- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Nucleocapsid-based Transcomplementation Cell Culture System of SARS-CoV-2 to Recapitulate the Complete Viral Life Cycle
Published: Vol 11, Iss 21, Nov 5, 2021 DOI: 10.21769/BioProtoc.4257 Views: 4472
Reviewed by: David PaulMirko CorteseChhuttan L MeenaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2021

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The ongoing COVID-19 pandemic is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). As this virus is classified as a biosafety level-3 (BSL-3) agent, the development of countermeasures and basic research methods is logistically difficult. Recently, using reverse genetics, we developed a BSL-2 cell culture system for production of transcription- and replication-component virus-like-particles (trVLPs) by genetic transcomplementation. The system consists of two parts: SARS-CoV-2 GFP/ΔN genomic RNA, in which the nucleocapsid (N) gene, a critical gene for virion packaging, is replaced by a GFP reporter gene; and a packaging cell line for ectopic expression of N (Caco-2-N). The complete viral life cycle can be recapitulated and confined to Caco-2-N cells, with GFP positivity serving as a surrogate readout for viral infection. In addition, we utilized an intein-mediated protein splicing technique to split the N gene into two independent vectors and generated the Caco-2-Nintein cells as a packaging cell line to further enhance the security of this cell culture model. Altogether, this system provides for a safe and convenient method to produce trVLPs in BSL-2 laboratories. These trVLPs can be modified to incorporate desired mutations, permitting high-throughput screening of antiviral compounds and evaluation of neutralizing antibodies. This protocol describes the details of the trVLP cell culture model to make SARS-CoV-2 research more readily accessible.
Keywords: SARS-CoV-2Background
The COVID-19 pandemic caused by SARS-CoV-2 still rages around the world, threatening public health and the global economy (Wang et al., 2020). Several reverse genetic systems have been reported for generating SARS-CoV-2 replicons and recombinant virus for basic virology research and antiviral development (Hou et al., 2020; Xie et al., 2020; Zhang et al., 2021a and 2021b). However, as SARS-CoV-2 is classified as BSL-3 pathogen, experiments involving authentic virus are restricted to BSL-3 laboratories, which hinders basic research and antiviral discovery. Pseudotyped virus and replicon systems of SARS-CoV-2 that can be used in BSL-2 laboratories have been developed but are limited to studying viral entry and replication, respectively (Nie et al., 2020; Zhang et al., 2021b). Therefore, a cell culture model for SARS-CoV-2 that could safely recapitulate the entire viral life cycle in a BSL-2 facility is urgently needed (Rome and Avorn, 2020).
Recently, we developed such a system in which transcription- and replication-component SARS-CoV-2 virus-like particles (SARS-CoV-2 trVLPs) can be generated (Ju et al., 2021). We replaced the N gene in the SARS-CoV-2 genome with a GFP reporter gene and then provided N in trans in Caco-2 cells ectopically expressing this gene (Caco-2-N). Transducing the Caco-2-N cells with the SARS-CoV-2 GFP/ΔN genome thus allows for the production of trVLPs, which can complete the entire life cycle exclusively in the Caco-2-N packaging cells. In normal cells, these trVLPs can only complete a single-round infection as the packaged viral genome lacks the N gene, which is critical for viral particle assembly. Thus, this system can be safely utilized in BSL-2 level laboratories for SARS-CoV-2 research. In addition, the GFP reporter provides a convenient surrogate readout for virus infection, which facilitates the use of this system for neutralizing antibody determination and high-throughput antiviral screening.
Genetic manipulation of this SARS-CoV-2 trVLPs system is not trivial due to the large size of the viral genome (~30 kb) and presence of multiple toxic elements (Almazan et al., 2014; Xie et al., 2021). Herein, we describe technical details of our cell culture system for production of trVLPs. Overall, the engineering process includes three parts: packaging cell line construction, viral genome-length RNA preparation, and trVLP recovery. Using lentiviral transduction, SARS-CoV-2 N protein can be stably expressed in Caco-2 cells to generate the packaging cell line necessary for trVLP production. Besides, an intein-mediated protein trans-splicing approach (Stevens et al., 2017) was utilized to minimize the chance of N gene recombination into the genome of trVLP. The genome-length viral RNA is prepared by in vitro transcription of a full-length cDNA template generated by in vitro ligation. Briefly, four fragments (A-B, C, D, and E) were designed to cover the entire genome of SARS-CoV-2 GFP/ΔN. Each fragment can be chemically synthesized and then amplified using PCR (Figure 1). After PCR amplification, the fragments, which are flanked by a type IIS restriction enzyme recognition site (BsaI), are digested and then ligated in vitro to assemble the full-length cDNA of the viral genome. As type IIS restriction enzymes recognize asymmetric DNA sequences and cleave at a defined distance outside of their recognition sequence, the fragments are unidirectionally assembled into the full-length cDNA. A T7 promoter and a poly(A) tail were engineered upstream of fragment A and downstream of fragment E, respectively, ultimately allowing for in vitro transcription to produce capped, polyadenylated viral RNA. This genome-length RNA can then be electroporated into the Caco-2-N packaging cell line and trVLPs subsequently collected from the supernatant. Then the trVLPs are amplified and titrated using a tissue-culture infectious dose 50% (TCID50) endpoint dilution assay following the Reed & Muench method (Lindenbach, 2009), and GFP expression is the proxy of trVLP infection. SARS-CoV-2 trVLP can be used for evaluating antivirals and neutralizing antibodies.
Figure 1. Overview of production of SARS-CoV-2 GFP/ΔN trVLPs. The N gene of SARS-CoV-2 was replaced with the GFP gene, and the cDNA genome divided into four fragments designated as A-B, C, D, and E. Each of these fragments was chemically synthesized and then PCR amplified and assembled by restriction enzyme digestion and in vitro ligation to create the full-length cDNA. The full-length RNA genome was generated by in vitro transcription of the full-length cDNA. This RNA genome can then be electroporated into the packaging cell line, Caco-2-N, to produce trVLPs. At 24 h post electroporation, the supernatant of electroporated cells is collected and can be used to inoculate Caco-2, Caco-2-N, or Caco-2-NIntein cells. trVLPs can infect and replicate in Caco-2-N or Caco-2-NIntein cells and can be secreted into the supernatant. However, trVLPs only complete a single-round infection in Caco-2 cells due to the absence of viral N protein.
Materials and Reagents
Materials
Electroporation cuvettes, 4-mm gap (Bio-Rad, catalog number: 1652088)
24-well plate (Thermo Fisher Scientific, catalog number: 142475)
96-well plate (Thermo Fisher Scientific, catalog number: 167008)
0.2-ml PCR tubes (Thermo Fisher Scientific, catalog number: N8010580)
1.5-ml tube (Corning, catalog number: MCT-150-C)
15-ml tube (Thermo Fisher Scientific, catalog number: 339650)
10-cm dish (Thermo Fisher Scientific, catalog number: 150466)
Cells
Caco-2 cells (ATCC, catalog number: HTB-37)
293T cells (ATCC, catalog number: CRL-3216)
Stbl3 competent cells (AlpaLife, catalog number: KTSM110L)
EPI300 competent cells (Lucigen, catalog number: C300C105)
Reagents
Dulbecco’s Modified Eagle Medium (DMEM; Gibco, catalog number: C11965500BT)
0.25% Trypsin-EDTA (Thermo Fisher Scientific, catalog number: 25200072)
Fetal bovine serum (BIOVISTECH, catalog number: SE100-011)
Penicillin/streptomycin, 10,000 U/ml (Thermo Fisher Scientific, catalog number: 15140122)
DNA transfection reagent Vigofect (Vigorous, catalog number: T001)
pMD2G (Addgene, catalog number: 12259)
psPAX2 (Addgene, catalog number: 12260)
Opti-MEMTM (Gibco, catalog number: 31985070)
5 kb DNA marker (Takara, catalog number: 3428A)
15 kb DNA marker (Takara, catalog number: 3582A)
PrimeSTAR® Max DNA Polymerase (Takara, catalog number: R045)
PrimeSTAR® GXL DNA Polymerase (Takara, catalog number: R050)
Restriction enzyme BsaI (NEB, catalog number: R0535L)
T4 DNA Ligase (NEB, catalog number: M0202L)
mMESSAGE mMACHINETM T7 transcription kit (Thermo Fisher Scientific, catalog number: AM1344)
E.Z.N.A® Plasmid DNA Mini Kit I (Omega, catalog number: D6943-02)
E.Z.N.A® Gel Extraction Kit (Omega, catalog number: D2500-02)
Phenol:chloroform:isoamyl alcohol 25:24:1 (pH 7.8; Solarbio, catalog number: P1012)
ReverTra Ace® qPCR RT Kit (Toyobo, FSQ-101)
TRIzolTM Reagent (Thermo Fisher Scientific, catalog number: 15596018)
CopyControlTM Induction Solution (Lucigen, catalog number: CCIS125)
Polybrene (Sigma, catalog number: TR-1003-G)
Dulbecco's Phosphate-Buffered Saline (PBS; Corning, catalog number: 21-031-CVR)
Equipment
Water bath (zhybioresources, catalog number: SYG-1210 )
Centrifuge (Eppendorf, catalog number: 5406000291)
Blue Light Gel Imager (Sangon Biotech, catalog number: G500312)
NanoDropTM One/Onec (Thermo Fisher Scientific, catalog number: 701-058108)
Gene Pulser Xcell Total Electroporation System (Bio-Rad, catalog number: 1652660)
Incubator shaker (Changzhou Huayi, catalog number: THZ-D)
Procedure
Timeline:
Propagation of plasmids bearing SARS-CoV-2 cDNA fragments
Steps 1-2, Chemical transformation (2 h) and colony screen (overnight).
Step 3, Plasmid preparation: 1 h.
Construction of Caco-2-N and Caco-2-Nintein cell line
Steps 1-2, Lentivirus packaging (3) and transduction (3 d).
Preparation of DNA fragment by PCR
Steps 1-3, PCR amplify fragments A-B (6 h), C, D and E (2-3 h).
Steps 4, Purification of PCR products by phenol-chloroform extraction: 1-2 h.
Generation of genome-length cDNA by restriction enzyme digestion and in vitro ligation
Steps 1-2, Fragments C, D, and E digestion (5 h) and purification (1-2 h).
Step 3, Fragments C, D, and E in vitro ligation: 24 h.
Step 4, PCR amplification of C-D-E (6 h) and purification (1-2 h).
Step 5, Fragments A-B and C-D-E digestion: 5 h.
Step 6, Full-length cDNA assembly by in vitro ligation: 48 h.
Generation of viral genome-length RNA by in vitro transcription (IVT)
Step 1, PCR amplification of N gene (1 h) and purification (1-2 h).
Steps 2-8, In vitro transcription: 1 d.
Electroporation and trVLP recovery
Steps 1-4, Electroporation: 2 h.
Steps 5-10, trVLP recovery: 3-4 d.
RT-PCR for verification of GFP gene of the trVLP
Step 1, trVLP infection (24 h) and viral RNA extraction: 1-2 h.
Step 2, RT-PCR for amplification of GFP gene (1-2 h) and analysis by agarose electrophoresis (30 min).
trVLP titration by TCID50
Steps 1-5, trVLP infection (24 h) and TCID50 calculation (1-2 h).
Propagation of plasmids carrying SARS-CoV-2 cDNA fragments
Plasmids
The genome of SARS-CoV-2 GFP/ΔN (derived from the Wuhan-Hu-1 strain. GenBank: MN908947 was divided into four fragments designated as A-B, C, D, and E, each obtained by PCR using a chemically synthesized SARS-CoV-2 genome as the template. The fragments were cloned into the pCCI, pMV, or pLVX vectors, resulting in four plasmids: pCCI-A-B, pMV-C, pLVX-D, and pLVX-E. The SARS-CoV-2 N gene with a Flag tag in the N terminal was cloned into the lentivirus vector pLVX-IRES-mCherry, resulting in pLVX-N-Flag-IRES-mCherry that was then was used to generate lentivirus. Caco-2 cells transduced with this lentivirus (Caco-2-N) serve as the packaging cell line for producing SARs-CoV-2 trVLPs, as the Caco-2-N cells complement the SARS-CoV-2 GFP/ΔN genome by providing N in trans. Meanwhile, N gene was also mutated at 212 amino acid (G212C) and split into two parts. These two parts are fused with intein elements Npu-N and Npu-C (Zettler et al., 2009) separately defining as NN-IntN and IntC-NC and further cloned into lentiviral vectors, resulting pLVX- NN-IntN-IRES-mCherry and pLVX-IntC-NC-IRES-puromycin, respectively. These two plasmids encoding either N- or C-terminal of N protein were transduced together to ligate the full-length N in Caco-2 cell for generating another packaging cell line Caco-2-Nintein.
Transformation
For each plasmid, thaw EPI 300 or Stbl3 competent cells on ice (see "Note" below) and add 2 μl (approximately 10 ng) of the respective plasmid to the competent cells. Mix by tapping the tube and incubate on ice for 30 min.
Note: For pMV- and pLVX-derived plasmids – including pMV-C, pLVX-D, pLVX-E, pLVX-N-Flag-IRES-mCherry, pLVX-NN-IntN-IRES-mCherry, and pLVX-IntC-NC-IRES-puromycin – use Stbl3 competent cells for transformation. For pCCI-derived plasmid pCCI-A-B, EPI300 competent cells for transformation.
Heat shocks the tube containing the cells and plasmid mixture in a 42°C water bath for 90 s, then immediately place back on ice for a 2 min incubation.
Add 1 ml LB medium to the tube and shake at 37°C for 45 min at 220 rpm using an incubator shaker to recover the cells.
After recovery, plate 100 μl of the cell culture onto an LB plate supplemented with the appropriate antibiotics (see "Notes" below) and incubate overnight at 37°C.
Notes:
For selection of Stbl3 bacteria transformed with pMV- and pLVX-derived plasmids, use LB agar plates supplemented with 50 μg/ml ampicillin.
For selection of EPI300 bacteria transformed with pCCI-A-B, use LB agar plates supplemented with 12.5 μg/ml chloramphenicol.
Preparation of plasmids carrying SARS-CoV-2 genes
After overnight incubation, for each plate, pick 2-3 bacterial colonies and inoculate 4 ml LB supplemented with the appropriate antibiotics (the antibiotic concentration remains the same as on the plates). Incubate overnight (12-16 h) at 37°C with shaking at 220 rpm.
For pMV- and pLVX-derived plasmids, the overnight culture can directly be used for plasmid preparation. For pCCI-A-B, the bacterial culture should be further induced as follows:
Add 1.5 ml of the overnight culture to a 50 ml flask or tube containing 13.5 ml LB supplemented with 12.5 μg/ml chloramphenicol and 15 μl of CopyControlTM Induction Solution (Lucigen, catalog number: CCIS125), and shake at 220 rpm for 5 h at 37°C.
Centrifuge the cell cultures by spinning at 4,000 × g for 5 min.
Discard the supernatant and remove as much residual liquid as possible.
Following the manufacturer's instructions, use the cell for plasmid extraction via the Plasmid Mini Kit I (Omega).
At the final step of plasmid extraction, add 50 μl deionized water to the mini-prep column and incubate at RT for 3 min, then centrifuge at 13,000 × g for 1 min to elute the plasmids.
Determine the concentration and quality of the plasmid prep using a NanoDrop. The plasmids can be used immediately or stored at -20°C until use.
Construction of Caco-2-N and Caco-2-Nintein cell lines
Lentivirus packaging
Prepare vesicular stomatitis virus G protein (VSV-G) pseudotyped lentivirus following this published protocol (Tiscornia et al., 2006). Briefly, use VigoFect DNA transfection reagent to transiently co-transfect HEK293T cells with the third-generation packaging plasmids pMD2G (catalog no. 12259; Addgene), psPAX2 (catalog no. 12260; Addgene), and the transfer vector pLVX-N-Flag-IRES-mCherry. Change the medium 12 h post transfection and collect supernatants at 36, 60, and 84 h post transfection. Pool all the supernatants, pass through a 0.45-μm filter, aliquot, and store at -80°C until needed.
Transduction
Seed 1 × 105 Caco-2 cells in a 24-well plate one day prior to infection to ensure 70-80% confluent next day.
When ready for transduction, remove the cell culture medium, add 1 ml of lentivirus supplemented with 10 μg/ml polybrene into the well, and mix by rocking the plate gently. Incubate at 37°C, 5% CO2 for 48 h.
Notes:
To generate the Caco-2-N cell line, lentivirus packaging pLVX-N-Flag-IRES-mCherry is used for transduction. To generate the Caco-2-Nintein cell line, lentivirus packaging pLVX-NN-IntN-IRES-mCherry and pLVX-IntC-NC-IRES-puromycin are mixed (1:1) and used for transduction of the Caco-2 cells.
The addition of polybrene increases transduction efficiency.
As a measure of transduction efficiency, monitor the mCherry signal daily using a fluorescent microscope. Generally, the mCherry signal can be readily detected at 48 h post infection.
At 48 h post transduction, check the mCherry signal and passage the transduced cells into a 6-well plate and incubate at 37°C, 5% CO2.
At another 24 h, check the mCherry signal and cell density to make sure at least 90% of cells are mCherry positive, harvesting cells that could be used as the packing cell line for production of trVLPs.
Note: Since these cells are not under selection, the N gene in these Caco-2-derived cell lines can be lost after several passages, which will decrease the packaging efficiency of trVLPs. Thus, re-transduce the Caco-2-N cell lines as needed to maintain the N gene expression level. Alternatively, use a cell sorter to harvest the first 30% of the mCherry-positive cells from the constructed Caco-2-N cell lines if the N expression level is not sufficient (the mCherry positive cells lower than 90%) for trVLP packaging.
Generation of SARS-CoV-2 trVLP cDNA by PCR
The template, primers, and DNA polymerase in the thermal cycling program for PCR are listed in Table 1. The sequences of primers used in this protocol are listed in Table 2.
Table 1. Components for PCR reactions to generate select DNA fragments
Table 2. Primers used in this protocol
Amplification of fragments A-B
Set up a 50 μl PCR reaction using PrimeSTAR® GXL following the manufacturer’s instructions. Prepare a 400 μl reaction master mix and aliquot it into eight 0.2-ml PCR tubes (50 μl/tube). The components for the PCR reaction are listed in Table 3.
Table 3. PCR reaction for amplification of fragment A-B
Incubate reactions in a thermal cycler according to the program shown in Table 4.
Table 4.Thermal cycling program for PCR reaction to amplify fragment A-B
Note: Usually, 30-40 μg of fragment A-B can be recovered from the 400 μl PCR reaction solution.
Examine the PCR products by gel electrophoresis. Mix 2 μl of PCR product and with 6× DNA loading buffer and load the mixture onto a 1% agar gel (Figure 2A).
Figure 2. Agarose gel electrophoresis verification of DNA and RNA fragments for trLVPs generation. (A) The genome sequence of trVLPs was divided into four fragments and each fragment PCR amplified. The purified PCR products of each fragment were determined by agarose gel. (B) PCR products of C-D-E and purified ligation products of full-length DNA (FL DNA). (C) Agarose gel analysis of SARS-CoV-2 N gene PCR products. (D) Agarose gel analysis of viral full-length RNA (FL-RNA) and (E) N gene mRNA generated by in vitro transcription using the FL DNA genome and N gene PCR products, respectively, as template. The black arrow indicates the FL RNA and N RNA, respectively.
Amplification of fragments C, D, and E
Set up a 50 μl PCR reaction according to the PrimeSTAR® Max (Takara) instructions. Prepare a 200 μl PCR reaction for each fragment and aliquot it into four 0.2 ml PCR tubes, 50 μl/tube. The components for the PCR reaction are listed in Table 5.
Table 5. PCR reaction for amplification of fragments C, D, and E
Incubate reactions in a thermal cycler according to the program shown in Table 6.
Table 6. Thermal cycling program for PCR reaction of fragments C, D, and E
Note: Usually, approximately 20 μg of each fragment can be recovered from the 200 μl PCR reaction solution.
Check the quality of the PCR products by gel electrophoresis. Mix 2 μl of PCR product and with 6× DNA loading buffer and load the mixture onto a 1% agar gel (Figure 2A).
Purification of PCR products by phenol-chloroform extraction
Pool the PCR products for each fragment into a 1.5-ml tube, add an equal volume of phenol:chloroform:isoamyl alcohol (25:24:1), and mix thoroughly and centrifuge at 13,000 × g for 1 min at 4°C.
Transfer the upper aqueous phase into a fresh 1.5-ml tube, add chloroform equal to the aqueous phase volume, mix thoroughly, and centrifuge at 13,000 × g for 1 min at 4°C.
Pipet the top layer into a new nuclease-free 1.5-ml tube and add 1/10th volume of 3 M sodium acetate (pH 5.2). Then add isopropanol in a volume ratio of 1:1, mix well and incubate at -20°C for at least 30 min.
Centrifuge at 13,000 × g for 15 min at 4°C to pellet the DNA.
Remove the supernatant, wash the pellet by adding 1 ml 75% ethanol, and centrifuge at 13,000 × g for 5 min at 4°C.
Remove the residual liquid completely, being careful not to remove any pellet.
Air dry the pellet until it is no longer visible and resuspend in 100 μl of nuclease-free water.
Measure the yield and quality of the recovered DNA by using a NanoDrop.
Check the quality of the purified DNA by gel electrophoresis (Figure 2A).
Generation of genome-length cDNA by restriction enzyme digestion and in vitro ligation
Fragments C, D, and E are first digested with Bsa I and ligated by T4 ligase to create the C-D-E ligation product, which is utilized as a template for PCR amplification. Next, the PCR products of fragment A-B and C-D-E are digested with BsaI and ligated to generate the full-length SARS-CoV-2 cDNA.
Fragments C, D, and E digestion
Set up an 80-μl digestion reaction for fragments C, D, and E, as shown in Table 7. Fragments C and E are digested with Bsa I.
Table 7. Fragments C, D, and E digestion reactions
Incubate the fragments C, D, and E digestion reactions at 37°C for 5 h or incubate overnight.
Digested DNA fragment purification from agarose gel
After digestion, add 16 μl 6× DNA loading buffer to each reaction tube, mix thoroughly, and load the mixture onto a 1% agarose gel and run at 180 V for 15 min.
Visualize the DNA fragment using a Blue Light Gel Imager. Avoid using UV light for visualization as it can cause DNA damage that will result in failure of downstream RNA transcription.
Cut the target bands (the sizes of the expected C, D, and E bands are 6,133 bp, 4,168 bp, and 1,636 bp, respectively) and extract the DNA fragment from the gel using a gel extraction kit (Omega) following the manufacturer's instructions.
At the final step, add 30 μl deionized water (pre-warmed at 65°C) onto the column and incubate at 37°C for 5 min.
Centrifuge at 13,000 × g for 1 min to elute the DNA.
Re-load the DNA elution onto the column and repeat Steps D2d and D2e to increase the recovery efficiency.
Measure quantity and quality of the recovered DNA using a NanoDrop.
Note: The recovery efficiency is approximately 30%.
In vitro ligation of fragments C, D, and E
Set up a 10-μl ligation reaction according to the components listed in Table 8. In this protocol, equal molar concentrations of fragments digested by BsaI are used.
Table 8. Fragments C, D, and E ligation reaction
Incubate the reaction system at 4°C for 24 h. The ligation products (without purification) are utilized as a template for amplifying fragment C-D-E.
PCR amplification and purification of fragment C-D-E
Set up a 50 μl PCR reaction using the ligation products from Step D3b as the template. Prepare a 400 μl reaction and aliquot it across eight 0.2-ml PCR tubes (50 μl/tube). The components are shown in Table 9.
Table 9. PCR reaction for amplification of fragment C-D-E
Incubate reactions in a thermal cycler according to the program shown in Table 10.
Table 10. Thermal cycling program for PCR amplification of fragment C-D-E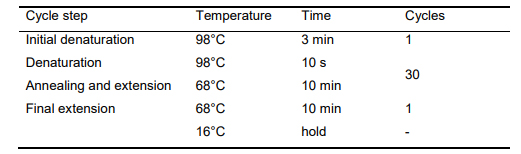
Purify the PCR products by phenol-chloroform extraction as described in Step C4.
Note: Approximately 40 μg of C-D-E fragment can be recovered from the 400 μl PCR reaction solution.
Load 2-μl onto a 1% agarose gel electrophoresis to check the quality of the purified PCR product, as shown in Figure 2B.
Fragments A-B and C-D-E digestion with BsaI
Set up a 120-μl digestion reaction for fragments A-B and C-D-E as shown in Table 11.
Table 11. Digestion reaction for fragments A-B and C-D-E
Incubate the digestion reaction at 37°C for 5 h.
After digestion, the digestion products are separated by agarose electrophoresis and recovered by gel extraction using a kit as described in Step D2.
At the final step of extraction, use 100 μl nuclease-free water for elution.
In vitro ligation of fragments A-B and C-D-E
Set up a 100-μl ligation reaction, using equal molar concentrations of the fragments to assemble the full-length cDNA. The reaction components are listed in Table 12.
Table 12. Fragments A-B and C-D-E ligation reaction
Incubate the reaction at 4°C for 24 h.
Add 1 μl of T4 ligase to the ligation reactions, mix well, and continue incubating at 4°C for another 24 h.
Purify the full-length cDNA ligation products by phenol-chloroform extraction as described in Section C. At the final step of purification, dissolve the DNA pellet in 10 μl nuclease-free water. The purified DNA can be used immediately or stored at -20°C.
Confirm the quality and size of the product by gel electrophoresis, loading 2 μl of purified ligation product onto a 1% agarose gel (Figure 2B).
Generation of viral genome-length RNA by in vitro transcription (IVT)
Preparation of N gene DNA.
In this protocol, N gene mRNA is electroporated into Caco-2-N cells along with genome-length viral RNA to increase trVLP production.
Set up a 50 μl PCR reaction according to the PrimeSTAR® MAX instructions. The PCR reaction components are shown in Table 13.
Table 13. PCR system for amplification of N gene
Incubate reactions in a thermal cycler following the program in Table 14.
Table 14. Thermal cycling program for PCR amplification of N gene
Check the quality of the PCR products by gel electrophoresis. Mix 2 μl PCR product with 6×DNA loading buffer and load the mixture onto a 1% agarose gel (Figure 2C).
Recover N gene PCR products by phenol-chloroform extraction as described in Step C4. At the final step of purification, dissolve the DNA pellets in 30 μl nuclease-free water.
Set up a 30 μl IVT reaction to generate full-length viral RNA and N gene mRNA using the Thermo mMESSAGE mMACHINE T7 transcription Kit, following the reaction setup shown in Table 15.
Table 15. IVT reaction for generating full-length viral RNA and N gene mRNA
Incubate the IVT reaction at 32°C for 5 h.
Add 1 μl DNase to the reaction and incubate at 37°C for 15 min to digest the DNA template.
Recover the viral RNA and N gene mRNA using lithium chloride precipitation following the manufacturer’s instructions in the mMESSAGE mMACHINE® T7 transcription Kit. At the final step of the precipitation, dissolve the RNA pellet in 30 μl nuclease-free water.
Measure the concentration of RNA using a NanoDrop. Usually, approximately 30 μg of viral RNA can be obtained.
Load 1 μg of RNA onto a 1% agarose gel to determine the quality of the genome-length RNA and N mRNA, as shown in Figures 2D and 2E.
Store the RNA at -80°C or use immediately.
Electroporation of the genome-length viral RNA and N gene mRNA into Caco-2-N cell for trVLP production
Seed 3 × 106 Caco-2-N cells in a 10-cm dish 2 days before electroporation to make sure sufficient cells are present for electroporation.
Prepare the following reagents and equipment before electroporation:
Cool the centrifuge to 4°C.
Pre-chill DPBS, Opti-MEM, and 4-mm cuvettes on ice.
Pre-warm a 10-cm dish containing 10 ml cell culture medium in a 37°C cell incubator.
Harvest Caco-2-N cells
Add 2 ml of 0.25% trypsin/EDTA to the Caco-2-N cells in the 10-cm dish and incubate at 37°C for approximately 8 min to detach the cells.
Add 2 ml of cell culture medium to the plate to stop digestion and pipet to generate a single-cell suspension.
Cell pretreatment and electroporation
Use a hemocytometer to determine the number of cells.
Transfer the appropriate volume of cell suspension containing 8 × 106 cells to a 15-ml tube and pellet the cells by centrifugation at 500 × g for 5 min at 4°C.
Discard the supernatant and wash cells by resuspending the pellet in 10 ml pre-chilled DPBS.
Precipitate cells by centrifugation at 500 × g for 5 min at 4°C.
Remove the supernatant and resuspend the pellet in 400 μl Opti-MEM.
Add 20 μg genome-length viral RNA and 10 μg N gene mRNA to the cell suspension, pipetting up and down to mix thoroughly.
Transfer the entire cell mixture into the pre-chilled 4-mm cuvette, put the cuvette into the electroporation chamber, applying a single pulse (270 V at 950 μF) using the GenePulser apparatus (Bio-Rad).
After electroporation, immediately pipet the electroporated cells into the pre-warmed 10 cm dish containing 10 ml cell culture medium. Gently rock the plate and incubate at 37°C, 5% CO2.
Monitor GFP signal daily using a fluorescent microscope. Generally, the GFP signal can be observed 12 h post electroporation, as shown in Figure 3A.
Prepare Caco-2-N cells for de novo infection
Seed 1 × 105 Caco-2-N cells into a 24-well plate 24 h before de novo infection with the generated trLVPs, incubating at 37°C, 5% CO2.
De novo infection
Check the GFP signal of the electroporated cells 24 h post electroporation
Collect 1 ml supernatant (containing trVLPs) from the electroporated cells (this will be considered P0).
Centrifuge the supernatant at 500 × g for 5 min to pellet cell debris.
Make sure the confluency of the Caco-2-N cells in the 24-well plate seeded above in Step F5 is approximately 70-80%. Remove the cell culture medium and add the collected supernatant containing trVLPs into the well, and incubate at 37°C, 5% CO2.
Monitor the GFP signal daily of the de novo infected cells (Figure 3B). Generally, the signal will be seen across the well 96 h post infection.
From the de novo infected cells, harvest supernatant containing trVLPs 4 days post infection (defined as P1 virus), centrifuge at 500 × g for 5 min at 4°C, and store as 1 ml aliquots at -80°C.
To amplify the virus, add 50 μl of P1 virus onto Caco-2-N cells in a 10-cm dish (the confluence is 70-80%) and incubate at 37°C, 5% CO2.
Monitor the GFP signal daily and harvest supernatants at 48 h post infection. Centrifuge at 500 × g for 5 min at 4°C. The collected virus is defined as P2 and can be stored as 1 ml aliquots at -80°C until use.
Figure 3. Fluorescence microscopy analysis of Caco-2-N cells infected with SARS-CoV-2 GFP/ΔN. (A) GFP and bright-field images of Caco-2-N cells at 12 h post electroporation. (B) Supernatants of Caco-2-N cells at 24 h post electroporation were collected and used to infect naive Caco-2-N cells in a 24-well plate. The GFP and bright-field images 24 h and 48 h post infection are shown.
RT-PCR verification of the GFP gene in SARS-CoV-2 GFP/ΔN trVLPs
To ensure the presence of the GFP gene in the SARS-CoV-2 GFP/ΔN trVLPs, viral RNA is extracted from the Caco-2-N cells infected by trVLPs and analyzed by RT-PCR (Figure 4A). A primer pair flanking the N region of ORF8 and the 3’UTR was designed for this purpose.
trVLP RNA extraction
Prepare Caco-2-N cells in a 24-well plate one day before infection so that the confluency at time of infection is 70-80%.
Infect cells by adding 50 μl trVLPs into the well and incubate at 37°C, 5% CO2.
48 h post infection, discard the supernatant from the trLVP-infected cells and add 500 μl of TRIzol reagent into the well, pipetting up and down to make a single-cell suspension.
Transfer the resultant cell lysate into a fresh 1.5 ml tube, add 100 μl chloroform, and vortex the tube or shake it by hand violently. Centrifuge at 13,000 × g for 15 min at 4°C.
Carefully transfer 200 μl of the upper aqueous phase (containing RNA) to a fresh 1.5 ml tube, mix well with 200 μl isopropanol, and incubate at -20°C for 30 min.
Centrifuge at 13,000 × g for 15 min at 4°C and remove the supernatant.
Add 600 μl 75% ethanol to gently wash the pellet and centrifuge at 13,000 × g for 3 min at 4°C.
Remove the supernatant and centrifuge the tube at 13,000 × g for 1 min at 4°C.
Remove the supernatant and open the tube lid to dry the pellet thoroughly.
Add 20 μl RNase-free H2O to dissolve the RNA sample.
RT-PCR
Use the Toyobo reverse transcription kit and the supplied random primers according to the manufacturer's direction to generate cDNA from the extracted viral RNA. A total of 10 μl cDNA is obtained that can then be used immediately or stored at -20°C until use.
Add 40 μl H2O to 10 μl cDNA and mix well.
Set up a 50 μl PCR reaction using the cDNA as the template following the setup shown in Table 16.
Table 16. PCR reaction for verification of GFP gene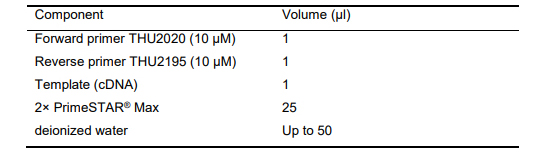
Incubate reactions in a thermal cycler following the program shown in Table 17.
Table 17. Thermal cycling program for PCR verification of GFP gene
Examine the size of the PCR products by gel electrophoresis. Mix 5 μl PCR product with 6×DNA loading buffer and load the mixture onto a 1% agarose gel (Figure 4B).
Figure 4. Analysis by RT-PCR of the GFP gene in trLVPs. RT-PCR validation of the GFP gene in SARS-CoV-2 GFP/ΔN trVLPs generated in Caco-2-N cells. The expected DNA size (A) and agarose electrophoresis analysis of the PCR products (B) are shown.
trVLP titration using a tissue-culture infectious dose 50% (TCID50) endpoint dilution assay
trVLP infectivity is quantified using a TCID50 endpoint method (Lindenbach, 2009).
Seed 1.5 × 104 Caco-2-N cells in 100 μl DMEM supplemented with 10% FBS per well in a 96-well plate one day before infection.
Thaw the trVLPs stored at -80°C and serially dilute 1:10 using DMEM with 10% FBS (12 μl trVLP sample into 108 μl DMEM with 10% FBS). Mix by vortexing for 5 s and remove 12 μl to add to another aliquot of 108 μl DMEM with 10% FBS (1:100 dilution). Repeat the dilution series out to a 108-fold dilution.
Add 100 μl of the diluted virions to each well of the seeded Caco-2-N cells in the 96-well plate on top of the existing media (to make a total of 200 μl per well), with eight replicates per dilution. Gently rock the plate to mix the virions well and incubate at 37°C, 5% CO2.
Check the GFP signal of each well 24 h post infection and count the number of GFP-positive wells for each concentration.
Calculate theTCID50 following the Reed & Muench method (Reed and Muench, 1938) using the equations described below:
TCID50/ml = (1 (ml)/the volume of original trVLPs (ml)) ×10^ (proportionate distance + Lower dilution).
Proportionate Distance = (Percentage next above 50% - 50%)/(Percentage next above 50% - Percentage next below 50%).
Lower dilution= 10^ (dilution in which position is next above 50%).
Acknowledgments
This work was funded by the National Natural Science Foundation of China (32041005 to QD), the Beijing Municipal Natural Science Foundation (M21001 to QD), and the Start-up Foundation of Tsinghua University (53332101319 to QD). X.J. was funded by the Postdoctoral Science Foundation of China (2021TQ0182) and the Shuimu Tsinghua Scholar Program.
The original research article in which this protocol was used is Ju et al. (2021).
Competing interests
Q.D. and X.J. have filed a patent application on the use of the SARS-CoV-2 transcomplementation system and its use for anti-SARS-CoV-2 drug screening.
References
- Almazan, F., Sola, I., Zuniga, S., Marquez-Jurado, S., Morales, L., Becares, M. and Enjuanes, L. (2014). Coronavirus reverse genetic systems: infectious clones and replicons. Virus Res 189262-270.
- Hou, Y. J., Okuda, K., Edwards, C. E., Martinez, D. R., Asakura, T., Dinnon, K. H., Kato, T., Lee, R. E., Yount, B. L. and Mascenik, T. M. (2020). SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 182(2): 429-446e414.
- Ju, X., Zhu, Y., Wang, Y., Li, J., Zhang, J., Gong, M., Ren, W., Li, S., Zhong, J., Zhang, L., Zhang, Q. C., Zhang, R. and Ding, Q. (2021). A novel cell culture system modeling the SARS-CoV-2 life cycle. PLoS Pathog 17(3): e1009439.
- Lindenbach, B. D., (2009). Measuring HCV infectivity produced in cell culture and in vivo. Methods Mol Biol 510329-336.
- Nie, J., Li, Q., Wu, J., Zhao, C., Hao, H., Liu, H., Zhang, L., Nie, L., Qin, H. and Wang, M. (2020). Quantification of SARS-CoV-2 neutralizing antibody by a pseudotyped virus-based assay. Nat Protoc 15(11): 3699-3715.
- Reed, L. and Muench, H. (1938). A simple method of estimating fifty percent end points. Amer J Hyg 27709-716.
- Rome, B. N. and Avorn, J. (2020). Drug Evaluation during the Covid-19 Pandemic. N Engl J Med 382(24): 2282-2284.
- Stevens, A. J., Sekar, G., Shah, N. H., Mostafavi, A. Z., Cowburn, D. and Muir, T. W. (2017). A promiscuous split intein with expanded protein engineering applications. Proc Natl Acad Sci U S A 114(32): 8538-8543.
- Tiscornia, G., Singer, O. and Verma, I. M. (2006). Production and purification of lentiviral vectors. Nat Protoc 1(1): 241-245.
- Wang, C., Horby, P. W., Hayden, F. G. and Gao, G. F. (2020). A novel coronavirus outbreak of global health concern. Lancet 395(10223): 470-473.
- Xie, X., Lokugamage, K. G., Zhang, X., Vu, M. N., Muruato, A. E., Menachery, V. D. and Shi, P. Y. (2021). Engineering SARS-CoV-2 using a reverse genetic system. Nat Protoc 161761-1784.
- Xie, X., Muruato, A., Lokugamage, K. G., Narayanan, K., Zhang, X., Zou, J., Liu, J., Schindewolf, C., Bopp, N. E. and Aguilar, P. V. (2020). An Infectious cDNA Clone of SARS-CoV. Cell Host Microbe 27(5): 841-848e843.
- Zettler, J., Schutz, V. and Mootz, H. D. (2009). The naturally split Npu DnaE intein exhibits an extraordinarily high rate in the protein trans-splicing reaction. FEBS Lett 583(5): 909-914.
- Zhang, X., Liu, Y., Liu, J., Bailey, A. L., Plante, K. S., Plante, J. A., Zou, J., Xia, H., Bopp, N. E. and Aguilar, P. V. (2021). A trans-complementation system for SARS-CoV-2 recapitulates authentic viral replication without virulence. Cell 184(8): 2229-2238e2213.
- Zhang, Y., Song, W., Chen, S., Yuan, Z. and Yi, Z. (2021). A bacterial artificial chromosome(BAC)-vectored noninfectious replicon of SARS-CoV. Antiviral Res 185104974.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Yu, Y., Ju, X. and Ding, Q. (2021). A Nucleocapsid-based Transcomplementation Cell Culture System of SARS-CoV-2 to Recapitulate the Complete Viral Life Cycle. Bio-protocol 11(21): e4257. DOI: 10.21769/BioProtoc.4257.
Category
Molecular Biology > DNA > Chromosome engineering
Microbiology > Microbial genetics > Recombination
Environmental science > Virus
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.