred, you should exclude those wells from subsequent analyses by unselecting them. You can also inspect the ports of the sensor cartridge post assay to identify wells in which the port contents failed to inject.
Click on the “Export” button to export the results in Microsoft Excel format.
Relevant data is located in the ‘Rate’ tab. The data consist of the columns ‘measurement’, ‘well’, ‘group’, ‘time’, ‘OCR’, ‘ECAR’, and ‘PER’, with rows representing the data for each well of the 96 well plate over the time points of the experiment.
To appreciate the variability in the data, it is important to undertake multiple independent experiments (i.e., biological replicates) using the same experimental conditions on different days with different parasite preparations. We typically undertake three independent experiments.
After the assay has been repeated three times independently, save each Excel results file (with the names rep1_intact, rep2_intact, and rep3_intact) to a folder with only these files inside.
This is how the rTgQCR11 intact MitoStress test data files (rep1_intact, rep2_intact, and rep3_intact) were created. Note that other conditions were tested on each plate but the names of irrelevant wells have been removed.
Go to https://bitbucket.org/ngMiaNG/seahorse-scripts/src/master/, download the files in the repository and save into a folder. This contains the “pre_R_script.py” Python script, the “treat_seahorse_data.r” R script, and the “README” and “requirements” text files as well as test data obtained from the rTgQCR11 Seahorse XFe96 experiments.
Read the “README” to understand the requirements and how the scripts work.
Run “pre_R_script.py” Python script from the command line via a terminal (e.g., CMD).
The script has inbuilt help that can be accessed by typing: python3 “pre_R_script.py” -h into the command line.
The general structure of the command to run the script is as follows: python3 “pre_R_script.py” -i “path to input directory” -o “path to output directory” -n “number of measurement cycles, 3 or 5” --intact. Note: the test data consists of 5 measurement cycles.
When --intact is specified, the “pre_R_script.py” Python script perform calculations of basal mOCR, maximal mOCR, spare capacity mOCR and basal ECAR using the Excel results files (rep1_intact, rep2_intact, and rep3_intact) as inputs.
After performing these calculations, the “pre_R_script.py” Python script calls upon the “treat_seahorse_data.r” R script to fit a linear mixed effects model to the data and perform statistical analyses (ANOVA and post hoc Tukey test).
Csv files containing the R analyzed results will be written to the folder specified as the output directory. This will consist of the estimated marginal means, standard error, degrees of freedom, and 95% confidence intervals from the fitted linear mixed effects model, and the adjusted p-values (Tukey).
These results can be used to plot graphs in GraphPad Prism. We typically present the data as column graphs of the basal mOCR and basal ECAR, and a graph of basal mOCR versus basal ECAR (Figure 7). The maximal mOCR and spare capacity are also calculated, and can be plotted if they show something noteworthy (Seidi et al., 2018).
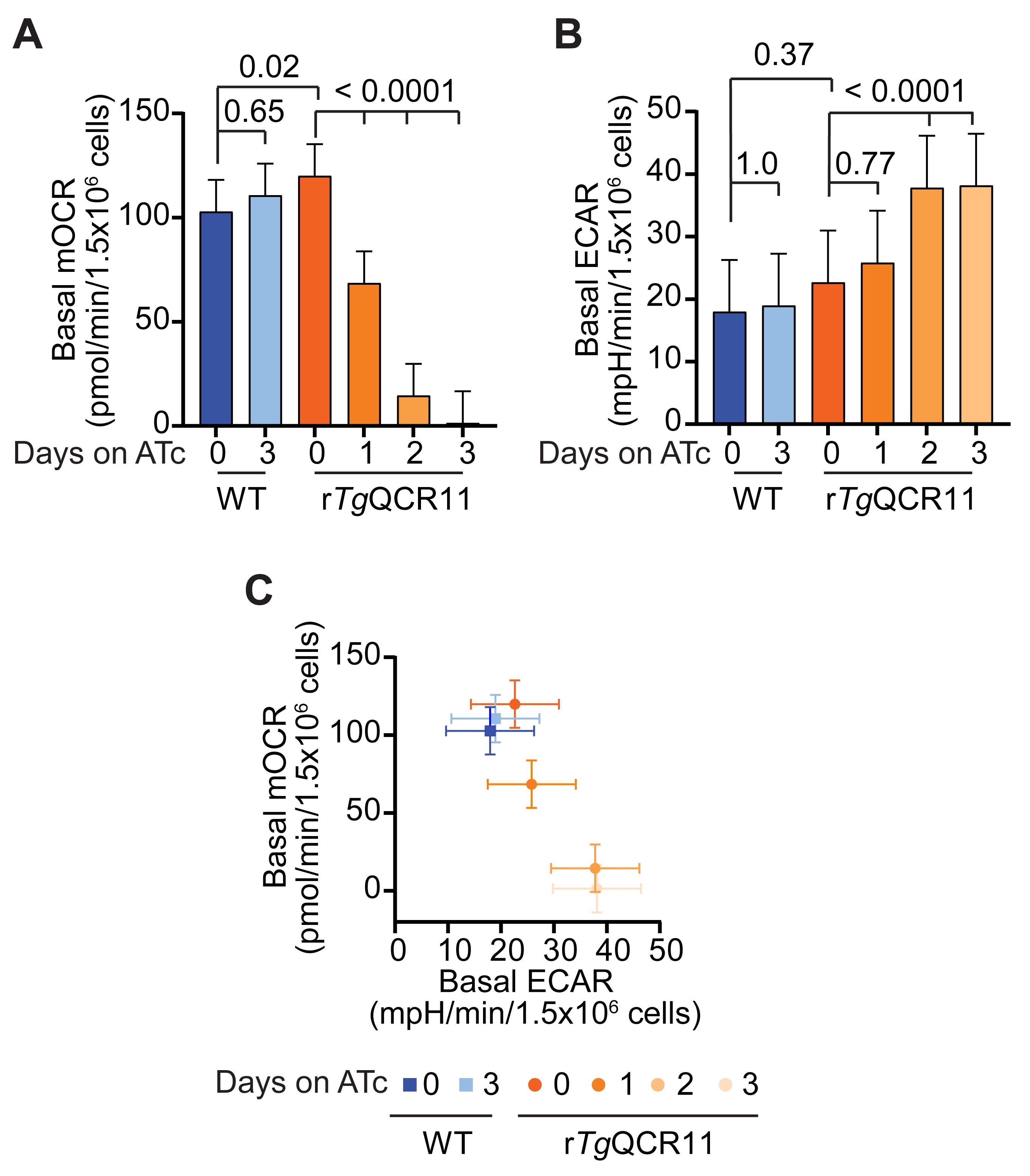
Figure 7. Example of analyzed basal mOCR and basal ECAR of intact T. gondii parasites in a MitoStress assay (Hayward et al., 2021). A-B. Column graphs depicting the (A) basal mOCR and (B) basal ECAR of WT (blue) and rTgQCR11 (orange) T. gondii parasites grown in the absence of ATc or in the presence of ATc for 1-3 days. A linear mixed effects model was fitted to the data and values depict the estimated marginal means ± 95% CI of three independent experiments. Tukey’s multiple pairwise comparison’s test was performed, with adjusted p-values shown. (C) Basal mOCR versus basal ECAR of WT (blue) and rTgQCR11 (orange) T. gondii parasites grown as described above. Data depict the estimated marginal mean mOCR and ECAR values ± 95% CI of three independent experiments fitted with a linear mixed effects model. Data are from Hayward et al. (2021).
Data analysis and calculation of substrate-elicited OCR
In the assay performed on permeabilized parasites described in Procedure Section C, the first OCR readings should be close to zero if the parasites have been starved effectively. If they are not, the starvation time or number of washes in Seahorse base medium may need to be increased. First, the substrate (malate or glycerol 3-phosphate plus FCCP) is injected (port A) to yield substrate-elicited OCR, then atovaquone is injected (port B) to assess non-mitochondrial OCR (Figure 6A and 6B). TMPD is injected (port C) to yield TMPD-elicited OCR, and the Complex IV inhibitor NaN3 is injected (port D) as validation that the TMPD-dependent OCR is mediated by the ETC upstream of Complex IV (Figure 6A and 6B).
The output from the Seahorse XFe96 assay is a trace of OCR over time for each condition tested, showing how the parasites responded to the various injections (Figure 6B-6D). Another way to depict the data from this experiment is to perform calculations on the data (Figure 6B) and present these data as column graphs (Figure 8). The first step in these calculations is to average the values obtained after injection of atovaquone to obtain the non-mitochondrial OCR (Figure 6B). The non-mitochondrial OCR can then be subtracted from each of the points after injection of malate/glycerol 3-phosphate, and each of the points after injection of TMPD, to yield the substrate-elicited mitochondrial OCR (mOCRSubstrate) and the TMPD-elicited mitochondrial OCR (mOCRTMPD), respectively (Figure 6B; Figure 8A-B). The ratio of these two values can then be taken to illustrate the extent to which TMPD is able to rescue the OCR defect (mOCRTMPD/Substrate) (Figure 8C).
While these calculations are quite simple and can be done manually in Excel, we have provided a Python script “pre_R_script.py” that performs these calculations and outputs three csv files containing mOCRSubstrate, mOCRTMPD, and mOCRTMPD/Substrate. The Python script then uses these csv files as inputs for subsequent analysis in the R software environment using the R script “treat_seahorse_data.r”. As described for analysis of the intact Seahorse XFe96 data in Data analysis Section A, we use a linear mixed effects model to analyze the data. Analysis of the estimated marginal means (previously referred to as least square means; Hayward et al., 2021) of the linear mixed effects model is also performed in R, and statistical differences are assessed using p-values adjusted using Tukey’s method. The Python script outputs the results of these analyses into 3 csv output files, and these analyzed data can then be used to create graphs in GraphPad Prism. We have provided three supplementary data files (rep1_perm, rep2_perm, and rep3_perm) for the permeabilized experiments using WT, rTgQCR11, and rTgApiCox25 parasites described in Hayward et al. (2021) that can be used as a test dataset (Figure 8). These can be downloaded from the “test_data” subfolder in a Bitbucket repository (https://bitbucket.org/ngMiaNG/seahorse-scripts/src/master/).
After the assay is finished, save the Wave results file to a USB drive or email the data to yourself.
Open the file using the Wave program.
Click on “Add - Overview” to view the plate map and results simultaneously.
Ensure that the “Background correction” box has been ticked – this subtracts the oxygen consumption rate of the blank wells from the experimental wells.
Note: If the OCR within the background wells is higher than the OCR within sample wells, negative values will result. This does sometimes happen in our experiments, particularly when knocking down an ETC protein. Our current analysis strategy includes these negative values in the analysis; see Data analysis Section C for further discussion of this point.
Inspect the data for any large outliers – occasionally, compounds do not inject properly from one of the ports or inject at incorrect times during the assay, leading to erroneous values in the data. If you have strong reason to believe an incorrect injection has occurred, you should exclude those wells from subsequent analyses by unselecting them.
Click on the “Export” button to export the results in Microsoft Excel format.
The relevant data in the Excel file is located in the ‘Rate’ tab. Notice that the data consists of the columns ‘measurement’, ‘well’, ‘group’, ‘time’, ‘OCR’, ‘ECAR’, and ‘PER’, with the rows representing the data for each well of the 96 well plate over the time points of the experiment.
After the assay has been performed three times independently, save Excel results files (named rep1_perm, rep2_ perm, and rep3_ perm) to a folder with only these files inside.
This is how the rTgQCR11 permeabilized test data files (rep1_perm, rep2_ perm, and rep3_ perm) were created. Note that other conditions were tested on each plate but the names of irrelevant wells have been removed.
Go to https://bitbucket.org/ngMiaNG/seahorse-scripts/src/master/, download the files from this repository and save into a folder. This contains the “pre_R_script.py” Python script, the “treat_seahorse_data.r” R script, and the “README” and “requirements” text files as well as test data obtained from the rTgQCR11 Seahorse XFe96 experiments.
Note: This is the same repository containing the files used to analyze the intact MitoStress Seahorse XFe96 experiments (Data analysis Section A).
Read the “README” to understand the requirements and how the scripts work.
Run “pre_R_script.py” Python script from the command line via a terminal (e.g., CMD).
The script has inbuilt help that can be accessed by typing: python3 “pre_R_script.py” -h into the command line.
The general structure of the command to run the script is as follows: python3 “pre_R_script.py” -i “path to input directory” -o “path to output directory” -n “number of measurement cycles, 3 or 5” --perm.
Note: The test data consists of 5 measurement cycles.
When --perm is selected, the “pre_R_script.py” Python script perform calculations of substrate dependent mOCR (sub_dependent_mOCR), TMPD dependent mOCR (TMPD_dependent_mOCR), and the ratio of TMPD to substrate dependent mOCR (ratio_TMPD_to_substrate) using the raw Excel results files (rep1_perm, rep2_ perm, and rep3_ perm) as inputs.
Note: In the calculation of ratio_TMPD_to_substrate, we typically exclude samples with substrate-dependent mOCR values less than 1, because these values disproportionately affect (and possibly overestimate) the ratio calculations.
After performing these calculations, the “pre_R_script.py” Python script calls upon the “treat_seahorse_data.r” R script to fit a linear mixed effects model to the data and perform statistical analyses (ANOVA and post hoc Tukey test).
Csv. files containing the R analyzed results will be written to the folder specified as the output directory. This will consist of the estimated marginal means, standard error, degrees of freedom, and 95% confidence intervals of the linear mixed effects model, as well as the statistical significance values from the post hoc Tukey test.
These results can be used to plot graphs in GraphPad Prism. Column graphs depicting the substrate dependent mOCR, TMPD dependent mOCR, and the ratio of TMPD to substrate dependent mOCR are typical ways we present the data (Figure 8).
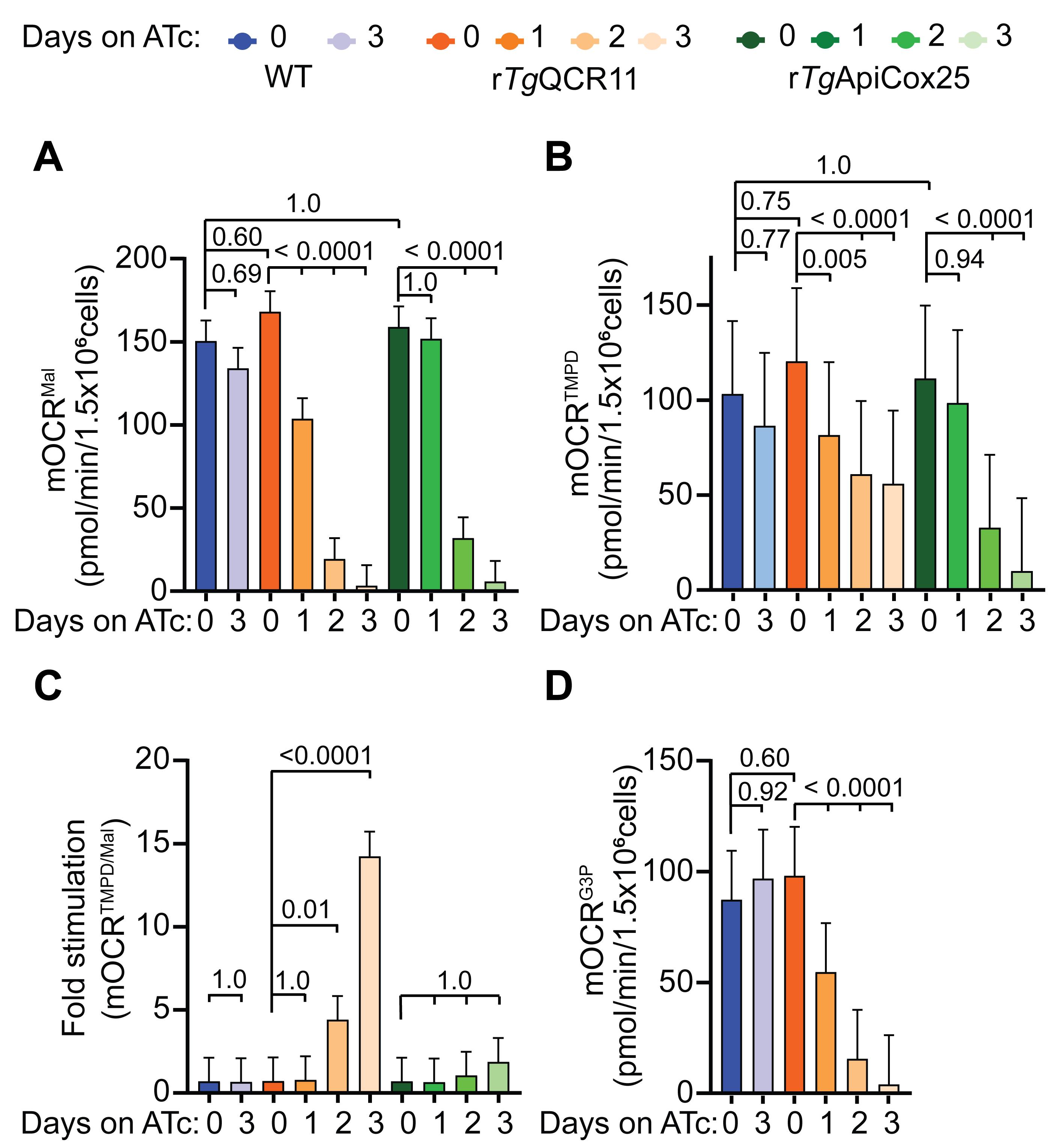
Figure 8. Example of analyzed mOCR of permeabilized parasites supplied various substrates (Hayward et al., 2021). A-D. Column graphs depicting the (A) malate dependent mOCR (mOCRMal), (B) TMPD dependent mOCR (mOCRTMPD), (C) fold stimulation of mOCR by TMPD relative to malate (mOCRTMPD/Mal), and (D) glycerol 3-phosphate dependent mOCR (mOCRG3P), of WT (blue), rTgQCR11 (orange), and rTgApiCox25 (green) T. gondii parasites grown in the absence of ATc or in the presence of ATc for 1-3 days. A linear mixed effects model was fitted to the data and values depict the estimated marginal means ± 95% CI from three independent experiments. Tukey’s multiple pairwise comparison’s test was performed, with adjusted p-values shown. The data in this figure were derived from a previous study (Hayward et al., 2021).
Comments on statistical analyses of the data
As mentioned in Data analysis Sections A and B, we use a linear mixed effects model to analyze the output data from both the intact MitoStress and permeabilized T. gondii Seahorse XFe96 assays. We use a linear mixed effects model because it helps in dealing with the rather complex data structure of these experiments. The different groups (e.g., WT and rTgQCR11) and days on drug (e.g., ATc days 0-3), contribute to the mean differences in OCR and ECAR that we wish to estimate. However, variation occurs between replicate wells and time point measurements within a plate (i.e., in technical replicates) and between replicate wells and time point measurements in experiments conducted on different days (i.e., in independent biological replicates). The linear mixed effects model allows us to separate the OCR and ECAR differences due to our factors of interest (‘fixed effects’) from the ‘random’ variation in the technical and biological replicates. To illustrate this point, we have plotted the basal mOCR of WT and rTgQCR11 parasites as boxplots, where each of the three independent experiments is shown in a different color and the values obtained in the different wells and timepoints of the experiment are also shown (Figure 9A). In these experiments, biological replicate 2 (rep_2, green) had consistently higher basal mOCR values across the different conditions (Figure 9A).
The linear mixed effects model compares conditions within a plate (i.e., within an experiment) to help account for this variation between experiments, and thereby measures the treatment effect as precisely as possible. To do this, we set the variation between plates (between experiments, or ‘biological replicates’) and wells (within experiments, or ‘technical replicates’) as random effects, and the variation between cell lines and days on drug (e.g., ATc) within the same experiment as fixed effects. The linear mixed effects model undertakes an analysis of variance in the data, and the statistical differences between the estimated marginal means are tested using adjusted p-values based on Tukey’s method. The results of this analysis suggest that, while ATc had no effect on the OCR of WT T. gondii parasites, knockdown of TgQCR11 by ATc resulted in significantly lower basal mOCR that was essentially zero by day 3 on ATc (Figure 9B).
Something to consider when using this approach is that ANOVA has an assumption of equal variances across the samples. When the box sizes of WT and rTgQCR11 parasites are compared, it is apparent that rTgQCR11 parasites grown for 2-3 days on ATc have smaller variance than the other conditions (Figure 9A), and so the assumption of equal variances is not strictly met by this dataset. One way to address this problem is to log transform the data before the linear mixed effects model analysis is conducted (the R-script can be easily modified to do this). When we plot the same basal mOCR data on a log scale (Figure 9C), the variance between conditions is somewhat more equal. Thus, log transforming the data prior to analysis better meets the ANOVA assumption of equal variances. When the log-transformed basal mOCR values are analyzed using the linear mixed effects model, the same general conclusion can be drawn from the data; specifically, that knockdown of TgQCR11 by ATc resulted in significantly lower basal mOCR that was essentially zero by day 3 (Figure 9D). While the means are generally the same, except for conditions in which negative values were excluded during the log transformation (see below), the 95% confidence interval error bars have changed. Furthermore, some comparisons that were statistically significant using the first approach are no longer so (e.g., WT day 0 vs rTgQCR11 day 0) (Figure 9D).
One key limitation of log transforming the data is that if the dataset contains negative values (as our rTgQCR11 dataset does), these values are excluded during the log transformation. In theory, negative mOCR values are not biologically possible as parasites do not generate oxygen. It is therefore likely that when negative values occur, the mOCR in those wells are just close to background and essentially zero. Since we often observe negative mOCR values when we knockdown proteins that function in the ETC (e.g., rTgQCR11 parasites grown for 3 days on ATc), we prefer not to simply exclude these values from the dataset as they are still meaningful (i.e., they reflect that the mOCR in those wells is close to zero). If we want these values to be included in an analysis using log-transformed data, then we need a way to make them non-negative before the log transformation is conducted. There is no clear or easy solution on how best to do this. One way is to assign the negative values an extremely small positive value instead; another is to identify the lowest negative value and add this value to all other values in the dataset (in effect, normalizing the data to the lowest negative value). However, neither of these approaches is particularly satisfactory, as the first would artificially reduce the variation in the condition containing the negative values (e.g., rTgQCR11 parasites grown for 3 days on ATc) and the second may lead to large negative outliers in the dataset overly influencing the rest of the dataset.
The approaches to data treatment and analysis that we describe in this section enable comparisons between different experimental conditions. We note that, depending on how the data are treated and analyzed, the calculated mOCR and ECAR values may differ, and researchers should be cautious in interpreting these as absolute values. Visualizing the dataset before conducting statistical analyses is important to appreciate the features of the data (e.g., the overall trend, distribution, variance, negative values). This will help inform which method of analysis is appropriate for the dataset, such as whether log transformation of the data is needed before using the linear mixed effects model. Consulting with a statistician about your particular dataset is also a good idea. And perhaps most importantly, being clear and open about how the data analysis was performed in the methods section of your publications will help improve the transparency and reproducibility of the Seahorse XFe96 experiments.
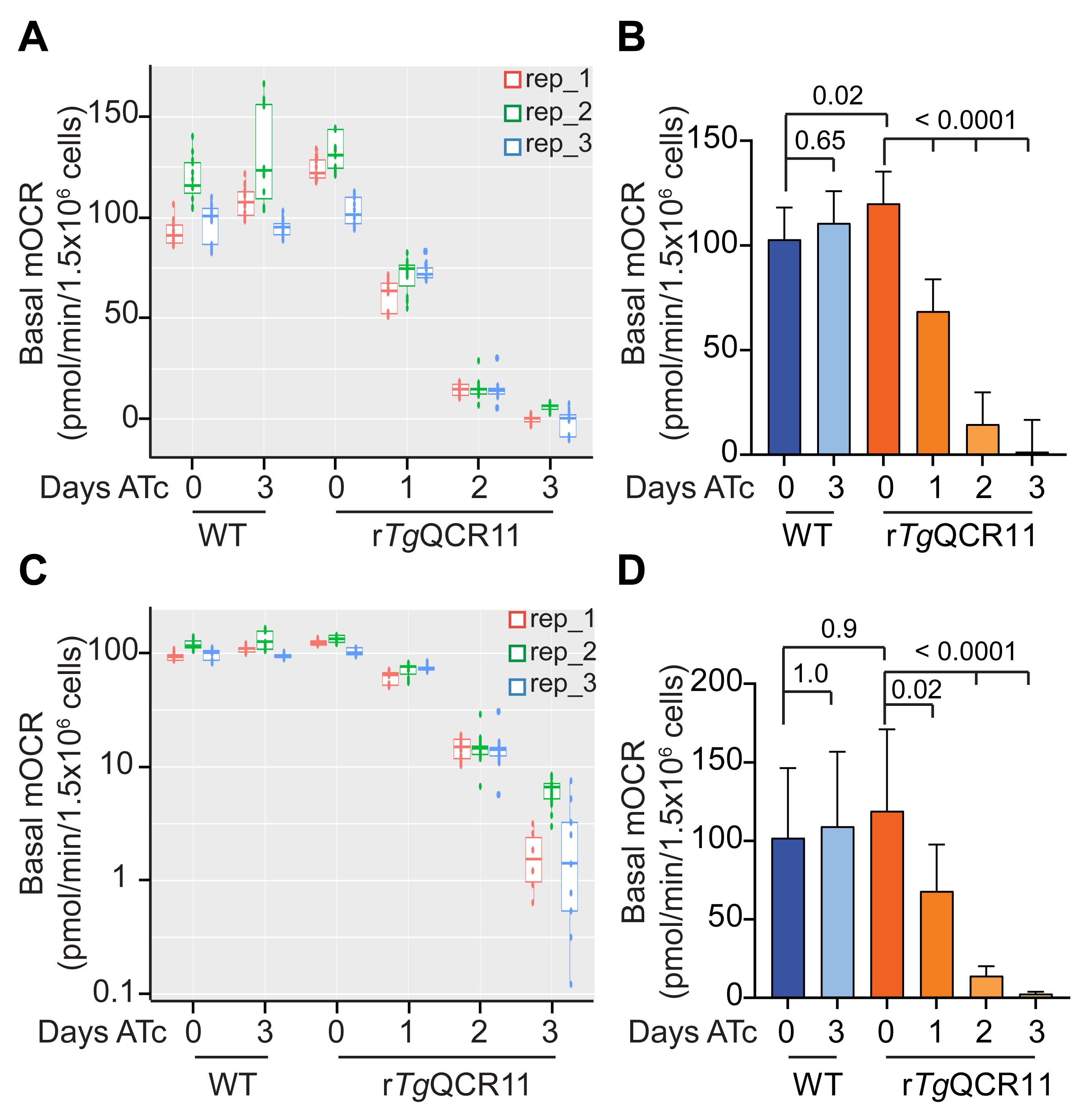
Figure 9. Comparison of two analysis strategies for basal mOCR data. A, C. Boxplots depicting the basal mitochondrial oxygen consumption rate (mOCR) of WT or rTgQCR11 T. gondii parasites grown in the absence or presence of ATc for 1-3 days, presented on a (A) linear or (C) log scale. Each of the three independent biological replicates are depicted (replicate 1 in red, replicate 2 in green, and replicate 3 in blue), with technical replicates (both different wells and different time point measurements) shown as dots. B, D. Column graphs depicting the basal mOCR of WT (blue) and rTgQCR11 (orange) T. gondii parasites grown in the absence or presence of ATc for 1-3 days. A linear mixed effects model was fitted to the (B) linear or (D) log transformed basal mOCR data. Values depict the estimated marginal means ± 95% CI of three independent experiments. Tukey’s multiple comparisons test was performed, with adjusted p-values shown. (B) is the same as Figure 7A , derived from Hayward et al. (2021), and is shown here for easy comparison to (D).
Recipes
Sodium bicarbonate (0.1 M)
Dissolve 420 mg of sodium bicarbonate in 50 mL of MilliQ water.
Filter sterilize using a 0.2 µm filter.
Store at RT. This solution is used to activate the Cell-Tak solution to coat the Seahorse XFe96 culture plate.
Glutamine stock (200 mM)
Dissolve 1.461 g of L-glutamine in ~45 mL of Seahorse XF base medium, adjust pH to 7.4, and adjust volume to 50 mL.
Filter sterilize using a 0.2 μm filter.
Store at -20°C until needed.
Glucose stock (1 M)
Dissolve 9 g of D-glucose in ~45 mL of Seahorse XF base medium, measure the pH and if necessary adjust to 7.4, and adjust volume to 50 mL.
Filter sterilize using a 0.2 μm filter.
Store at 4°C until needed.
Base medium with glucose (5 mM) and glutamine (1 mM) (BM+Glc+Gln)
Add 250 μL of Glucose stock (1 M) and 250 μL of Glutamine stock (200 mM) to 50 mL of Seahorse XF base medium.
We recommend making this solution fresh each time the assay is performed.
FCCP stock (50 mM)
Dissolve 10 mg of FCCP in 787 μL of DMSO.
Aliquot smaller volumes and store at -20°C until needed.
Atovaquone stock (10 mM)
Dissolve 10 mg of atovaquone in 2.726 mL of DMSO.
Aliquot smaller volumes and store at -20°C until needed.
Digitonin stock (5% w/v)
Dissolve 100 mg of digitonin in 2 mL of MilliQ water.
Can be kept at 4°C until needed.
Digitonin will precipitate at colder temperatures so briefly heat at ~90°C to bring digitonin back into solution immediately before use.
Mitochondrial Assay Solution (MAS) buffer (1×, 500 mL)
Mannitol 20.039 g, 220 mM
Sucrose 11.981 g, 70 mM
KH2PO4 680 mg, 10 mM
MgCl2 508 mg, 5 mM
HEPES 238 mg, 2 mM
EGTA 190 mg, 1 mM
Fatty acid free BSA*, 0.2% (w/v)
Dissolve all reagents but BSA in ~400 mL of MilliQ water; adjust pH to 7.4 with potassium hydroxide (KOH); make up to 500 ml. Filter sterilize using a 0.2 μm filter for long-term storage at 4°C.
*Note: Add 100 mg of fatty acid free BSA to 50 mL of MAS buffer on the day of the assay.
Malate stock (320 mM)
Dissolve 858 mg of L-malic acid in ~10 mL of MAS buffer.
Adjust pH to 7.4 with potassium hydroxide (KOH); make up to 20 mL.
Aliquot smaller volumes and store at -20°C until needed.
Glycerol 3-phosphate stock (200 mM)
Dissolve 741 mg of sn-Glycerol 3-phosphate bis(cyclohexylammonium) salt in ~5 mL of MAS buffer.
Adjust pH to 7.4 with potassium hydroxide (KOH); make up to 10 mL. Aliquot smaller volumes and store at -20°C until needed.
TMPD stock (500 mM)
Dissolve 411 mg of N,N,N′,N′-Tetramethyl-p-phenylenediamine in 5 mL of ethanol.
Aliquot smaller volumes and store at -20°C until needed.
Ascorbate stock (200 mM)
Dissolve 704 mg of L-ascorbic acid in ~10 mL of MAS buffer.
Adjust pH to 7.4 with potassium hydroxide (KOH); make up to 20 mL.
Aliquot smaller volumes and store at -20°C until needed.
Sodium azide stock (110 mM)
Dissolve 143 mg of sodium azide in 20 mL of MAS buffer.
Aliquot smaller volumes and store at -20°C until needed.
Acknowledgments
This work was supported by an Australian Government Research Training Program Scholarship to J.A.H., an Australian National University Research School of Biology innovation grant to E.R. and G.G.v.D., and a National Health and Medical Research Council Ideas grant (GNT1182369) to G.G.v.D.
Competing interests
The authors declare that no competing interests exist.
References
- Alday, P. H. and Doggett, J. S. (2017). Drugs in development for toxoplasmosis: advances, challenges, and current status. Drug Des Devel Ther 11: 273-293.
- Aw, Y. T. V., Seidi, A., Hayward, J. A., Lee, J., Makota, F. V., Rug, M. and van Dooren, G. G. (2021). A key cytosolic iron-sulfur cluster synthesis protein localizes to the mitochondrion of Toxoplasma gondii. Mol Microbiol 115(5): 968-985.
- Brooks, C. F., Johnsen, H., van Dooren, G. G., Muthalagi, M., Lin, S. S., Bohne, W., Fischer, K. and Striepen, B. (2010). The toxoplasma apicoplast phosphate translocator links cytosolic and apicoplast metabolism and is essential for parasite survival. Cell Host Microbe 7(1): 62-73.
- Clark, L. C., Jr., Wolf, R., Granger, D. and Taylor, Z. (1953). Continuous recording of blood oxygen tensions by polarography. J Appl Physiol 6(3): 189-193.
- Divakaruni, A. S., Paradyse, A., Ferrick, D. A., Murphy, A. N. and Jastroch, M. (2014). Analysis and interpretation of microplate-based oxygen consumption and pH data. Methods Enzymol 547: 309-354.
- Divo, A. A., Geary, T. G., Jensen, J. B. and Ginsburg, H. (1985). The mitochondrion of Plasmodium falciparum visualized by rhodamine 123 fluorescence. J Protozool 32(3): 442-446.
- Doggett, J. S., Nilsen, A., Forquer, I., Wegmann, K. W., Jones-Brando, L., Yolken, R. H., Bordon, C., Charman, S. A., Katneni, K., Schultz, T., et al. (2012). Endochin-like quinolones are highly efficacious against acute and latent experimental toxoplasmosis. Proc Natl Acad Sci U S A 109(39): 15936-15941.
- Evers, F., Cabrera-Orefice, A., Elurbe, D. M., Kea-Te Lindert, M., Boltryk, S. D., Voss, T. S., Huynen, M. A., Brandt, U. and Kooij, T. W. A. (2021). Composition and stage dynamics of mitochondrial complexes in Plasmodium falciparum. Nat Commun 12(1): 3820.
- Fisher, N., Meunier, B. and Biagini, G. A. (2020). The cytochrome bc1 complex as an antipathogenic target. FEBS Lett 594(18): 2935-2952.
- Fry, M. and Beesley, J. E. (1991).Mitochondria of mammalian Plasmodium spp. Parasitology 102 Pt 1: 17-26.
- Fry, M. and Pudney, M. (1992). Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4'-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem Pharmacol 43(7): 1545-1553.
- Hayward, J. A. and van Dooren, G. G. (2019). Same same, but different: Uncovering unique features of the mitochondrial respiratory chain of apicomplexans. Mol Biochem Parasitol 232: 111204.
- Hayward, J. A., Rajendran, E., Zwahlen, S. M., Faou, P. and van Dooren, G. G. (2021). Divergent features of the coenzyme Q:cytochrome c oxidoreductase complex in Toxoplasma gondii parasites. PLoS Pathog 17(2): e1009211.
- Huet, D., Rajendran, E., van Dooren, G. G. and Lourido, S. (2018). Identification of cryptic subunits from an apicomplexan ATP synthase. Elife 7: e38097.
- Lin, S. S., Kerscher, S., Saleh, A., Brandt, U., Gross, U. and Bohne, W. (2008). The Toxoplasma gondii type-II NADH dehydrogenase TgNDH2-I is inhibited by 1-hydroxy-2-alkyl-4(1H)quinolones. Biochim Biophys Acta 1777(11): 1455-1462.
- Maclean, A. E., Bridges, H. R., Silva, M. F., Ding, S., Ovciarikova, J., Hirst, J. and Sheiner, L. (2021). Complexome profile of Toxoplasma gondii mitochondria identifies divergent subunits of respiratory chain complexes including new subunits of cytochrome bc1 complex. PLoS Pathog 17(3): e1009301.
- McFadden, D. C., Tomavo, S., Berry, E. A. and Boothroyd, J. C. (2000). Characterization of cytochrome b from Toxoplasma gondii and Q(o) domain mutations as a mechanism of atovaquone-resistance. Mol Biochem Parasitol 108(1): 1-12.
- Mitchell, P. (1975). The protonmotive Q cycle: a general formulation. FEBS Lett 59(2): 137-139.
- Mookerjee, S. A., Goncalves, R. L. S., Gerencser, A. A., Nicholls, D. G. and Brand, M. D. (2015). The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta 1847(2): 171-181.
- Moreno, S. N., Zhong, L., Lu, H. G., Souza, W. D. and Benchimol, M. (1998). Vacuolar-type H+-ATPase regulates cytoplasmic pH in Toxoplasma gondii tachyzoites. Biochem J 330 ( Pt 2): 853-860.
- Mühleip, A., Kock Flygaard, R., Ovciarikova, J., Lacombe, A., Fernandes, P., Sheiner, L. and Amunts, A. (2021). ATP synthase hexamer assemblies shape cristae of Toxoplasma mitochondria. Nat Commun 12(1): 120.
- Pappas, G., Roussos, N. and Falagas, M. E. (2009). Toxoplasmosis snapshots: global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. Int J Parasitol 39(12): 1385-1394.
- Pfefferkorn, E. R., Borotz, S. E. and Nothnagel, R. F. (1993). Mutants of Toxoplasma gondii resistant to atovaquone (566C80) or decoquinate. J Parasitol 79(4): 559-564.
- Plitzko, B. and Loesgen, S. (2018). Measurement of Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) in Culture Cells for Assessment of the Energy Metabolism. Bio-protocol 8(10): e2850.
- Sakata-Kato, T. and Wirth, D. F. (2016). A Novel Methodology for Bioenergetic Analysis of Plasmodium falciparum Reveals a Glucose-Regulated Metabolic Shift and Enables Mode of Action Analyses of Mitochondrial Inhibitors. ACS Infect Dis 2(12): 903-916.
- Saleh, A., Friesen, J., Baumeister, S., Gross, U. and Bohne, W. (2007). Growth inhibition of Toxoplasma gondii and Plasmodium falciparum by nanomolar concentrations of 1-hydroxy-2-dodecyl-4(1H)quinolone, a high-affinity inhibitor of alternative (type II) NADH dehydrogenases. Antimicrob Agents Chemother 51(4): 1217-1222.
- Salunke, R., Mourier, T., Banerjee, M., Pain, A. and Shanmugam, D. (2018). Highly diverged novel subunit composition of apicomplexan F-type ATP synthase identified from Toxoplasma gondii. PLoS Biol 16(7): e2006128.
- Schmidt, O., Pfanner, N. and Meisinger, C. (2010). Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol 11(9): 655-667.
- Seidi, A., Muellner-Wong, L. S., Rajendran, E., Tjhin, E. T., Dagley, L. F., Aw, V. Y., Faou, P., Webb, A. I., Tonkin, C. J. and van Dooren, G. G. (2018). Elucidating the mitochondrial proteome of Toxoplasma gondii reveals the presence of a divergent cytochrome c oxidase. Elife 7: e38131.
- Shah-Simpson, S., Pereira, C. F., Dumoulin, P. C., Caradonna, K. L. and Burleigh, B. A. (2016). Bioenergetic profiling of Trypanosoma cruzi life stages using Seahorse extracellular flux technology. Mol Biochem Parasitol 208(2): 91-95.
- Smith, N. C., Goulart, C., Hayward, J. A., Kupz, A., Miller, C. M. and van Dooren, G. G. (2021). Control of human toxoplasmosis. Int J Parasitol 51(2-3): 95-121.
- Srivastava, I. K., Rottenberg, H., and Vaidya, A. B. (1997). Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J Biol Chem 272(7): 3961-3966.
- Symersky, J., Osowski, D., Walters, D. E. and Mueller, D. M. (2012). Oligomycin frames a common drug-binding site in the ATP synthase. Proc Natl Acad Sci U S A 109(35): 13961-13965.
- Tjhin, E. T., Hayward, J. A., McFadden, G. I. and van Dooren, G. G. (2020). Characterization of the apicoplast-localized enzyme TgUroD in Toxoplasma gondii reveals a key role of the apicoplast in heme biosynthesis. J Biol Chem 295(6): 1539-1550.
- Vercesi, A. E., Rodrigues, C. O., Uyemura, S. A., Zhong, L. and Moreno, S. N. (1998). Respiration and oxidative phosphorylation in the apicomplexan parasite Toxoplasma gondii. J Biol Chem 273(47): 31040-31047.
- Zeng, J. M., Hapuarachchi, S. V., Shafik, S. H., Martin, R. E., Kirk, K., van Dooren, G. G. and Lehane, A. M. (2021). Identifying the major lactate transporter of Toxoplasma gondii tachyzoites. Sci Rep 11(1): 6787.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hayward, J. A., Rajendran, E., Makota, F. V., Bassett, B. J., Devoy, M., Neeman, T. and van Dooren, G. G. (2022). Real-Time Analysis of Mitochondrial Electron Transport Chain Function in Toxoplasma gondii Parasites Using a Seahorse XFe96 Extracellular Flux Analyzer. Bio-protocol 12(1): e4288. DOI: 10.21769/BioProtoc.4288.
Category
Microbiology > Microbial cell biology > Cell-based analysis > Reporter assay
Cell Biology > Cell-based analysis > Mitochondrial respiration
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.