- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Wounding Zebrafish Larval Epidermis by Laceration
Published: Vol 11, Iss 24, Dec 20, 2021 DOI: 10.21769/BioProtoc.4260 Views: 3635
Reviewed by: Rajesh D GunageMatthew SwireKatie KoczirkaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Wound healing is a critical process for maintaining the integrity of tissues, driven in large part by the active migration of cells to cover damaged regions. While the long-term tissue injury response over hours and days has been extensively studied, the rapid early migratory response of cells to injury in vivo is still being uncovered, especially in model systems such as zebrafish larvae, which are ideal for live imaging with high spatiotemporal resolution. Observing these dynamics requires a wounding method that prompts a robust wound response and is compatible with immediate live imaging or other downstream applications. We have developed a procedure for wounding the epidermis in the tailfin of larval zebrafish, which we term “tissue laceration”. In this procedure, the tailfin is impaled with a glass needle that is then dragged through the tissue, which generates a full-thickness wound that elicits a dramatic migratory wound response within seconds from cells up to several hundred micrometers away from the wound. Laceration generates a larger wound response in the first few minutes following wounding compared to other mechanical wounds such as tail transection, and laceration does not require specialized equipment compared to laser wounding methods. This procedure can be used to interrogate the processes by which epidermal cells far away from the wound are able to rapidly detect injury and respond to the wound.
Background
Wound healing is an active area of research in cell and regenerative biology, with a major focus on elucidating the mechanisms underlying early (tens of minutes) and long-term (hours and days) responses to damage. Zebrafish larvae have emerged as a key model system to study wound healing, due to their suitability for live in vivo imaging and their ease of manipulation (Mateus et al., 2012; Yoo et al., 2012; Enyedi et al., 2016; Miskolci et al., 2019). The early response of zebrafish larvae to wounding is particularly rapid and effective, and involves contraction of the wound site through an actomyosin purse string, as well as active migration of epidermal cells to the site of injury, with both of these processes initiating within seconds after wounding (Gault et al., 2014). Compared to other model systems, zebrafish epidermis has the additional advantage that the purse string and migratory wound responses are spatially segregated into distinct cell types, with the outer layer of the epidermis closing by purse string contraction, while the basal layer of the epidermis closes via cell migration (Gault et al., 2014). This suggests that zebrafish might be used to specifically investigate the contribution of active cell migration to wound healing at short timescales in a living animal.
However, existing techniques for injuring larval zebrafish differ in the extent to which they promote cell migration or purse string contraction. For example, transection of the larval tail is a commonly used and straightforward wounding technique that produces a clean straight-edged wound (Yoo et al., 2012; Briona and Dorsky, 2014; Zeng et al., 2018; Franco et al., 2019). Tail transection elicits a wound response that appears to involve a significant degree of purse string contraction relative to cell migration (Mateus et al., 2012; Kennard and Theriot, 2020). Laser injury can produce both migration and purse-string contraction (Gault et al., 2014); however, this approach requires specialized equipment, the nature of the damage to the tissue is complex and appears to involve distinct dynamics and regulation compared to purely mechanical wounds (Vogel and Venugopalan, 2003; Datta et al., 2017; Miskolci et al., 2019).
To explore the rapid migration of epidermal cells to injury, we developed a new wounding procedure termed “tissue laceration”, in which the larval tailfin is impaled and torn with a glass needle, producing an irregular ragged-edge wound. Although this wound geometry is more variable than other wounding methods, laceration nevertheless results in a quantitative, reproducible wound response that includes significant migration of epidermal cells up to several hundred micrometers away from the wound. In comparison to tail transection, tissue laceration leads to more cell migration, especially within the first 5 minutes after injury (Kennard and Theriot, 2020). Glass needles are transparent, can bend without breaking, and come to a very fine point, which make it easier to produce the complex laceration wound geometry on a sample in the middle of timeseries acquisition. Bulkier, opaque tools such as a scalpel or a syringe can be more challenging to use precisely on the microscope. This method does not require specialized equipment beyond what is already available in most labs that work with zebrafish, and is flexible enough to be adapted to different situations, such as at a stereomicroscope or on a confocal in the middle of an acquisition. With practice, this method is a reliable way to obtain a strong and consistent migratory response to interrogate the contribution of cell migration to wound healing in vivo.
Materials and Reagents
Solid borosilicate glass rods, 1 mm diameter (Sutter Instruments, catalog number: BR-100-10)
35 mm Glass-bottom dishes (Cellvis, catalog number: D35-20-1.5N or D35C4-20-1.5N)
Flame-polished Pasteur pipettes
Modeling clay
Deep well Petri dishes, 100 × 15 mm (Fisherbrand, catalog number: FB0875712)
20 ml glass scintillation vials with caps
1.5 ml flip-cap tubes
Tungsten needles (e.g., Roboz, catalog number: RS-6063)
Zebrafish larvae
Tricaine (Sigma, catalog number E10521). Store at room temperature
1 M Tris solution, pH 9 (solid Tris: Fisher BioReagents, catalog number: BP153-1)
Low-melt agarose (Invitrogen, catalog number: 16520050). Store at room temperature
E3 medium. Store at room temperature (see Recipes)
Equipment
Embryo manipulation tools:
Curved pin manipulator (Fine Science Tools, catalog number: 26007-03) and small spatula manipulator (Fine Science Tools, catalog number: 26007-05) mounted in a holder (Fine Science Tools, catalog number: 26018-17)
Scalpel handle and blades (#11-blade)
Needle puller (Sutter Instruments, model: P-87)
Stereomicroscope typically used for dissection (we used a Zeiss Stemi 508)
Inverted microscope for higher-resolution imaging (we have tried this protocol with a Nikon Ti2 and a Leica DM6000B)
Heat block for 1.5 ml tubes, maintained at 38°C
Swing arm lamp mounted near the inverted microscope (Dazor Lighting Technology, catalog number: 6134)
Procedure
Preparing glass needles
We found success with using solid borosilicate glass rods, which provided a balance between strength and flexibility. We have also tried using etched Tungsten needles (e.g., Roboz, catalog number: RS-6063), but we found that the tips were very fragile and easily broke on the glass bottom surface of the dish during laceration, while glass needles were flexible and thus did not break as easily.
Reproducing the exact tip shape with a different needle puller will be challenging; the parameters we optimized on our needle puller can be used as a starting point and are shared below. Needles that were too short had a tendency to break rather than bend on contact with the glass bottom surface; needles that were too long and thin were too flexible and did not efficiently tear the tailfin. As with any micromanipulation procedure, trial and error are always required to optimize for your precise setup.
Prepare a storage container for needles by rolling out two “worms” of modeling clay that can fit inside a Petri dish in parallel lines (Figure 1A). The needles can be gently pressed into these worms to be held in place for storage.
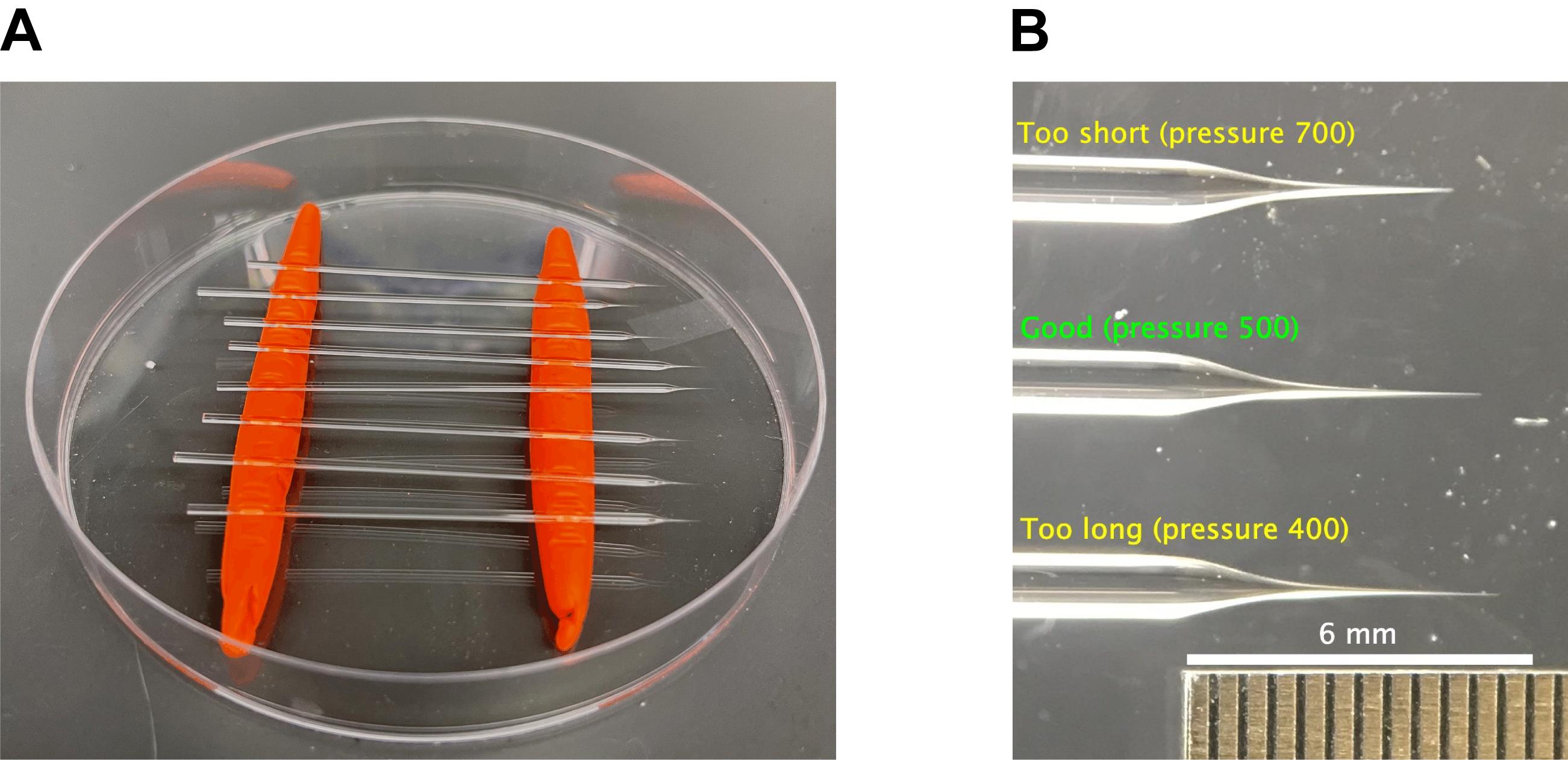
Figure 1. Example of laceration needles and needle holder. (A) Needle holder fashioned in a Petri dish with clay “worms” to secure the needles, with examples of needles used for laceration. (B) Magnified view of needle tips of different lengths. Needles of different lengths were obtained by adjusting the pressure setting as indicated, keeping all other parameters in protocol Step A3 the same. From top to bottom: too short (pressure 700), just right (pressure 500), and too long (pressure 400). Ruler shown for scale; individual ticks are spaced 0.5 mm apart.Set up the pulling program on your needle puller. Precise needle pulling parameters are not easy to reproduce between machines due to differences in filament properties, local humidity, and many other factors. Sutter Instruments has produced a series of useful tutorial videos with practical advice on pulling needles, e.g., https://youtu.be/s4Os3FIDYxc. As a rough starting point, we pulled needles on a Sutter Instruments P-87 needle puller (Flaming-Brown type) installed with a 3 × 3 mm box filament and a ramp calibration value of 665 using the following parameters: Heat 665 (set to ramp value); Pull 55; Velocity 60; Time 250; pressure 500. The needle was formed in a single pull, which took at least 10 s of heating – longer than a pull for a typical microinjection needle. Examples of a good needle, as well as needles with tips that are too long or too short, are shown in Figure 1B. However, the true test of the usefulness of a needle is its behavior during laceration: if it bends too easily, making it difficult to tear the tissue, it is too long, while if the tip is brittle and breaks very easily while positioning it for laceration, it may be too short.
Insert borosilicate rods into the needle puller and pull according to the instructions of your needle puller. Remove needles and place them in the needle storage container.
Needles were not sterilized beyond the heat of the filament used when pulling the needle and were reused for several weeks, or until tissue debris stuck to the needle, or if the solution contained poorly soluble drugs such as blebbistatin, which might adsorb to the glass. Generally, needles broke from use within this timeframe.
Preparation of low-melt agarose in E3 medium
Prepare a 60× stock solution of E3 (see Recipes).
Dilute E3 to 1× with Reverse osmosis water (RO water) and add low-melt agarose to a concentration of 1.2% w/v. This 1.2% concentration was chosen to tolerate some dilution during the mounting process while maintaining sufficient rigidity. We have routinely worked with agarose concentrations up to 2% with similar efficacy.
Microwave the E3-agarose mixture until fully dissolved, using low to medium power to prevent boiling over.
Divide into aliquots of ~6-7 ml (we used 20 ml glass scintillation vials with screw caps but other microwaveable containers may be used), which can be stored for several months at 4°C.
Prior to experiments, re-melt an aliquot in the microwave using short pulses of a few seconds, interspersed with gentle shaking until the agarose is clearly dissolved. Caution, the vial can get quite hot!
Pipet 720 µl of this medium into 1.5 ml tubes, and keep the tubes at 38-40°C to prevent the agarose from solidifying. These working vials can be used for up to 1 week, after which time the agarose near the top of the tube starts to polymerize. Note that 38°C is an elevated temperature for zebrafish larvae. In our experience the agarose cools rapidly upon removing from the heat block and the stress to the zebrafish larvae upon immersion is minimal. If elevated temperatures are a concern for your experiment, you may be able to use lower temperatures, which will shorten the amount of time available for mounting the larvae before the agarose sets. We have not tried using lower temperatures.
Handling of zebrafish larvae
Zebrafish from 30-120 h post-fertilization (hpf) have been successfully wounded with this protocol.
Maintain zebrafish stocks according to standard procedures (Westerfield, 2007) and guidance from your institution’s department of comparative medicine, or another office responsible for the welfare of research animals.
The afternoon before embryos are to be fertilized, place 1-2 male and 1-3 female zebrafish of breeding age (at least 3 months old depending on rearing conditions) into a spawning tank. Fish will spawn the following morning after lights come on in the zebrafish facility.
Return adults to their housing after spawning. Collect the fertilized eggs by pouring the spawning water through a mesh strainer and rinsing the eggs into a Petri dish with system water.
Split embryos into multiple Petri dishes of system water, at a density of no more than 60 embryos per deep-welled Petri dish, or 50 per regular Petri dish. Inspect embryos at the end of each day, including the day of fertilization, to remove dead embryos and prevent fungal growth. Transferring embryos into fresh system water can be beneficial for minimizing microbial growth. Antifungal agents like methylene blue can also be added, but these agents will also frequently stain cell types within the larval epidermis, which can be undesirable for downstream imaging.
Screen embryos for desired genetic phenotypes (e.g., fluorescent markers) according to your specifications.
Mounting larvae
For most of the work in this paper, we opted for a quick and flexible procedure of mounting in agarose followed by cutting a window out of the agarose to allow access to the tailfin with a microneedle. Alternatively, devices like the zWEDGI can be used for immobilization of the larva while the tail remains free (Huemer et al., 2017a and 2017b). These devices help with consistent mounting of the larvae in the correct position, and also allow independent control of the composition of the media on the anterior and posterior side of the larvae, but require more time devoted upfront to assembly. Detailed protocols for use of these devices have been previously published (Huemer et al., 2017b). See Video 1 for a demonstration of the techniques described in this section.
 Video 1. Demonstration of mounting and laceration of larval zebrafish.
Video 1. Demonstration of mounting and laceration of larval zebrafish.If larvae are still in their chorions, dechorionate the larvae and allow them to straighten for a few minutes. Then, transfer them with a fire-polished glass Pasteur pipette to a Petri dish containing E3 + tricaine. Allow a few minutes to anesthetize the larvae, which can be confirmed with a gentle puff from a pipette or a gentle tap with a curved pin manipulator.
While larvae are being anesthetized, add 30 µl of 25× tricaine solution to the side of one of the aliquots of E3 + low melt agarose and flick gently to mix while minimizing bubbles. Mark this aliquot with a felt tip pen and throw it out at the end of the day.
Gather anesthetized larvae in the Pasteur pipette and transfer them into the cap of the marked tube of agarose. Working quickly, carefully remove any excess liquid from the fish and then close the cap and invert the tube to suspend the larvae in the agarose. Flip the tube right side up and check that all larvae are in the agarose, as opposed to stuck to the walls or still in the cap – it may require shaking the tube to get the larvae into the main solution of agarose.
Gather the larvae and agarose using the Pasteur pipette and pipette ~500 µl of solution into the well of a 35 mm glass-bottom dish. Use the curved pin manipulator to position and mount the fish in the orientation shown in Figure 2A, as described here: Position the tailfins near the center of the dish to allow maximum room for maneuvering, and mount the larva on its right side so that the tailfin is in-plane with the glass bottom of the dish; this requires practice and patience. Press gently on the larva on the yolk sac and yolk extension using the side of the curved pin manipulator to move it around in the agar. Avoid touching the larva with the point of the curved pin manipulator, which can damage the larva. Also avoid directly touching the tip tailfin as much as possible, to prevent inadvertent damage before imaging. The large size of the yolk sac will tend to cause larvae to tilt relative to the glass surface, so larvae must frequently be prodded and re-oriented until the agarose starts to set, roughly a few minutes after being placed in the dish, depending on ambient temperature. You can tell the agarose is starting to set when the surface holds deformations. We typically mount 4 larvae at a time to ensure consistent mounting, though at first only one or two should be attempted at a time, and with practice more larvae can be simultaneously mounted. Larvae that are successfully mounted in a plane parallel to the glass bottom will require a smaller imaging volume and therefore lead to lower phototoxicity and less computer memory for data storage.
Once the agarose starts to solidify, leave it a few more minutes to fully solidify (indicated by an opaque appearance), then cover it in E3 + tricaine solution or other solution of interest.
Using a #11 scalpel blade, cut a window around the tailfin of the larvae, as shown in Figure 2B-2D. This blade was chosen in order to have a clear view of the tip to avoid inadvertently damaging the larva.
Using the tip of the blade, drag it through the agarose to make a full-thickness cut through the agarose perpendicular to the anterior-posterior axis, over the trunks of the larvae (Figure 2B). This cut should be positioned about halfway between the tailfin and the yolk extension, but can be more posterior than this; if the cut is too far up the larvae, then the pumping of blood flow can induce periodic movement of the tailfin that can cause motion artifacts during imaging. Care must be taken not to injure the larvae: carefully lift the blade up and over each larva, staying as close as possible to the larvae, and then back down to touch the glass bottom surface on the other side.
Next make a cut parallel to this one that is posterior to the larva.
Finally make two cuts parallel to the anterior-posterior axis of the larvae on either side, to join up with the other cuts and completely cut out a rectangle of agarose from around the tailfin.
Using the small spatula manipulator, insert the spatula between two larvae in the cut above the trunk and gently pry the agarose off the tailfin and out of the dish (Figure 2C). If the agarose is concentrated enough, the whole block should come out evenly (Figure 2D). Sometimes this procedure breaks the agarose into small pieces, which should be completely removed to avoid interference with the needle during wounding.
Using the curved pin manipulator, trace around the tailfin to check for residual agarose: if all agarose is removed, tracing around should not cause the tailfin to move, while residual agarose will catch the tailfin and cause it to bend. Generally tracing around the tailfin will also serve to fully remove the residual agarose.
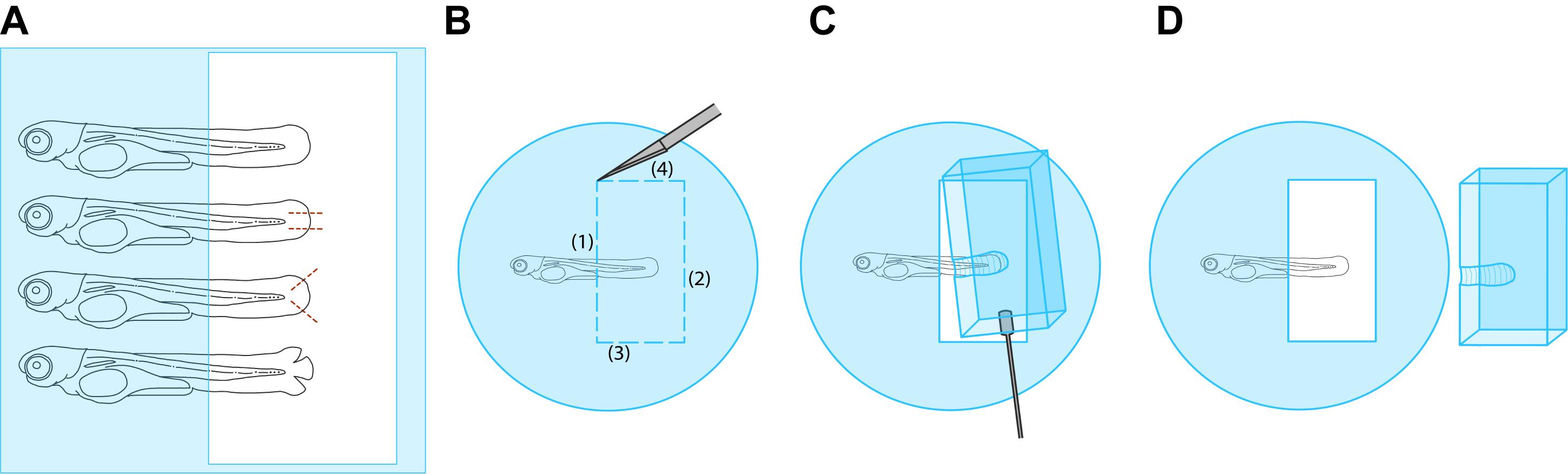
Figure 2. Schematic of mounted and wounded larvae. (A) Four larvae are drawn in an appropriate orientation following mounting. Portions of the larvae that are shaded in blue are embedded in agarose. The position of the window to be cut out of the agarose is indicated by the unshaded rectangle around the tailfins. Different stages of the wounding process are shown. From top to bottom: unwounded; wounded with 2 parallel lacerations, as in (Kennard and Theriot, 2020); wounded according to a revised method with lacerations at 45° angles to the anterior-posterior axis (this work, see main text); wounded tailfin immediately after laceration. Positions of lacerations are indicated with dashed lines. (B-D) Step-by-step illustration of cutting out an agarose window. (B) Cuts are made in the agarose using a scalpel in the order shown by the numbers 1-4. In the first cut, the scalpel is lifted around the larva to avoid cutting it. (C) Using a small spatula manipulator, the block of agarose is removed from around the larva. (D) the block of agarose is fully removed from the dish and discarded. Note the block will have a divot in it formerly occupied by the larva.
Laceration
This is also a flexible procedure, which can be done in several ways: first, wounding on the microscope allows for imaging the same larvae immediately before and after wounding, which is necessary for observing the events within the first minute or so after wounding. With practice, this can be done quickly and reproducibly by hand. Finally, a sample can be wounded manually at a stereomicroscope and then moved to the confocal for imaging. This approach has the advantage of increased visibility and control provided by a stereomicroscope, at the expense of missing the events in the first few minutes after wounding.
Alternatively, a micromanipulator can also be used to hold the needle in place, which confers greater stability to the needle at the expense of reduced control (depending on how elaborate your micromanipulator controller is) and increased setup and alignment time. This protocol discusses the manual wounding strategies in depth, and briefly touches on some considerations for micromanipulator wounding. See Video 1 for a demonstration of wounding at a stereomicroscope.
Manual laceration at a stereomicroscope
Take a needle and hold it like a pen; your middle finger, as well as fingers from your other hand, can be used for further stabilization. Effectiveness of laceration appears to be best when the needle is held as close to straight up-and-down as is feasible, without blocking the view of the larvae with your fingers.
With the needle, impale the larvae at a position dorsal or ventral to the terminus of the notochord and drag the needle in a posterior direction, tearing the tailfin (Figure 2A).
In subsequent investigations, we have found that pulling the needle at a ~45° angle in the X-Y plane, following the orientation of the actinotrichia, produces a cleaner wound that is more likely to be consistent from larva to larva, and less likely to rip a large chunk out of the tailfin (Figure 2A).
Repeat this laceration at a position on the other side of the notochord, so that there are two tears in the tailfin that are ideally symmetric about the dorsal/ventral axis.
The larva is now ready for imaging or other downstream analysis of the wound response.
Note: This procedure can also be used with unmounted (anesthetized) larvae in a Petri dish for higher throughput. In this case, holding the curved pin manipulator in your non-dominant hand, rest the point of the tool above (dorsal to) the larva and lower it until it is securely pinning the larva against the bottom of the dish just posterior to the yolk sac. Holding the needle in your dominant hand, wound the larva as described; the wounding procedure will tug the larva in the posterior direction, which will be resisted by the manipulator tool. With practice, this can be done in just a few seconds and allow wounding many unmounted larvae in a Petri dish in a short period of time.
Manual laceration at an inverted microscope
This procedure is done on an inverted scope to allow for ease of access; it has been practiced successfully on both a Leica DM6000B and a Nikon Ti2. This procedure is easier with lower magnification lenses, though it has been successfully and routinely applied up to 60× magnification. It is highly recommended to practice at 10× until comfortable.
If desired, acquire a few images of the tailfin prior to the injury, then stop or pause the acquisition for wounding. Make sure to select the eyepieces in the optical path rather than the camera for the next steps.
Caution: If using a confocal, make sure your lasers are off or your laser safety interlock is functioning properly to prevent inadvertent exposure of your eyes to direct laser light through the eyepieces.
Move the transmitted light turret out of the optical path to provide more room for manipulation; to provide light, an external lamp on a swing arm stand can be moved in position in the optical path (resting on some part of the turret if required). Using this light source has the additional advantage of illuminating the sample like a low-NA condenser, which provides a greater depth of field for wounding than is typically used for acquisition.
Hold a needle in one hand between your thumb and your index and middle fingers. Move your arms over the sample, positioning the needle so it is in the optical path, being careful not to block the optical path with your fingers. Bringing your other hand in from the other side of the sample, you can further stabilize the needle using your other fingers, as well as resting your hands on stable parts of the microscope body. Make sure that there is no tension in your hands, to avoid excessive shaking of your fingers.
Depending on the optical setup, it may be helpful to bring the needle in from the front or the back of the sample.
In general, the needle needs to be positioned at an angle to prevent your hands from blocking the optical path. However, the effectiveness of tissue impalement and injury appears to be greater when the needle is as close to vertical as is feasible.
Carefully bring the needle into focus. First observe the position of the needle relative to the sample with the naked eye to make sure you are holding it close to the larva, then carefully reposition your body to look through the eyepieces without moving your hands too much. Gradually lower the needle towards the sample, gently swaying the needle back and forth across the field of view to provide a visual cue of when the needle is coming into focus. Often it is helpful to center a location further up the shaft of the needle within the optical path, rather than the tip of the needle; as the needle lowers into focus, you will be unlikely to miss the whole shaft, whereas a small error in positioning of the tip might cause you to move the needle outside of the field of view, or accidentally impale the larva prematurely. On the other hand, if you focus on a point too far up the shaft, you may risk lowering the needle too far and breaking the tip.
Once the tip of the needle is in focus and in the field of view, proceed to lacerate the tailfin as described above (“Manual wounding at a stereomicroscope”). Hand tremor is more noticeable in this setup than with the stereomicroscope, and it takes practice to impale with the same degree of control afforded under the stereomicroscope. Often the majority of time is spent positioning the needle “close enough” (given the degree of hand tremor), and then committing to that position and impaling the tailfin, regardless of what happens next. In practice, we have found that many interesting events following wound healing are robust to variation in the geometry of the wound.
After laceration, swing the lamp out of the optical path, replace the transmitted light turret, and switch the optical path back to the camera rather than the eyepieces, and resume image acquisition.
Movement of the tailfin during acquisition is a greater problem in this procedure than in typical zebrafish imaging where the entire larva is embedded in agarose. Especially at higher magnifications, the larva may have shifted relative to its position prior to wounding (in X, Y, and Z). This is especially true in the first few frames as the tailfin “relaxes” back to its original position after being tugged during the wounding procedure. Re-focusing of the sample after wounding may be necessary. If possible, it can be beneficial to set up a larger volume in Z during acquisition than necessary, to accommodate such sample movement. Additional image registration approaches can also help to correct for this movement post hoc (Kennard and Theriot, 2020).
Wounding with a micromanipulator
The procedure above can also be carried out with the needle placed in a micromanipulator. This approach will greatly reduce imprecision due to hand tremor. However, micromanipulators require practice and care to align properly in the optical path prior to imaging, and can add a lot of extra setup time to an experiment. Micromanipulators also present additional opportunities to accidentally break a needle during sample exchange, which requires realignment of the setup with a new needle. Furthermore, most micromanipulators are designed for small precise motions, rather than the larger, forceful motions needed to impale and tear the tailfin. Although micromanipulators can be positioned precisely, they nevertheless do not guarantee precise wound positioning because the tailfin is quite flexible and does not stay in place as it is tugged during laceration. We have found that the manual procedures described above are a better fit for our experimental needs.
Despite these caveats, if laceration with a micromanipulator is desired, we have found in preliminary trials that the 3-axis hydraulic joystick type manipulator controller (Narishige MO-202U) provides enough degrees of freedom and range of motion with intuitive control for effective laceration, although an even larger range of motion would be ideal.
Data analysis
The flexibility of the laceration protocol allows it to be combined with a variety of different assays, each with distinct analysis. For example, it is possible to compare the effect of different treatments on the response of basal epidermal cells to laceration (see Kennard and Theriot, 2020 for an example of this type of analysis, including statistical details). Other analyses will vary based on the desired assay or endpoint. For microscopic imaging purposes, each larva is generally considered an independent biological replicate.
Due to the variability in wound geometry inherent to laceration, no formal criteria have been established for excluding larvae based on the shape of the wound. Occasionally – especially when the investigator is still refining their laceration technique – a slip of the needle can lead to an inadvertently extreme wound, resulting in significant damage to the notochord, muscle, or other tissues more anterior to the tailfin. In these circumstances, the wound response can be much more severe than is typically observed during laceration, and it may be worthwhile to exclude such replicates from further analysis (see Kennard and Theriot, 2020 for examples of typical responses). This extreme damage is infrequent, so criteria for defining this type of extreme wound have not been established. One possible indication is that material – such as blood cells – appears to be leaking out of the tailfin when observed with brightfield microscopy following laceration. The laceration protocol outlined here should not damage vascularized tissue, so bleeding suggests that the wound is more serious than desired, and this particular replicate should be excluded from further analysis.
Notes
Advantages and limitations of variable wound geometry
Although with practice laceration can produce consistent results, this technique leads to more variable wound geometries than other methods, such as scalpel wounding or laser wounding (see Figure 3 for a representative sample of wound geometries following laceration). In our experience, we find that this can be an advantage, since if the wound location is controlled too precisely, biological differences in e.g., anterior-posterior developmental patterning can lead to systematic biases in wound response depending on the position of the wound (cf Lopez-Baez et al., 2018 figure 1 supplement 1e). We have found that the response of cells not immediately adjacent to a laceration wound (~20-300 µm away) is largely consistent despite variation in wound geometry (Kennard and Theriot, 2020). Thus, we believe laceration is well-suited for interrogating the behavior of these more-distant cells.
A major limitation of the variable wound geometry caused by laceration is that wound healing progress is harder to measure. Simple circular wounds are commonly used for wound healing studies because the progression of wound healing can be easily measured by the size of the wound as a fraction of the original wound size (Abreu- Blanco et al., 2012; Gault et al., 2014). In contrast, it is not obvious from a single image whether a laceration wound is closed or is still healing. Instead, dynamic metrics such as the cell velocity as a function of distance from the wound and time since injury can indicate if wound healing processes are still underway (Kennard and Theriot, 2020).
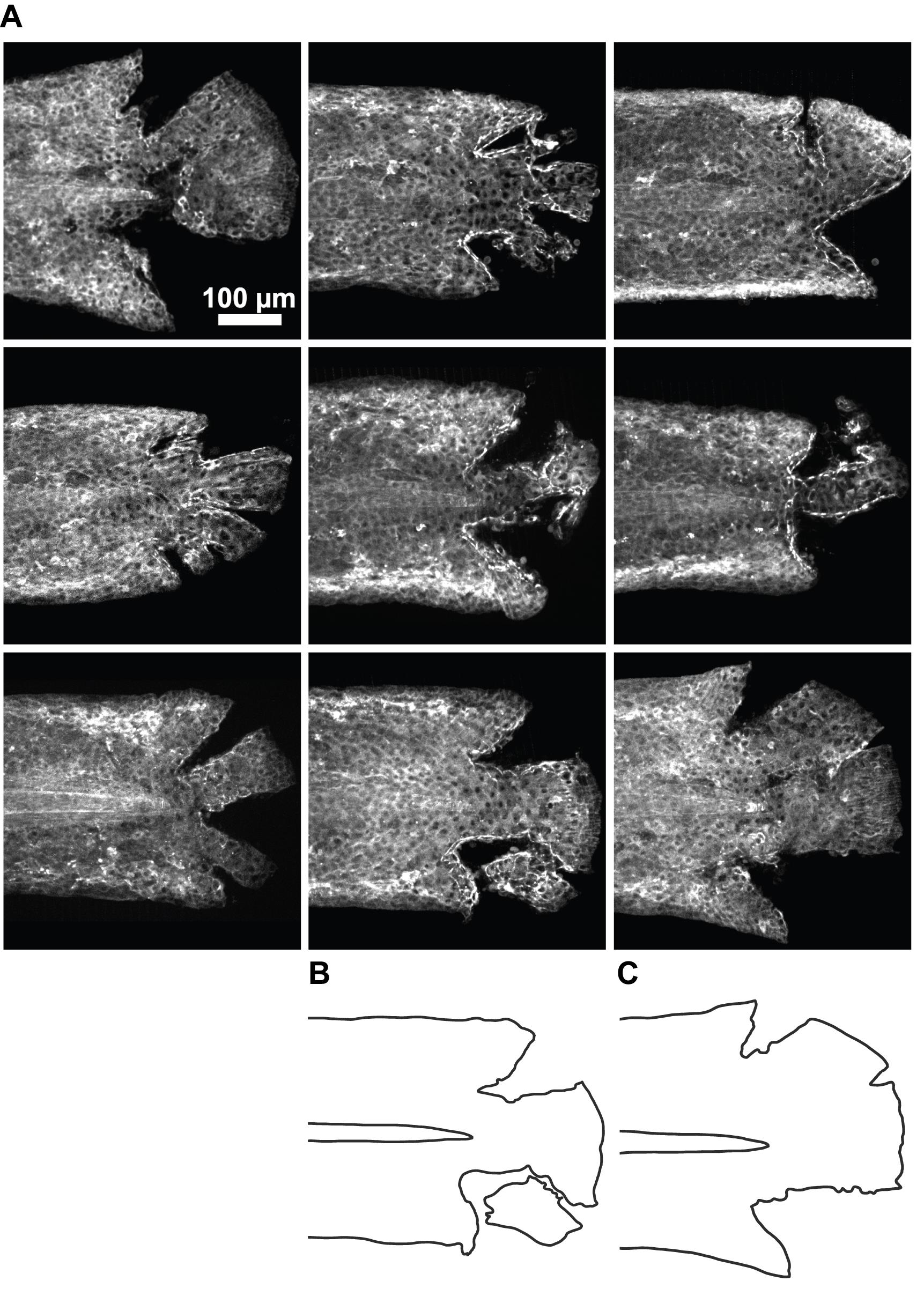
Figure 3. Demonstration of the variability in wound geometry following laceration. (A) Tailfins from 9 representative larvae are shown immediately after laceration. Larvae are 3dpf and express myosin light chain kinase fused to mApple under a heat-shock inducible promoter (hsp90:myl9-mApple) (Lou et al., 2015). Shown are maximum-intensity projections of z-stacks acquired on a spinning disk confocal using a 20× NA 0.95 water-immersion objective (Nikon). To draw attention to particularly salient variation in wound geometry, drawings of the wound geometry for two larvae are shown below each larva, indicating how lacerating the tailfin can lead to clean rips (C) or removal of a chunk of tailfin (B).Potential issues
One of the most common failure modes is that the tip of the needle breaks against the glass bottom surface, due to too much pressure being applied on the needle. This can lead to glass fragments remaining in the field of view during your acquisition, potentially obscuring events of interest. If not much of the tip is broken off, one may be tempted to continue to use the needle. However, we find that these blunter needles are more likely to break again because the remaining portion of the tip is thicker and more brittle than the part that broke off, and with each subsequent break the tip becomes larger and less capable of impaling the tissue and producing a clean tear. We recommend having several backup needles on hand so one can quickly dispose of the broken needle in a sharps bin and carry on with a fresh needle.
Possible application to regeneration studies
Our primary interest in this technique was to observe the rapid migratory wound response occurring within an hour after wound healing, for which the simplicity and flexibility of this procedure were advantageous. We have not formally investigated whether this wounding technique could also be used for studying regeneration on a timescale of hours or days. Supporting this possibility in principle, we have observed that more than 100 lacerated wildtype larvae were all able to regenerate an intact tailfin within a week (unpublished observations). It would be interesting to observe how the complex geometry of a laceration wound is restructured to reform an intact tailfin. One technical challenge that has not been evaluated is the suitability of the agarose window technique (Step D) for imaging longer than a few hours. If long-term imaging is desired, a dedicated imaging chamber such as the zWEDGI device may be more appropriate (Huemer et al., 2017a and 2017b).
Recipes
E3 medium (E3 + tricaine)
E3 (1× concentration: 5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4) was prepared as a 60× stock according to the cited protocol, without autoclaving or the addition of methylene blue (Cold Spring Harbor, 2011). It was kept for several weeks at room temperature.
A 25× tricaine stock solution (4 mg/ml stock concentration) was prepared by dissolving tricaine into water and adding 2% (v/v) of 1 M Tris, pH 9. Additional Tris solution was titrated in to bring the solution to a final pH of 7. This stock solution was dated and stored in the fridge for up to 6 months.
On the day of imaging, a solution of 1× E3 + tricaine (referred to as E3 + tricaine) was freshly prepared by diluting the 60× E3 and the 25× tricaine stocks to 1× with MilliQ water. This solution was discarded at the end of each day.
Acknowledgments
ASK and ECL were supported by an NIGMS training grant (T32GM008294). ECL was also supported by an NSF GRFP (DGE-1656518). JAT acknowledges support from the Howard Hughes Medical Institute and the Washington Research Foundation.
This protocol was derived from a previous publication: Kennard and Theriot, 2020 (DOI: 10.7554/eLife.62386).
Competing interests
The authors declare that they have no competing interests.
Ethics
Work with zebrafish was approved by the University of Washington Institutional Animal Care and Use Committee, protocol number 4427-01.
References
- Abreu-Blanco, M. T., Verboon, J. M., Liu, R., Watts, J. J., and Parkhurst, S. M. (2012). Drosophila embryos close epithelial wounds using a combination of cellular protrusions and an actomyosin purse string. J Cell Sci 125(4): 5984-5997.
- Briona, L. K. and Dorsky, R. I. (2014). Spinal cord transection in the larval zebrafish. J Vis Exp(87): e51479.
- Datta, R., Wong, A., Camarata, T., Tamanna, F., Ilahi, I. and Vasilyev, A. (2017). Precise Cellular Ablation Approach for Modeling Acute Kidney Injury in Developing Zebrafish. J Vis Exp(124): e55606.
- Cold Spring Harbor. (2011). E3 medium (for zebrafish embryos).Cold Spring Harb Protoc pdb.rec066449.
- Enyedi, B., Jelcic, M. and Niethammer, P. (2016). The Cell Nucleus Serves as a Mechanotransducer of Tissue Damage-Induced Inflammation. Cell 165(5): 1160-1170.
- Franco, J. J., Atieh, Y., Bryan, C. D., Kwan, K. M. and Eisenhoffer, G. T. (2019). Cellular crowding influences extrusion and proliferation to facilitate epithelial tissue repair. Mol Biol Cell 30(16): 1890-1899.
- Gault, W. J., Enyedi, B. and Niethammer, P. (2014). Osmotic surveillance mediates rapid wound closure through nucleotide release. J Cell Biol 207(6): 767-782.
- Huemer, K., Squirrell, J. M., Swader, R., LeBert, D. C., Huttenlocher, A. and Eliceiri, K. W. (2017a). zWEDGI: Wounding and Entrapment Device for Imaging Live Zebrafish Larvae. Zebrafish 14(1): 42-50.
- Huemer, K., Squirrell, J. M., Swader, R., Pelkey, K., LeBert, D. C., Huttenlocher, A. and Eliceiri, K. W. (2017b). Long-term Live Imaging Device for Improved Experimental Manipulation of Zebrafish Larvae. J Vis Exp(128): e56340.
- Kennard, A. S. and Theriot, J. A. (2020). Osmolarity-independent electrical cues guide rapid response to injury in zebrafish epidermis. Elife 9: e62386.
- Lopez-Baez, J. C., Simpson, D. J., L, L. L. F., Zeng, Z., Brunsdon, H., Salzano, A., Brombin, A., Wyatt, C., Rybski, W., Huitema, L. F. A., Dale, R. M., et al. (2018). Wilms Tumor 1b defines a wound-specific sheath cell subpopulation associated with notochord repair. Elife 7: e30657.
- Lou, S. S., Diz-Munoz, A., Weiner, O. D., Fletcher, D. A. and Theriot, J. A. (2015). Myosin light chain kinase regulates cell polarization independently of membrane tension or Rho kinase. J Cell Biol 209(2): 275-288.
- Mateus, R., Pereira, T., Sousa, S., de Lima, J. E., Pascoal, S., Saude, L. and Jacinto, A. (2012). In vivo cell and tissue dynamics underlying zebrafish fin fold regeneration. PLoS One 7(12): e51766.
- Miskolci, V., Squirrell, J., Rindy, J., Vincent, W., Sauer, J. D., Gibson, A., Eliceiri, K. W. and Huttenlocher, A. (2019). Distinct inflammatory and wound healing responses to complex caudal fin injuries of larval zebrafish. Elife 8: e45976.
- Vogel, A. and Venugopalan, V. (2003). Mechanisms of pulsed laser ablation of biological tissues. Chem Rev 103(2): 577-644.
- Westerfield, M. (2007). The Zebrafish Book, 5th edition: A Guide for the Laboratory Use of Zebrafish (Danio rerio). (5th edition). University of Oregon Press, Eugene, OR.
- Yoo, S. K., Freisinger, C. M., LeBert, D. C. and Huttenlocher, A. (2012). Early redox, Src family kinase, and calcium signaling integrate wound responses and tissue regeneration in zebrafish. J Cell Biol 199(2): 225-234.
- Zeng, Z., Lopez-Baez, J. C., Lleras-Forero, L., Brunsdon, H., Wyatt, C., Rybski, W., Hastie, N. D., Schulte-Merker, S. and Patton, E. E. (2018). Notochord Injury Assays that Stimulate Transcriptional Responses in Zebrafish Larvae. Bio-protocol 8(23): e3100.
Article Information
Copyright
![]() Kennard et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Kennard et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Kennard, A. S., Prinz, C. K., Labuz, E. C. and Theriot, J. A. (2021). Wounding Zebrafish Larval Epidermis by Laceration. Bio-protocol 11(24): e4260. DOI: 10.21769/BioProtoc.4260.
- Kennard, A. S. and Theriot, J. A. (2020). Osmolarity-independent electrical cues guide rapid response to injury in zebrafish epidermis. Elife 9: e62386.
Category
Developmental Biology > Cell growth and fate > Regeneration
Cell Biology > Tissue analysis > Injury model
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.