- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Selection of Vaccinia Virus Recombinants Using CRISPR/Cas9
Published: Vol 11, Iss 24, Dec 20, 2021 DOI: 10.21769/BioProtoc.4270 Views: 3441
Reviewed by: ASWAD KHADILKARBruno HernaezIsmar HagaJonas Albarnaz
Original research article
The authors used this protocol in:

Nov 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
The engineering of poxvirus genomes is fundamental to primary and applied virology research. Indeed, recombinant poxviruses form the basis for many novel vaccines and virotherapies but producing and purifying these viruses can be arduous. In recent years, CRISPR/Cas9 has become the favoured approach for genome manipulation due to its speed and high success rate. However, recent data suggests poxvirus genomes are not repaired well following Cas9 cleavage. As a result, CRISPR/Cas9 is inefficient as an editing tool, but very effective as a programmable selection agent. Here, we describe protocols for the generation and enrichment of recombinant vaccinia viruses using targeted Cas9 as a selection tool. This novel use of Cas9 is a simple addition to current homologous recombination-based methods that are widespread in the field, facilitating implementation in laboratories already working with poxviruses. This is also the first method that allows for isolation of new vaccinia viruses in less than a fortnight, without the need to incorporate a marker gene or manipulation of large poxvirus genomes in vitro and reactivation with helper viruses. Whilst this protocol describes applications for laboratory strains of vaccinia virus, it should be readily adaptable to other poxviruses.
Graphic abstract:
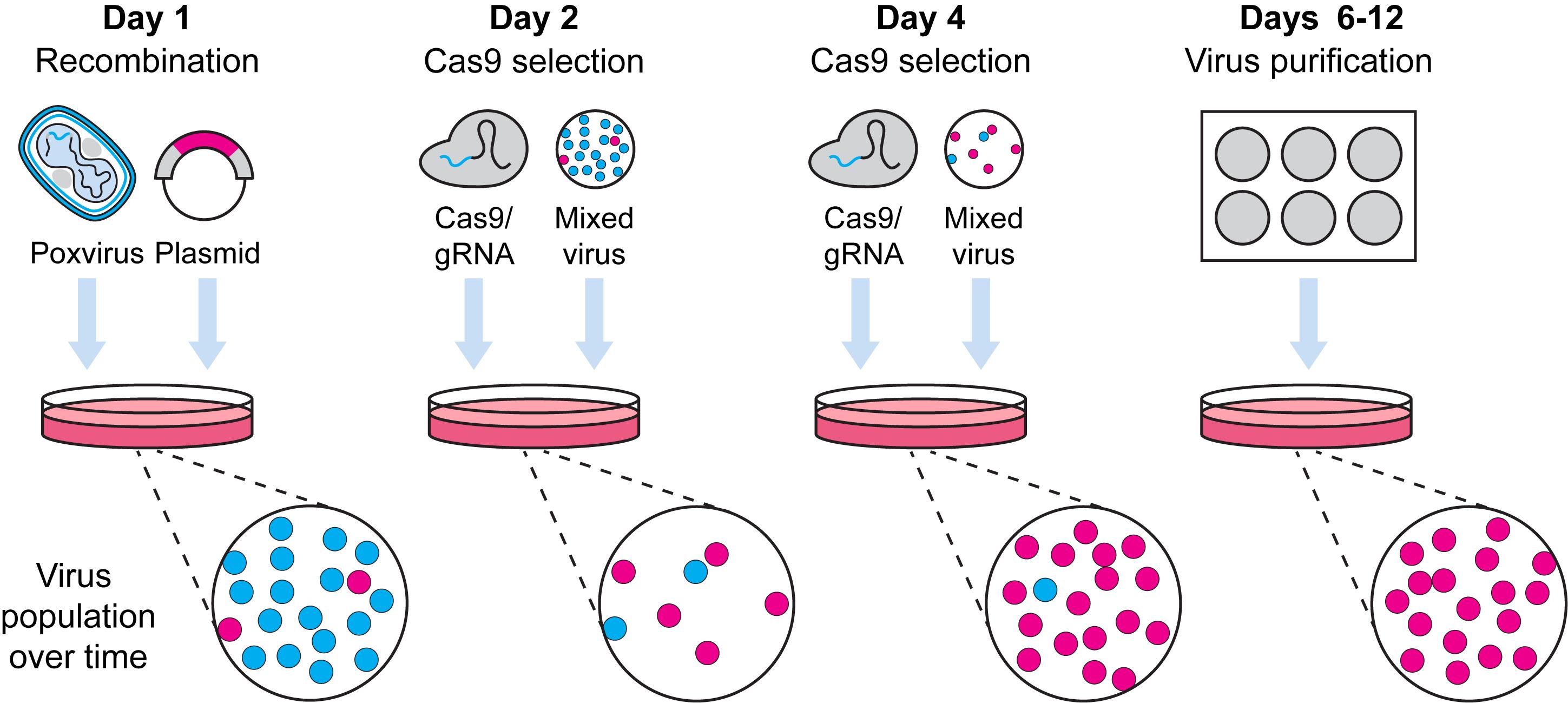
Pipeline for Cas9 selection of recombinant poxviruses.
Background
Poxviruses are important research viruses, both in the context of infection models and as vectors for many virotherapeutics. A significant part of their usefulness lies in their genomes, which are amenable to substantial manipulation (Smith and Moss, 1983). This allows researchers to incorporate foreign genes, such as those required for immunomodulation or markers for virus tracking, to remove viral genes, as is often required for pathogenesis studies, and to introduce point-mutations of interest. However, existing processes for manipulating poxvirus genomes, including homologous recombination or transient-dominant selection, can be inefficient and time-consuming.
CRISPR/Cas9 editing has wide applications that includes modifying viruses with double-stranded DNA genomes (Russell et al., 2015; Yuen et al., 2015; King and Munger, 2019). These methods can be thought of as using a two-step, “cut and paste” approach for DNA manipulation – Cas9 nuclease is first directed to target sites by guide RNA molecules (gRNAs) and forms double-stranded DNA breaks, then cellular enzymes repair these sites introducing mutations. In the absence of repair templates, DNA repair proceeds via non-homologous end joining, which is error-prone and can lead to the incorporation of random indels. Alternatively, when a repair template having ends that share identity with sequences that flank the target site is present, specific genetic changes can be made. However, for poxviruses that replicate in the cell cytoplasm, repair of Cas9 cleavage is very inefficient (Gowripalan et al., 2020). This has a profound antiviral effect and allows the repurposing of targeted Cas9 as a selection tool rather than as one that generates gene edits.
Here, we describe a method for Cas9 selection of recombinant vaccinia viruses (VACV) (Figure 1). To make a complete protocol, we start by sharing our method for generating VACV by homologous recombination. There are many other variations of this method and any of these should be equally effective. This is followed by our method for using Cas9/gRNA complexes to target parent virus genomes, removing these and leading to enrichment of the desired recombinant. Multiple rounds of selection further increase the selective pressure and, over time, we observe recombinant viruses becoming the dominant population. The process is robust, allowing for generation of both marker-containing and marker-free viruses. Our protocol is for laboratory strains of VACV but should be adaptable to other poxviruses, the proviso being that a cell type that is permissive for the virus and is highly transfectable can be found for selection. It should be noted that the requirement for high transfection efficiency is greater for Cas9 selection than for the initial recombination.
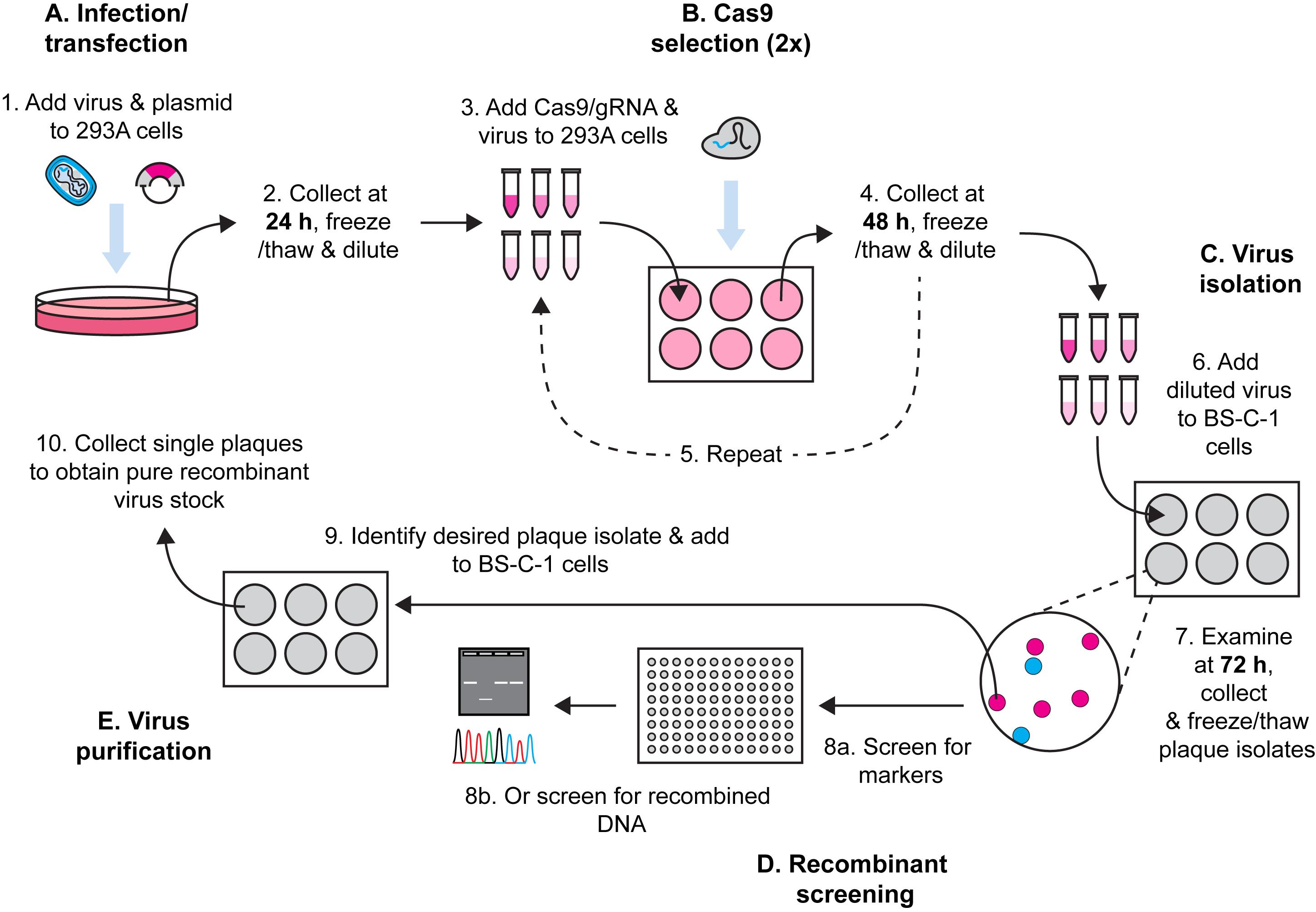
Figure 1. Workflow for generation of recombinant VACVs using Cas9 selection.
Materials and Reagents
6-well plates, flat bottom (Corning® Costar, catalog number: CLS3516)
96-well plates, flat bottom (Corning® Costar, catalog number: CLS3595)
Pipette tips
Serological pipettes
2.0 ml cryogenic vials (Corning®, catalog number: 430659)
2.0 ml Sarstedt tubes (Sarstedt, catalog number: 72.693.005)
5 ml Sarstedt tubes (Sarstedt, catalog number: 60.9921.532)
30 ml Sarstedt tubes (Sarstedt, catalog number: 60.9922.216)
8-strip Sarstedt PCR tubes (Sarstedt, catalog number: 72.991.002)
Dulbecco’s Modified Eagle’s Medium – high glucose (Millipore Sigma, catalog number: D5671)
Dulbecco’s Modified Eagle’s Medium – high glucose, no glutamine, no phenol red (Thermo Fisher Scientific, catalog number: 31053028)
L-Glutamine (200 mM) (Thermo Fisher Scientific, catalog number: 25030081)
Fetal bovine serum (FBS) (Millipore Sigma, catalog number: 12003C-500 mL)
Trypsin-EDTA (0.5%), no phenol red (Thermo Fisher Scientific, catalog number: 15400054)
Dulbecco’s Phosphate Buffered Saline (Millipore Sigma, catalog number: D8537)
LipofectamineTM 2000 (Thermo Fisher Scientific, catalog number: 11668019)
293A (Human embryonic kidney epithelial) cells (Thermo Fisher Scientific, catalog number: R70507)
BS-C-1 (African Green monkey kidney epithelial) cells (ATCC, catalog number: CCL-26)
VACV WR (NIH TC-adapted; ATCC VR1354), often referred to as Western Reserve (source was B. Moss, National Institutes of Health). Other replication competent strains should behave similarly.
VACV mCherry (derivative of VACV WR) (Gowripalan et al., 2020)
Cas9 nuclease, S. pyogenes with 10× Cas9 nuclease reaction buffer (New England Biolabs, catalog number: M0386M)
gRNAs purchased as Ultramer® RNA Oligos, stored at -80°C (Integrated DNA Technologies, see Note 1)
Plasmids for recombination synthesized prior to experimentation and stored at -20°C (see Note 2)
NucleoSpin Plasmid Mini Kit (Machery-Nagel, catalog number: 740588.250)
Carboxymethyl cellulose (CMC) sodium salt (Millipore Sigma, catalog number: C4888-500G)
100% ethanol (Ajax AR Grade, catalog number: AJA214)
Sharpie® Ultra Fine Permanent Marker (Sharpie, catalog number: 37001)
Taq DNA polymerase with ThermoPol® Buffer (New England Biolabs, catalog number: M0267X)
Proteinase K (Fungal) (Invitrogen, catalog number: 25530-015)
Stocks are typically made at 10 mg/ml in 50 mM Tris pH 8 and stored at -20°C and dilutions are made in nuclease-free water.dNTP Mix (Bioline, catalog number: BIO39053)
PCR primers purchased as Custom DNA oligos and stored at -20°C (Integrated DNA Technologies)
Gel loading dye, Purple (6×) (New England Biolabs, catalog number: B7024S)
UltraPureTM Agarose (Thermo Fisher Scientific, catalog number: 16500500)
SYBRTM Safe DNA Gel Stain (Thermo Fisher Scientific, catalog number: S33102)
D0 media (see Recipes)
D2 media (see Recipes)
D2 media – no phenol red (see Recipes)
D10 media (see Recipes)
D10 media – no phenol red (see Recipes)
4% CMC/D2 – no phenol red (see Recipes)
Equipment
Class 2 biosafety cabinet (Euroclone, model: Safemate Vision 1.2 ABC)
Incubator with CO2 (Thermo Fisher Scientific, model: Heracell 150i CO2)
-80°C freezer (Thermo Fisher Scientific, model: Forma 705 -80°C)
Branson Analog Sonifier S-450 Sonicator (Branson, model: S-450; 102 with 3” cup)
Olympus CKX53 inverted microscope (Olympus, model: N5731900)
Dry-block heater with digital control (Major Science, catalog number: MD-02N)
Veriti® PCR thermocycler (Applied Biosystems, catalog number: 4375786)
Table-top centrifuge (Eppendorf, model: Centrifuge 5424)
Procedure
Infection and transfection
Perform all work in a class 2 biosafety cabinet.
Seed 1 × 106 293A cells/well in 2 ml D10 media (Recipes 4 and 5) in a 6-well plate (Note 3).
We found this cell number to be ideal for achieving 70% confluency and active cell growth, the desired conditions for transfection.Incubate cells overnight at 37°C, 5% CO2.
The next day, thaw VACV stock at room temperature.
Note: This should be the parent vaccinia stock that is to be modified.
Sonicate virus stock 3 times (20 s each, 5 s break in between) on ice.
Dilute virus stock to 5 × 104 pfu/ml in D0 media (see Recipe 1), allowing 1 ml for each well. This gives in the order of 0.05 pfu/cell, based on the number of cells plated.
Aspirate media from 293A cells and replace with diluted virus.
Incubate cells for 1 h at 37°C, 5% CO2.
Note: Rocking of plates during virus inoculations such as these is routine in some laboratories but is unnecessary at any stage of this protocol.
Dilute 2 µg recombinant plasmid DNA in 120 µl D0 media and incubate at room temperature for 10 min (Table 1). Label tube “Mix 1”.
Note recombinant plasmids can be synthesized using any desired method (see Note 2).
Recommended: It is advised to perform transfections in duplicate, so as to have two distinct lineages of virus and ensure acquisition of desired recombinant.
Table 1. Preparation of DNA for transfection (Mix 1)
Component Volume (µl) Per well 3 wells Plasmid DNA 2 µg 6 µg D0 media Up to 120 Up to 360 Total 120 360 Prepare “Mix 2” by adding 4 µl LipofectamineTM 2000 reagent to 200 µl D0 media and leave for 5 min (Table 2).
Table 2. Preparation of LipofectamineTM 2000 mixture (Mix 2)
Component Volume (µl) Per well 3 wells LipofectamineTM 2000 (Thermo Fisher Scientific) 4 12 D0 media 196 588 Total 200 600 Combine “Mix 1” and “Mix 2” to a final volume of 320 µl/well and incubate at room temperature for 20-30 min.
After 1 h incubation, remove virus-containing media from cells and discard.
Add 680 µl fresh D0 media to each well.
In a dropwise manner, add 320 µl of DNA/LipofectamineTM mix to each well and rock the plate gently.
Incubate plate for 4 h at 37°C, 5% CO2.
Remove media and replace with 2 ml D2 media (see Recipes 2 and 3).
Incubate plate for 24 h at 37°C, 5% CO2.
At 24 h post-infection, collect cells and supernatant by pipetting up and down vigorously and transferring to a 2 ml cryogenic vial.
Note: If strongly adherent cells are used, cells can be scraped into the media.
Freeze cells and supernatant rapidly using a dry-ice/ethanol bath, and thaw using a 37°C waterbath or heat block. Cycle through freezing and thawing three times to release virus (or store at -80°C till ready for Cas9 selection).
Proceed to Section B for selection of recombinant viruses.
Cas9 selection
Perform all work in a class 2 biosafety cabinet.
Seed 1 × 106 293A cells/well in a 6-well plate and incubate overnight at 37°C, 5% CO2.
Allow for 6 wells per tube of collected cells from A (see Note 4).
For selection, prepare ribonucleoprotein (RNP) complexes of Cas9 and gRNA by mixing the components in Table 3.
Note: gRNAs should be kept on ice and used in aliquots to prevent repeated freeze-thaw cycles.
Table 3. Preparation of Cas9/gRNA RNP complexes
Component Volume (µl) Per well 6 wells Cas9 nuclease, S. pyogenes (NEB) 0.67 4 10× Cas9 nuclease reaction buffer (NEB) 0.67 4 gRNA (2 µg/µl) 1.00 6 Nuclease-free water 4.33 26 Total 6.67 40 Incubate Cas9/gRNA RNP mix for 10 min at room temperature, then add D0 media for a total of 120 µl per well (e.g., for 6 wells this is 680 µl of D0, for a total volume of 720 µl).
During the incubation in step B4, prepare LipofectamineTM 2000 mix as in Table 2 to a final volume of 200 µl per well.
Combine Cas9/gRNA mix and LipofectamineTM 2000 giving a final volume of 320 µl (120 µl RNP + 200 µl LipofectamineTM 2000) per well and let sit for 20-30 min.
Remove media from cells and replace with 680 µl fresh D0 media.
In a dropwise manner, add 320 µl of Cas9/LipofectamineTM 2000 transfection mix to each well.
Incubate for 4 h at 37°C, 5% CO2.
Using the freeze-thawed tubes of virus from A (which should include the desired recombinant), sonicate 3 times for 20 s each on ice, with 5 s breaks.
Prepare 1/5 serial dilutions of virus in 1.25 ml D0 media, changing tip between each tube. Start with a 1/25 dilution (Figure 2).
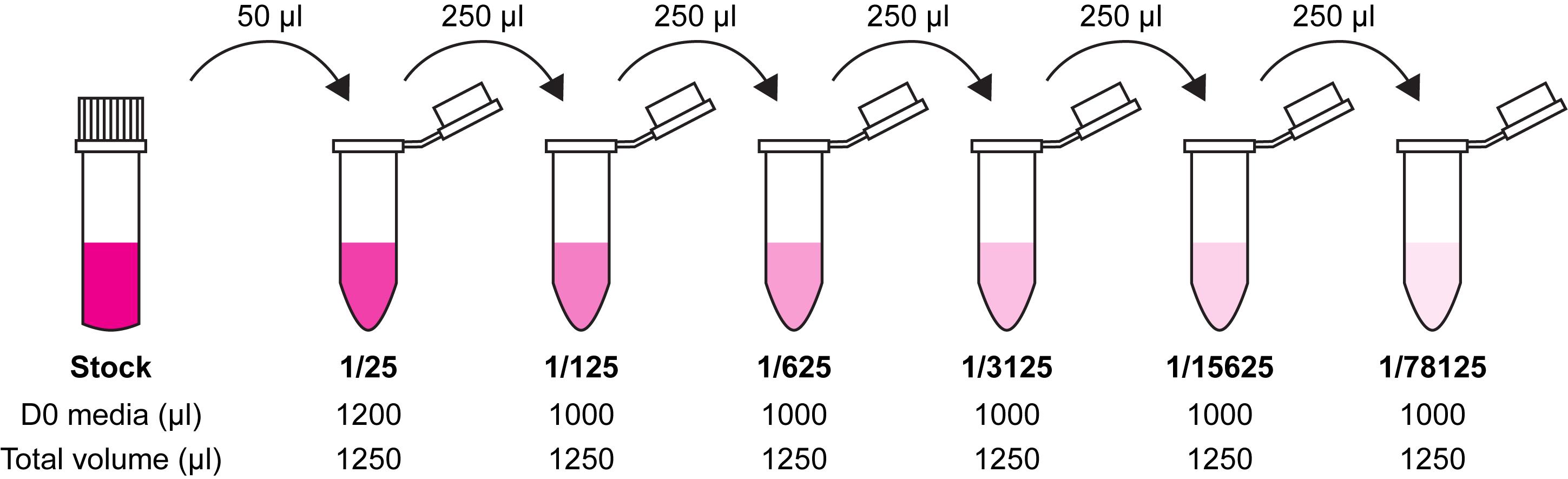
Figure 2. Preparation of 1/5 serial dilutions for plating out virus. Take 50 µl of virus stock after freeze-thawing and use to make a 1/25 virus dilution in D0 media. Take 250 µl of this stock and add to a further tube containing 1 ml D0 media. Continue until 6 dilutions have been prepared, up to 1/78125.Aspirate media from cells at 4 h post-transfection and add 1 ml of each virus dilution.
Incubate for 1 h at 37°C, 5% CO2.
Replace virus with 2 ml fresh D2 media (or D2 without phenol red if you wish to observe fluorescence) and incubate for 48 h at 37°C, 5% CO2.
Optional: If a visual marker is included in the recombinant virus design, wells with higher dilutions can be examined by microscopy to identify and count plaques that have the marker and those with parent phenotype to assess frequency of desired recombinant.
Note: If enough virus with the desired marker is seen at this stage a single round of selection might be adequate and steps B18-B21 can be skipped.
Use an inverted microscope to look for evidence of virus infection (cytopathic effect, CPE). Starting from the most- to least-infected, identify the first well in which the CPE is not uniform across the monolayer (Figure 3). Collect cells and supernatant from this well by pipetting up and down vigorously (scraping first if necessary) and transfer to a 2 ml cryogenic vial.
Optional: Wells on either side can be harvested similarly and kept as back-ups.
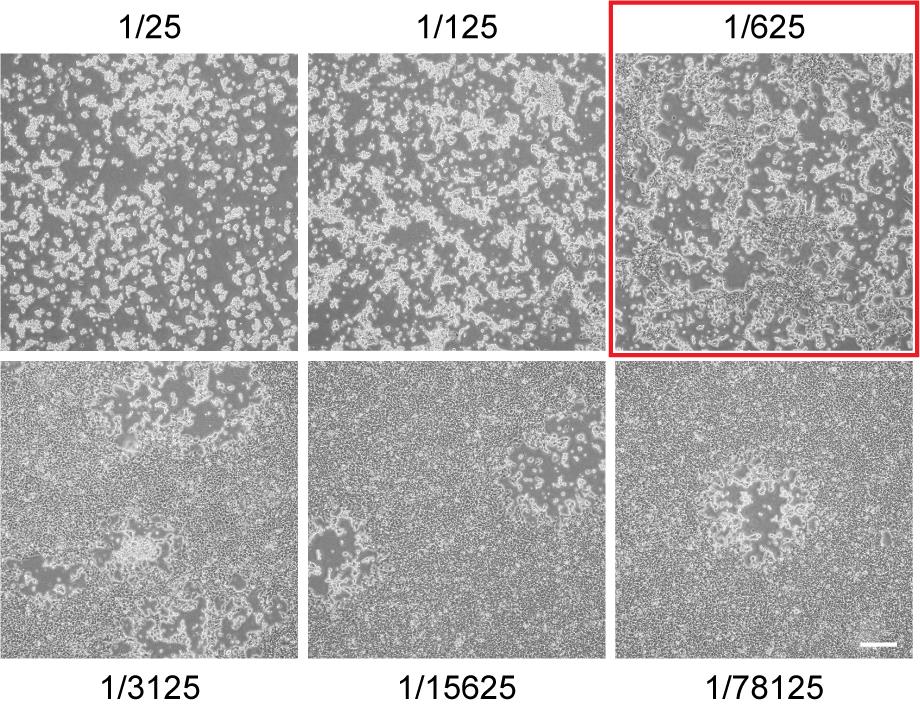
Figure 3. Cytopathic effect as an indicator of appropriate well selection at 48 h post-VACV infection. Cytopathic effect seen on a Cas9 selection plate with the well chosen to harvest indicated with the red box. In this instance, the chosen dilution is 1/625 but this varies across experiments. Scale bar in bottom right frame = 300 µm.Freeze-thaw 3 times and sonicate 3 times as above.
Prepare new plates of Cas9/gRNA-transfected 293A cells by repeating steps B1-B9.
Note: Ideally the cells should be prepared to be ready to transfect on the day the virus is harvested from the first round of selection.
Use virus collected in Step B16 to prepare serial 5-fold dilutions in 1.25 ml D0 media, starting with a 1/25 dilution (Figure 2).
At 4 h post-transfection, remove media and replace with 1 ml of each virus dilution.
Complete the incubations and harvesting of virus as in steps B13-B16.
Optional: Where there is colour or another marker in the final virus and there are well-separated plaques of the desired phenotype visible on these cultures, it is possible to proceed directly to step C9 and collect the progeny of individual plaques instead of harvesting a whole well.
Freeze stocks at -80°C until ready to use.
Note: A third round of selection can be performed if recombinant viruses are not adequately enriched after this stage.
Proceed to Section C for identification and purification of recombinants.
Isolation of recombinant viruses
Perform all work in a class 2 biosafety cabinet.
Plate BS-C-1 cells to confluency in a 6-well plate in 2 ml D10 media (6 wells per plaque to be tested; see Note 5).
The following day, take virus collected after 2 rounds of Cas9 selection and sonicate 3 times as above.
Serially dilute collected virus 1/10 in 1.2 ml D0 media. See volumes listed in Table 4.
Table 4. Preparation of 1/10 virus dilutions
Component Volume (µl) Tube A Tube B Tube C Tube D Tube E Tube F Virus (source) 120
(Stock)120
(Tube A)120
(Tube B)120
(Tube C)120
(Tube D)120
(Tube E)D0 media 1,080 1,080 1,080 1,080 1,080 1,080 Total 1,200 1,200 1,200 1,200 1,200 1,200 Remove media from cells and add 1 ml virus stock to each well.
Incubate plate at 37°C for 1 h, 5% CO2.
After 1 h, aspirate media and replace with 2 ml 4% CMC/D2 (see Recipe 6).
Incubate for 48 h at 37°C, 5% CO2.
Locate plaques using an inverted microscope. If the desired recombinant has fluorescent or other visible markers, use an appropriate method to identify plaques that contain the marker.
Mark location of desired plaques using an ultra-fine permanent marker on the bottom of the 6-well plate.
Plaques should be taken from the most dilute well possible and should be well-spaced from other plaques. It is recommended to mark a minimum of 10 plaques.
Return plate to biosafety cabinet.
Insert a pipette into the well above the marked area and aspirate 10 µl, whilst scraping to collect virus from a plaque. Virus collected by this method is referred to hereafter as a “plaque isolate”.
Transfer plaque isolate to a 2 ml Sarstedt tube containing 500 µl D10 media.
Repeat for all marked plaques.
Plaques can be stored at -80°C.
Proceed to Section D to screen these isolates by PCR (necessary for marker-free recombinants), or directly to Section E if a visual marker allows identification of plaques with recombinant virus.
Screening of plaque isolates by PCR (marker-free recombinants)
Plate BS-C-1 cells to confluency in wells of a 96-well plate (one well per plaque to be tested) (see Note 6).
Take 10-50 µl from each plaque isolate in C and add to 200 µl D2 media.
Remove media from cells and replace with the diluted virus.
Incubate plate for 3 days at 37°C, 5% CO2.
Examine cells for presence of CPE and/or expression of markers under a microscope.
Remove media from wells with CPE and wash with 150 µl PBS, without disturbing cells.
Lyse cells by adding 10 µg/ml Proteinase K (Invitrogen) in 100 µl 1× ThermoPol® buffer (New England Biolabs) to each well and incubating for 5 minutes at room temperature.
Freeze plate at -80°C, then thaw at room temperature.
Heat samples using a 56°C heating block for 20 min.
Heat samples at 85°C for 10 min to inactivate Proteinase K, using a heating block.
Note: It is essential that the sample is at 85°C for the full 10 min.
Take 2 µl of each sample to perform a PCR.
Design primers to distinguish recombinant and parent viruses (see example in Figure 4).
Optional: Incorporate novel restriction sites during recombinant plasmid design to aid in the identification of recombinants with close similarity to the parent virus.
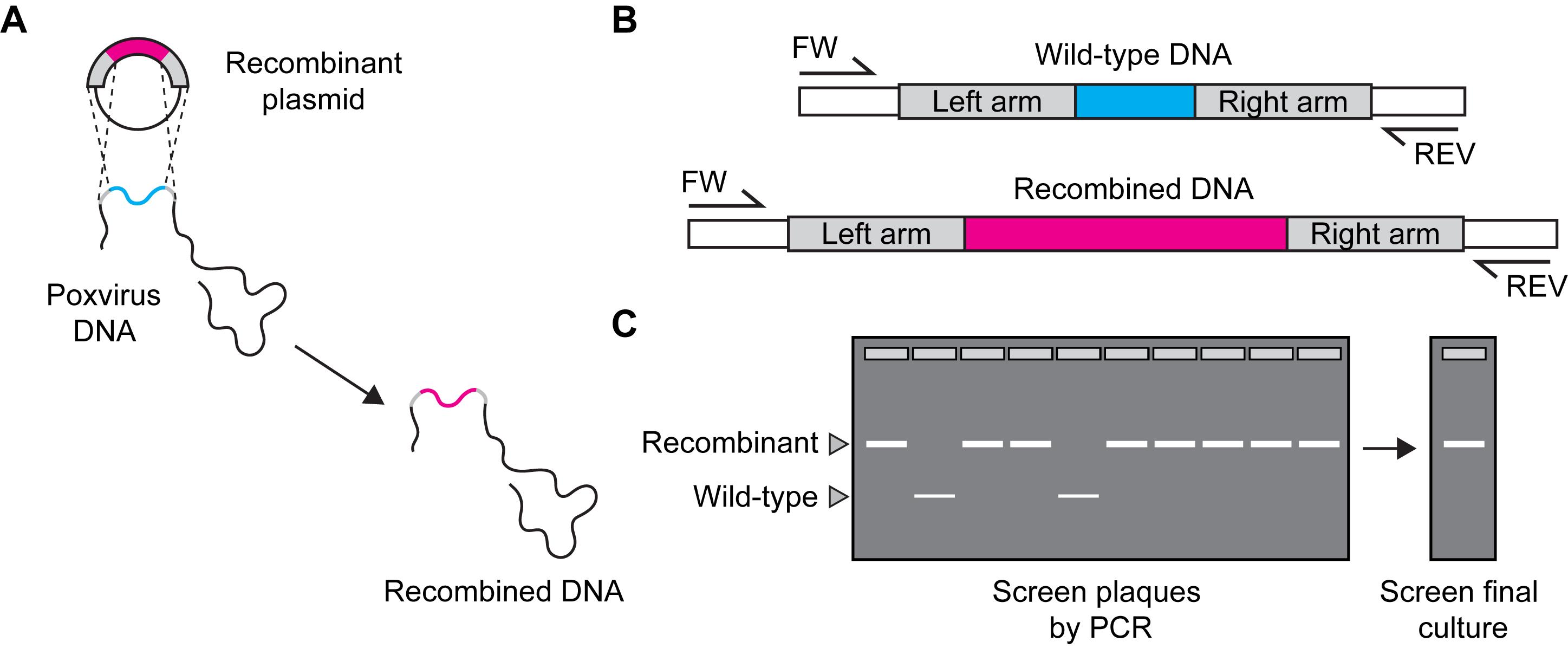
Figure 4. Confirming identity of recombinant viruses by PCR. A. To check for recombination events. B. Design forward (FW) and reverse (REV) primers to differentiate wild-type and recombinant viruses. C. Screen individual plaque isolates and final cultures to check for wild-type (WT) virus. Choose a strategy to unambiguously differentiate between parent and recombinant virus. In this example, a second primer pair that only amplifies the foreign DNA can be of value.Prepare PCR mixtures as follows (Table 5):
Table 5. Preparation of PCR master mix
Component Volume (µl) Per reaction 4 reactions 10× ThermoPol® buffer (NEB) 2.5 10 dNTPs 0.5 2 Primers (10 µM stock) 0.5 (each) 2 (each) Template DNA 2 - Taq DNA polymerase (NEB) 0.125 1 Nuclease-free water Up to 25 Up to 100 Total 25 100 Perform PCRs using a thermocycler set to conditions appropriate for amplification.
Note: Optimise thermocycling conditions to your specific primers and template size prior to virus screening.
Run 10 µl of each sample with 2 µl 6× Loading dye on a 1-2% UltraPureTM agarose gel with SYBRTM Safe staining to check for DNA amplification.
Optional: If plaques after marker-screening or PCR only show the desired virus, or if the edit to the genome is too small to be detected by a differential set of PCR primers, sequencing of PCR products that include the edited region of the genome can be done to further validate or identify plaques of interest.
Proceed to Section E for purification of recombinant viruses.
Further plaque purification to obtain recombinant virus seed stocks
Perform all work in a class 2 biosafety cabinet.
Seed BS-C-1 cells to confluency in a 6-well plate (6 wells per plaque isolate to be purified).
Serially dilute plaque isolates, which were identified by marker expression or in Section D as containing the desired recombinant, in 1.2 ml D0 media (Table 4).
Aspirate media from BS-C-1 cells and add 1 ml of each virus dilution.
Incubate plate at 37°C for 1 h, 5% CO2.
Remove media and replace with 2 ml 4% CMC/D2.
Incubate plate for 48-72 h at 37°C, 5% CO2.
Examine plates under a microscope for plaque formation.
Collect plaque isolates into 500 µl D10 media as in C and freeze-thaw 3 times.
Note: If using a marker-free virus, the identity of each plaque isolate should be confirmed by PCR and/or sequencing.
Repeat steps E1-E9. Each repetition is considered a ‘round’ of plaque purification.
If picking multiple plaques at each step, label numerically to trace lineage, i.e., if using plaque isolate 1 from the 1st round, plaque isolate 3 from the 2nd round, and plaque isolate 4 from the 3rd round, label could read “Plaque pick 1-3-4” etc.
Conventionally, 3 rounds of plaque purification are adequate for a virus stock to be considered pure. However, we have found that virus from the first plaques picked after selection (Section C) are sometimes entirely free of parent virus by PCR and in these cases one or two additional rounds of purification may be sufficient.
Recommended: For marker-free viruses, take 1-2 µl of the final purified virus and add to BS-C-1 cells in a 96-well plate. Culture for 3 days and extract DNA as above (Section D). Use PCR to check for any contaminating parent virus (Figure 3C).
This plaque suspension can be considered a seed stock, or it can be expanded using 100 µl to infect a 25 cm2 tissue culture flask of cells that are preferred for growing general VACV stocks. Allow growth for 3 days as usual (37°C, 5% CO2).
Data analysis
The data produced and analyzed in the process of making recombinant viruses by this protocol are all qualitative. Our experience is that at least 10 plaque isolates or picks should be assayed in the first instance after the selection steps. If trouble-shooting is required, we recommend the use of experiments similar to those in our original study, for example testing whether Cas9/gRNA reduces fluorescence, cytopathic effect, or virus titres in virus-infected cells. In these cases at very least a test and a control gRNA are required with at least three replicates (Gowripalan et al., 2020). We used one- or two-way ANOVA with Tukey’s post-test for pair-wise comparisons as appropriate where there were more than two conditions. If comparing one test and one control gRNA, a t-test is adequate. In our original study to obtain estimates of virus frequencies, 100 plaques were classified using microscopy. For marker-free systems, 20-100 plaques were assayed via PCR and sequencing (Gowripalan et al., 2020).
Notes
gRNAs should consist of a 20 bp sequence complementary to the gene target known as the cRNA, followed by a common tracrRNA sequence (76 bases, 5’-GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGC-3’) for a total of 96 bases. cRNAs can be designed or validated using online platforms, such as “CRISPR-Cas9 guide RNA design checker” (Integrated DNA Technologies).
The design of plasmids for engineering VACV by recombination has been published, and typically follows general molecular biology principles (Broder and Earl, 1997; Wong et al., 2011; Marzook and Newsome, 2019). For instance, recombination arms should be 500 bp or greater, GC content should ideally be between 40% and 60%, and promoters should be appropriate for expression in your context. Plasmids can be marker-free or contain markers for observation. This includes fluorophore expression or chromogenic models, such as the β-galactosidase system. The thymidine kinase gene (J13R) is a commonly used recombination site. We prepared plasmids using the NucleoSpin Plasmid Mini Kit (Machery-Nagel).
293A cells were chosen for this work because they support VACV replication and can be transfected with high efficiencies. Other HEK293 clones would also be appropriate. When using poxviruses, which do not grow in HEK293 cells, cell lines that support both virus replication and high transfection efficiencies should be chosen. Inhibition of virus growth by Cas9 can be tested and optimised by a) targeting a fluorescent gene in a recombinant VACV and reading out reduced fluorescence or b) looking for reduced CPE and virus production if only the non-recombinant virus is present (Gowripalan et al., 2020).
Once an estimate of virus titre has been established in your experiment, you can use fewer virus dilutions, i.e., instead of 6 serial dilutions, 3 may be sufficient to achieve well-separated virus plaques in at least one well.
We use BS-C-1 cells for this step because they are large cells that are well contact inhibited, which makes them ideal for the identification and collection of virus from plaques.
Growing mini-cultures of virus isolates in 96 well plates to generate PCR templates was developed because our experience of attempting to PCR from viral genomes directly from plaque isolates was unreliable. This can occur in some cases because not enough virus was collected, but in others probably due to contaminants in the culture media. This step adds two days, but this time is offset by the ability to screen out plaque isolates that do not grow, and by the greater reliability of subsequent PCRs. As a result, overall efficiency is improved.
Recipes
D0 media
500 ml Dulbecco’s Modified Eagle’s Medium – high glucose
10 mM L-Glutamine
D2 media
500 ml Dulbecco’s Modified Eagle’s Medium – high glucose
10 mM L-Glutamine
2% (v/v) heat-inactivated FBS
D2 media – no phenol red
500 ml Dulbecco’s Modified Eagle’s Medium – high glucose, no glutamine, no phenol red
10 mM L-Glutamine
2% (v/v) heat-inactivated FBS
D10 media
500 ml Dulbecco’s Modified Eagle’s Medium – high glucose
10 mM L-Glutamine
10% (v/v) heat-inactivated FBS
D10 media – no phenol red
500 ml Dulbecco’s Modified Eagle’s Medium – high glucose, no glutamine, no phenol red
10 mM L-Glutamine
10% (v/v) heat-inactivated FBS
4% CMC/D2
2 g CMC
50 ml PBS
450 ml D2 media (or D2 media – no phenol red if observing fluorescence)
Mix 2 g CMC with 50 ml PBS and autoclave.Top up to 500 ml with appropriate D2 media.
Acknowledgments
We thank Sarah Croft for helpful comments on the manuscript. This work was funded by grants and fellowships from the NHMRC: APP1104329, APP1084283 and APP1126599 and ARC:DP190101325. The original research that led to this protocol has been described previously (Gowripalan et al., 2020).
Competing interests
The authors declare no competing interests.
References
- Broder, C. C. and Earl, P. L. (1997). Design and construction of recombinant vaccinia viruses. Methods Mol Biol 62: 173-197.
- Gowripalan, A., Smith, S., Stefanovic, T. and Tscharke, D. C. (2020). Rapid poxvirus engineering using CRISPR/Cas9 as a selection tool. Commun Biol 3(1): 643.
- King, M. W. and Munger, J. (2019). Editing the human cytomegalovirus genome with the CRISPR/Cas9 system. Virology 529: 186-194.
- Marzook, N. B. and Newsome, T. P. (2019). Construction and Isolation of Recombinant Vaccinia Virus Expressing Fluorescent Proteins. Methods Mol Biol 2023: 73-92.
- Russell, T. A., Stefanovic, T. and Tscharke, D. C. (2015). Engineering herpes simplex viruses by infection-transfection methods including recombination site targeting by CRISPR/Cas9 nucleases. J Virol Methods 213: 18-25.
- Smith, G. L. and Moss, B. (1983). Infectious poxvirus vectors have capacity for at least 25 000 base pairs of foreign DNA. Gene 25(1): 21-28.
- Wong, Y. C., Lin, L. C., Melo-Silva, C. R., Smith, S. A. and Tscharke, D. C. (2011). Engineering recombinant poxviruses using a compact GFP-blasticidin resistance fusion gene for selection. J Virol Methods 171(1): 295-298.
- Yuen, K. S., Chan, C. P., Wong, N. M., Ho, C. H., Ho, T. H., Lei, T., Deng, W., Tsao, S. W., Chen, H., Kok, K. H., et al. (2015). CRISPR/Cas9-mediated genome editing of Epstein-Barr virus in human cells. J Gen Virol 96(Pt 3): 626-636.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Gowripalan, A., Smith, S. A. and Tscharke, D. C. (2021). Selection of Vaccinia Virus Recombinants Using CRISPR/Cas9. Bio-protocol 11(24): e4270. DOI: 10.21769/BioProtoc.4270.
Category
Microbiology > Microbial genetics > DNA
Microbiology > in vivo model > Viruses
Molecular Biology > DNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.