- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

APEX-mediated Proximity Labeling of Proteins in Cells Targeted by Extracellular Vesicles
Published: Vol 11, Iss 21, Nov 5, 2021 DOI: 10.21769/BioProtoc.4213 Views: 5022
Reviewed by: Pavan VedulaFarah HaqueAnthony FlamierChao Wang
Original research article
The authors used this protocol in:
Sep 2021

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Extracellular vesicles (EVs) are thought to mediate intercellular communication through the delivery of cargo proteins and RNA to target cells. The uptake of EVs is often followed visually using lipophilic-dyes or fluorescently-tagged proteins to label membrane constituents that are then internalized into recipient cells (Christianson et al., 2013; De Jong et al., 2019). However, these methods do not probe the exposure of EV cargo to intracellular compartments, such as the cytoplasm and nucleus, where protein or RNA molecules could elicit functional changes in recipient cells. In this protocol, we employ an EV cargo protein-APEX fusion to detect proximity interactions with recipient cell cytoplasmic/nuclear targets. This approach results in the biotinylation of proteins in close contact with the reporter fusion and thus permits profiling of biotinylated proteins affinity purified on immobilized streptavidin beads.
Graphic abstract:
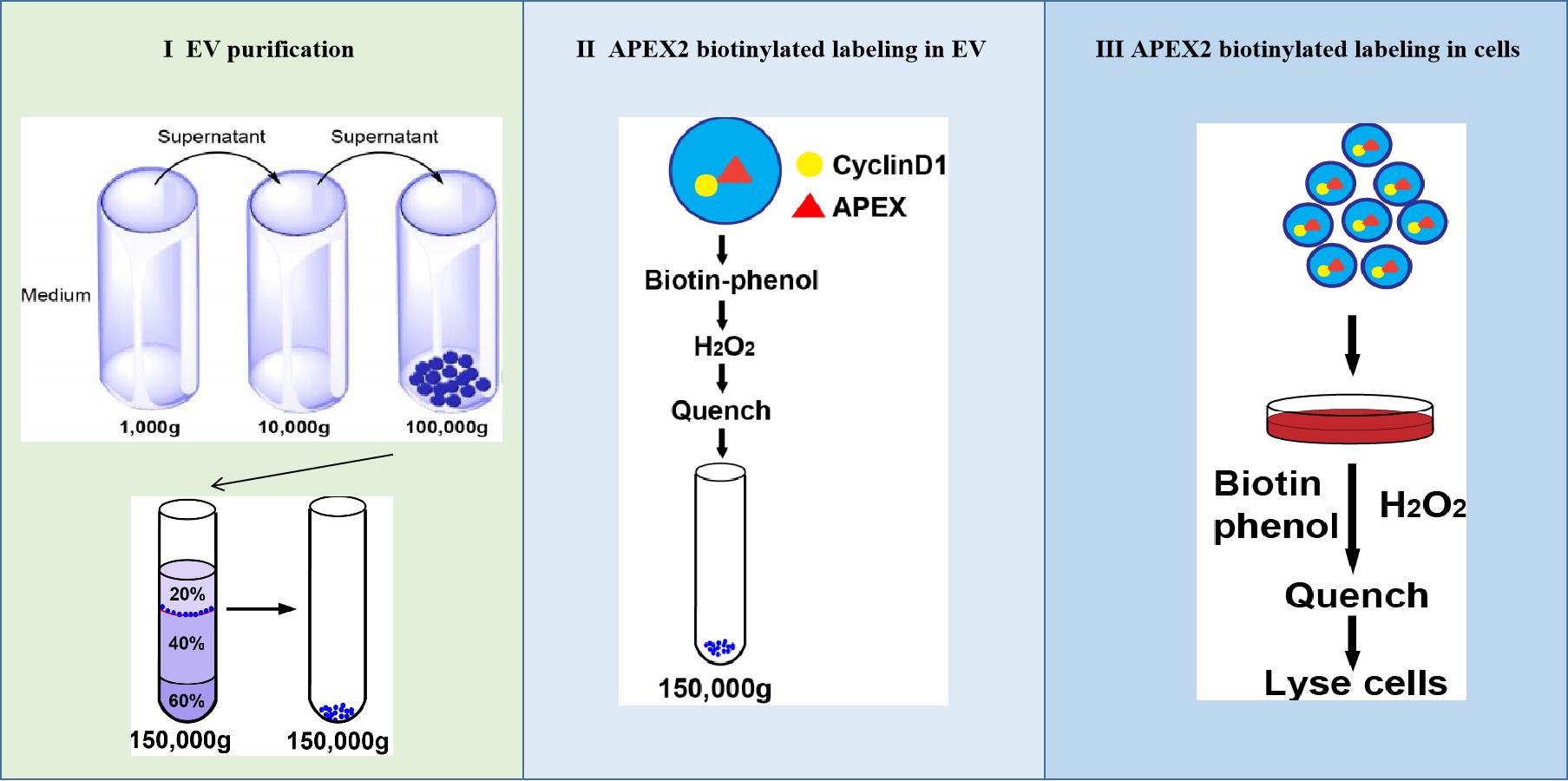
Schematic showing three steps of APEX-mediated proximity labeling of proteins in cells targeted by EVs.
Background
Extracellular vesicles (EVs) are secreted by cells, circulate in body fluids, and ultimately generate functional changes through interaction with or uptake into recipient cells (Raposo and Stoorvogel, 2013). Protein, RNA, and possibly DNA are packaged by EVs and may be delivered into target cells to elicit changes in gene expression and cell behavior (Budnik et al., 2016; Van Niel et al., 2018; Shurtleff et al., 2016; Temoche-Diaz et al., 2019; Song et al., 2021). Most of the current methodologies used to visualize EV uptake, however, do not identify the molecular targets of molecules released into recipient cells. An approach to define such targets should be of broad utility.
APEX biotinylation labeling is a newly-developed technique that reveals the subcellular proteomes of many landmarks in the nucleus and cytoplasm (Chen and Perrimon, 2017). Using hydrogen peroxide (H2O2) as an electron donor, the enzyme APEX catalyzes the oxidation of the substrate biotin-phenol (BP) (Hung et al., 2016). The biotin-phenoxyl radical is a highly reactive, short-lived (<1 ms) species that conjugates to other proteins that are proximal to the APEX active site. Biotinylated protein products may then be isolated by streptavidin affinity purification and identified using conventional mass spectrometry (MS) techniques (Lobingier et al., 2017).
We used this approach to investigate the contacts made by the protein cyclinD1 when it is delivered to mouse embryonic cells (mESCs) from EVs produced by differentiating neural progenitor cells (Song et al., 2021). The use of a fusion of APEX to other EV cargo proteins should prove useful to identify molecular contacts within target cells.
Materials and Reagents
Syringe, 10 ml (BD, catalog number: 302995)
Syringe filter (0.45 µm; Thermo Scientific, catalog number: 44525-PP)
Falcon tube (Fisher Scientific, catalog number: 08-771-23)
35 mm dishes (tissue culture dish) (Corning, catalog number: CLS430165)
10 cm dishes (tissue culture dish) (Corning, catalog number: CLS430167)
15 cm dishes (tissue culture dish) (Corning, catalog number: CLS430597)
Open-Top Thinwall Ultra-Clear Tube (38.5 ml), 25 × 89 mm (Beckman Coulter, catalog number: 344058)
Note: On text "38.5 ml ultra-clear tube".
Open-Top Thinwall Ultra-Clear Tube (13.2 ml), 14 × 89 mm (Beckman Coulter, catalog number: 344059)
Note: On text "13.2 ml ultra-clear tube".
Open-Top Thinwall Ultra-Clear Tube (5 ml), 13 × 51 mm (Beckman Coulter, catalog number: 344057)
Note: On text "5 ml ultra-clear tube".
Transfer pipettes (Fisherbrand, catalog number: 13-711-7M)
293T cells (ATCC, catalog number: CRL-3216)
N2A cells (ATCC, catalog number: CCL-131)
mESCs (R1, ATCC, catalog number: SCRC-1011)
Retinoic acid (RA) (Sigma, catalog number: R2625)
Fetal bovine serum (FBS) (VWR, catalog number: 89510-194)
Exosome-depleted FBS (System Biosciences (SBI), catalog number: EXO-FBS-250A-1)
Puromycin (Sigma-Aldrich, catalog number: P8833-100MG)
pcDNA3 APEX-nes (Addgene, catalog number: 49386)
XPack CMV-XP-MCS-EF1α-Puro Cloning Lentivector (System Biosciences)
psPAX (Addgene, catalog number: 12260)
pMD2.G (Addgene, catalog number: 12259)
Lipofectamine 2000 (Life Technologies, catalog number: 11668019)
OPTI-MEM (Thermo Scientific, catalog number: 31985062)
Sucrose (Fisher Chemical, catalog number: S5-3)
Biotin-phenol (Sigma-Aldrich, catalog number: SML2135)
Protease inhibitor cocktail (100×) (Sigma-Aldrich, catalog number: P8340)
Streptavidin-HRP (Thermo Fisher Scientific; catalog number: 21130)
H2O2 (Thermo Fisher, catalog number: 34062)
Sodium ascorbate (Sigma-Aldrich, catalog number: A7631)
Sodium azide (Sigma-Aldrich, catalog number: 26628-22-8)
Trolox (Sigma-Aldrich, catalog number: 238813-5G)
Biotin (Thermo Fisher Scientific, catalog number: 29129)
DTT (Gold Biotechnology, catalog number: DTT25)
DPBS (Dulbecco's phosphate-buffered saline; Thermo Fisher, catalog number: 14190144)
Streptavidin-agarose beads (Sigma, catalog number: 16-126)
NovexTM WedgeWellTM 10% Tris-glycine mini gels, 10-well (Thermo Fisher, catalog number: XP10200BOX)
Ponceau S (Thermo Fisher Scientific, catalog number: XP00100PK2)
Coomassie G250 (Sigma, catalog number: 1.15444)
DMEM/F12 culture medium (Thermo Fisher Scientific, catalog number: 11320033)
DMEM culture medium (Life Technologies, catalog number: 10566-024)
Neurobasal culture medium (Thermo Fisher Scientific, catalog number: 21103049)
B-27TM Supplement (50×) (Thermo Fisher Scientific, catalog number: 17504044)
N-2 Supplement (100×) (Thermo Fisher Scientific, catalog number: 17502048)
L-glutamine (Thermo Fisher Scientific, catalog number: 25030081)
Non-essential amino acids (100×) (Thermo Fisher Scientific, catalog number: 11140050)
β-mercaptoethanol (0.1 M) (Sigma, catalog number: M3148)
DMEM + 10% FBS (see Recipes)
RA (10 μM) + 1% exosome-depleted FBS in DMEM (see Recipes)
Buffer C (see Recipes)
Quencher solution (see Recipes)
4× Loading buffer (20 ml) (see Recipes)
N2B27 medium (1 L) (see Recipes)
RIPA (see Recipes)
Buffer D (see Recipes)
TBS-T (see Recipes)
Equipment
Sorvall RC 6+ centrifuge (Thermo Scientific, model: 46910)
Fixed angle rotor F14S-6X250y FiberLite (Thermo Scientific, catalog number: 78500)
Ultracentrifuge (Beckman Coulter, model: Optima XE-90, catalog number: A94471)
Swinging-bucket rotor SW 32 Ti and bucket set (Beckman Coulter, catalog number: 369694)
Swinging-bucket rotor SW 28 Ti and bucket set (Beckman Coulter, catalog number: 342204)
Swinging-bucket rotor SW 55 Ti and bucket set (Beckman Coulter, catalog number: 342194)
Swinging-bucket rotor SW 41 Ti and bucket set (Beckman Coulter, catalog number: 331362)
ChemiDoc MP Imaging System (Bio-Rad Laboratories, model: ChemiDoc MP 10)
NanoSight NS300 instrument equipped with a 405-nm laser (Malvern Instruments, Malvern, United Kingdom)
Refractometer (Fisher Scientific)
Bath sonicator (Covaris, model: S220)
Software
Nanosight NTA 3.1 software (Malvern Instruments)
Excel (Microsoft, 2016)
GraphPad Prism 7
ImageLab software v4.0
PEAKS Studio X+
Vsn R package
Procedure
Part I: EV purification
Plasmid construction
Clone PCR fragments of cyclin D1 and APEX from pcDNA3 APEX-nes into the XPack CMV-XP-MCS-EF1α-Puro Cloning Lentivector.
PCR cyclinD1 from the cDNA of mESCs by using the following primers:
CyclinD1-F: acgGGATCCCATGGAACACCAGCTCCTG
CyclinD1-R: acgGAATTCGATGTCCACATCTCGCACG
Insert cyclinD1 PCR fragments into pcDNA3 APEX-nes through BamHI and EcoRI to get pcDNA3 cyclinD1-APEX-nes plasmid.
PCR cyclinD1-Flag-APEX from the above vector by using the following primers:
acgCTCGAGTATGGAACACCAGCTCCTG
acgGGATCCtGATGTCCACATCTCGCACG
Insert cyclinD1-Flag-APEX PCR fragments into XPack CMV-XP-MCS-EF1α-Puro through XhoI and BamHI to get XPack CMV-XP-cyclinD1-Flag-APEX -EF1α-Puro plasmid.
Confirm vectors by using Sanger DNA sequencing (UC Berkeley DNA sequencing facility).
Lentivirus package and transfection
Vector transfection
Seed 293T cells at early passage (P1 in our experiments) on 10 cm culture dish and culture to get 60-70% confluency.
Mix 6 µg of transfer vector (XPack CMV-XP-cyclinD1-APEX-EF1α-Puro), 3.9 µg of psPAX, and 2.1 µg of pMD2.G in 0.5 ml of OPTI-MEM (total DNA 12 µg).
In another tube, mix 36 µl of lipofectamine in 0.5 ml of OPTI-MEM.
Mix the DNA and the lipofectamine, and incubate at room temperature for 20 min.
Change the culture medium into 5 ml OPTI-MEM.
Add the DNA and liposome mixture (1 ml) to the culture dish.
After 8 h, remove OPTI-MEM media and discard into 100% bleach.
Incubate cells in fresh growth medium (see Recipe 1) for 2 days.
Harvest virus
Centrifuge the medium at 1,000 × g for 5 min at 4°C.
Filter the medium, which has the cyclinD1-APEX lentivirus, slowly through a 0.45 µm filter into a 15 ml falcon tube.
Transduce 50% confluency N2A cells with lentivirus
Add 1.5 ml cyclinD1-APEX lentivirus to N2A cells in 35 mm dish and culture for 24 h.
Replace medium with growth medium (see Recipe 1) containing 5 µg/ml puromycin.
Culture cells in 5 µg/ml puromycin for 4 days to select N2A cells stably expressing cyclinD1-APEX.
Cell culture and differentiation
Cell culture
Culture cyclinD1-APEX stably expressing N2A cells in four 15 cm dishes in 30 ml cell culture growth medium (see Recipe 1) until cells reach 80% confluency (the cell number is ~3 × 107cells per dish).
Note: The surface available for cell growth in these dishes is 151.9 sq. cm. The total volume of cell culture growth medium used for these dishes is 30 ml per plate.
Cell differentiation
Split cells in the four 15 cm dishes to a total of fourteen 15 cm dishes, each containing approximately 8 × 106 cells, and incubate in 30 ml of RA (10 µM) containing medium with 1% exosome-depleted FBS (see Recipe 2).
Note: The maximum volume of medium that can be processed in the steps below is 420 ml at each time point.
Incubate cells for 6 days. Feed the cells with RA-containing medium (see Recipe 2) every 3 days. Collect medium afterward at day 6.
Collect conditioned medium and sediment EVs
Collect the conditioned medium from the fourteen 15 cm plates into separate containers.
Note: Approximately 420 ml of total condition medium should be collected.
Centrifuge conditioned medium at 1,000 × g for 15 min at 4°C using Sorvall RC 6+ centrifuge with a fixed rotor of F14S-6X250y FiberLite (or equivalent centrifuge) in 250 ml tube to remove floating cells (Low Spin Speed).
Decant the supernatant fraction into a new container immediately after the centrifuge finishes.
Note: It is important to remember not to disturb the pellet.
Use a Sorvall RC 6+ Centrifuge (with F14S-6X250y FiberLite fixed angle rotor) to centrifuge the supernatant at 10,000 × g for 15 min at 4°C and sediment large EVs and cellular debris (medium speed spin).
After the centrifuge stops, immediately but carefully decant the supernatant fraction into a new container.
Note: It is important to remember not to disturb the pellet.
Transfer 32 ml of the collected supernatant into one 38.5 ml ultra-clear tube. Keep transferring until twelve 38.5 ml ultra-clear tubes are filled.
Use SW-28 and SW32 Ti rotors at ~100,000 × g (28,000 RPM) for 1.5 h at 4°C at maximum acceleration/brake to centrifuge.
Gently remove supernatant and resuspend the pellet fraction by pipetting two to three times in 200 µl of phosphate buffered saline (PBS, pH 7.4).
Pipette the resuspended pellet fraction into a new 5 ml ultra-clear tube.
Note: After combining the pellet fractions from 12 tubes, ~3 ml of resuspend pellet may be collected.
Dilute resuspension from the above step with PBS to a total volume of ~4.8 ml.
Use SW-55 rotor to centrifuge sample at approximately 150,000 × g (38,500 RPM) for 1 h at 4°C and with maximum acceleration and brake.
Gently remove the supernatant. Add 100 µl of PBS into the tube and incubate at room temperature for 30 min.
Resuspend pellet by gently pipetting.
Add 900 µl of 60% sucrose. Mix thoroughly until a homogenous suspension is made.
Note: Sucrose is dissolved in buffer C (see Recipe 3). Use the refractometer to measure the sucrose concentration. The read of the refractometer should be 60.
First, carefully add 2 ml of 40% sucrose and then carefully place 1 ml of 20% sucrose on top. Add the sucrose medium very slowly and smoothly to avoid disturbing the layer between the cushion. Three separate layers of the different densities of sucrose should be apparent.
Note: This step requires extreme caution. Dispense sucrose slowly to the side of the clear tube, and let it trickle down the wall of the tube.
Use the SW-55 rotor to centrifuge sample at approximately 150,000 × g (38,500 RPM) for 16 h, at 4°C and with minimum acceleration with no brake.
Note: It is important to have no brake during deceleration. Having the brake on can cause disruption of the gradient.
Once the centrifugation stops, aspirate ~0.8 ml of supernatant from top to bottom. Collect the 20%/40% sucrose interface (Figure 1) (the total volume at this step is ~1 ml) into a new 5 ml ultra-clear tube.
Note: Be careful not to disrupt the second gradient of the 40%/60% sucrose interface.

Figure 1. EVs at 20%/40% interface after overnight centrifugationDilute up to ~4.8 ml of PBS in the 5 ml ultra-clear tube.
Centrifuge at ~150,000 × g (38,500 RPM) for 1 h in an SW-55 rotator at 4°C and with maximum acceleration and deceleration.
Gently decant the supernatant fraction and add 100 µl of PBS into the tube to incubate at room temperature for 30 min.
Resuspend the EV pellet by gently pipetting.
EV measurement by NTA
Estimate EV sizes and quantities using the NanoSight NS300 instrument equipped with a 405-nm laser; analyze the data in the scatter mode.
Dilute collected vesicles as described above (D21) at 1,000× with filtered PBS.
Introduce samples into the chamber automatically, at a constant flow rate during five repeats of 60-s captures, and at camera level 13 in scatter mode with Nanosight NTA 3.1 software.
Estimate the EV size at the detection threshold using the Nanosight NTA 3.1 software, after which export “experiment summary” and “particle data” (Figure 2).
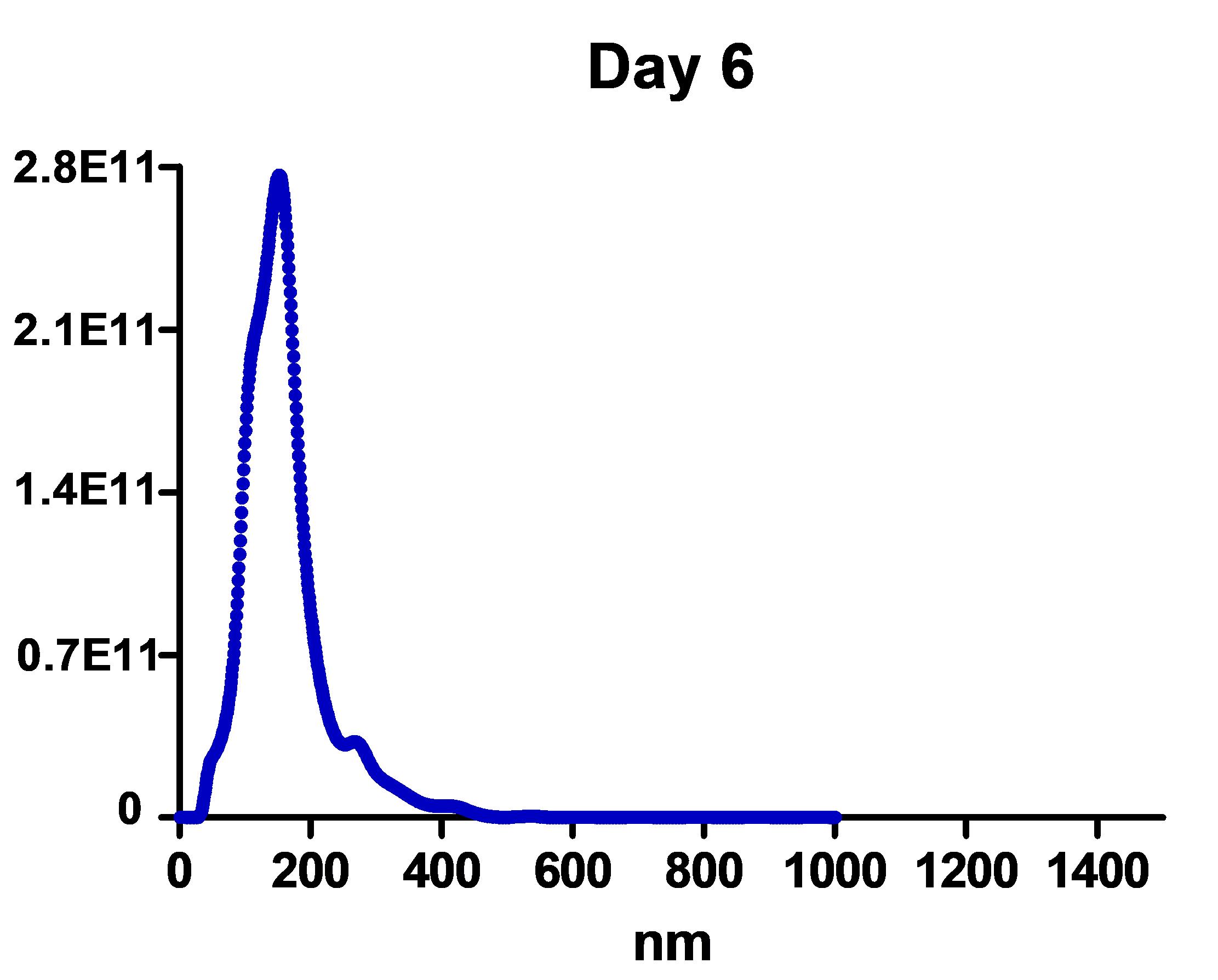
Figure 2. Nanoparticle tracking analysis of the size distribution and the number of purified EVs from the 420-ml medium of N2A cells treated with RA for 6 days.Calculate the particle numbers in each size category from the particle data, and pool, bin, and count “true” particles with track length >3 with Excel.
Note: ~5 × 108 to ~2.5 × 109 per 1 µl particle number is normally counted in step D21 EV sample.
Incubate biotin-phenol (500 μM) with ~1 × 1010 purified EVs (see Step D21 in Part I) for 30 min at 37°C in a total mixture volume <50 μl in an Eppendorf tube. Prepare two samples at this step.
Note: Keep in a small volume to ensure there is less of the biotin-phenol residual after the quencher solution wash.
Transfer the mixture into two 13.2 ml ultra-clear tubes.
Initiate APEX labeling by adding 1 mM (0.003%) H2O2 in one tube.
Note: For the other tube, perform the same treatment as follows, but do not add H2O2. This serves as a negative control.
After 1 min, add 12 ml quencher solution (see Recipe 4) immediately.
Centrifuge at ~110,000 × g (31,500 RPM) for 1 h in an SW-41 rotor and 4°C with maximum acceleration and brake.
Use quencher solution (see Recipe 4) to sediment and wash EVs once. Once stopped, aspirate the supernatant and add 12 ml quencher solution (see Recipe 4).
Centrifuge at ~110,000 × g (31,500 RPM) for 1 h in an SW-41 rotor at 4°C and with maximum acceleration and deceleration.
Gently decant the supernatant fraction. Add 40 µl of PBS into the tube and incubate at room temperature for 30 min.
Resuspend the EV pellet fraction in 40 µl PBS by gently pipetting.
Transfer the two EV samples into new Eppendorf tubes.
Add 4× SDS-loading buffer (see Recipe 5) to prepare samples for SDS PAGE and streptavidin-HRP blotting.
Incubate samples at 95°C for 10 min.
Load 15 µl of samples per lane in a 10% Tris-glycine mini gels.
Run gels at a constant voltage of 150 V for 1 h.
Transfer proteins from gels onto a PVDF membrane with constant amps of 0.6 A for 1.5 h.
Block membrane(s) with 5% BSA in TBST overnight in the cold room (4°C).
Incubate membrane(s) with the Streptavidin-HRP (1:10,000) for 1 h at room temperature.
Wash three times with TBST for 8 min of each wash.
Add the HRP substrate following the manufacturer's specifications. Develop signal using Chemidoc Imaging System.
Note: The remaining EVs can be used for the following steps after proving that the APEX biotinylated labeling works well in EVs.
APEX reaction in receipt mES cells
Prepare two 35 mm dishes of ES cells at a density of 4 × 104-5 × 104/cm2.
Incubate cylinD1-APEX EVs with mESCs in N2B27 medium (see Recipe 6) for 2 days at a concentration of ~5 × 109 EV/ml medium.
Pre-warm N2B27 medium (see Recipe 6) at 37°C for 30 min and then add 2 μl of the biotin-phenol stock solution (500 mM) into 2 ml of N2B27 medium to make a 500 μM biotin-phenol solution.
Incubate the above solution with cells for 30 min at 37°C.
Immediately prior to use, dilute 1 mM (0.003%) H2O2 into the medium for each dish for a 1-min labeling reaction at room temperature. Use the other dish without H2O2 as a negative control.
Quench the reaction by immediately washing cells thoroughly with room temperature quencher solution three times (see Recipe 4).
Note: Ensure that the washes are performed using the quencher solution instead of purely DPB.
Lyse cells in 1 ml RIPA (see Recipe 7) supplemented with 10 mM sodium ascorbate, 1 mM sodium azide, and 1× protease inhibitors.
Sonicate cell lysate in a bath sonicator for 10 min on ice and then centrifuge at 10,000 × g for 10 min at 4°C.
Add 40 μl of concentrated streptavidin-agarose beads to the supernatant fraction (800 μl), and then rotate the mixture overnight at 4°C.
Note: Wash 40 μl of streptavidin-agarose beads once with 1 ml of RIPA.
Centrifuge above sample at 3,000 × g for 10 min at 4°C. Discard the supernatant fraction.
Note: Ensure all the following buffers and samples are kept on ice.
Add 1 ml of RIPA buffer (see Recipe 7) to streptavidin-agarose beads and rotate the mixture at 4°C for 10 min.
Centrifuge above sample at 3,000 × g for 10 min at 4°C. Discard the supernatant fraction.
Note: The process from 10 to 11 is named the beads wash in the following steps.
Wash beads once again by RIPA buffer (see Recipe 7).
Wash beads once with buffer D (see Recipe 8).
Elute biotinylated proteins from the beads by heating the sample in a 4× SDS-loading buffer (see Recipe 5) supplemented with 2 mM biotin and 20 mM DTT for 10 min at 95°C.
Load 15 μl of sample per lane in 10% Tris-glycine mini gels. Run at a constant voltage of 150 V for 1 h.
Ponceau S stain gels at room temperature for 3 min (Figure 3).

Figure 3. Streptavidin-HRP blotting after cyclinD1-APEX EV incubation with mESCNote: Treat EV pre-incubation mESC with biotin-phenol with H2O2 (B+H) or not (B). Detect biotinylated protein by blotting with streptavidin-horseradish peroxidase (SA-HRP). Ponceau S staining (left of panel) of the same membrane serves as a loading control.
Wash gel three times with ddH2O.
Evaluate biotinylated proteins by blotting with streptavidin-HRP (Figure 3).
Note: The same procedure in Part II, Step 11 from d to h.
Mass Spectrometry (MS) analysis
Electrophorese heated samples from Part III, Step A16 in a 10% Tris-glycine mini gel for ~3 min.
Stain the proteins with Coomassie and excise stained bands from the gel with a fresh razor blade.
Submit samples to a Spectrometry Facility (we used the Taplin Mass at Harvard Medical School) for in-gel tryptic digestion of proteins followed by liquid chromatography and mass spectrometry analysis according to their standards.
MS data analysis
After raw MS data is acquired, perform peptide identification, quantification, and filtering on the Peaks Studio X+ platform with the default settings via the Proteome Discoverer database.
Implement Variance stabilization normalization (Vsn) with the justvsn function from the R/Bioconductor-package Vsn to normalize the quantified protein data.
Perform T-test analyses to examine significant changes in protein abundances between two different groups.
Data analysis
Export immunoblot images using ImageLab software v4.0 as .tif format.
Use ImageJ to open and process immunoblot images, including rotating, cropping, adjusting brightness, and contrast when necessary.
Prepare figures with GraphPad Prism or preferred program (Figure 2).
Recipes
DMEM + 10% FBS (500 ml)
50 ml FBS
450 ml DMEM
RA (10 μM) + 1% exosome-depleted FBS in DMEM (500 ml)
5 ml exosome-depleted FBS
RA (10 μM)
Add DMEM to 500 ml
Buffer C
20 mM Tris-HCl pH 7.4
137 mM NaCl
Quencher solution
10 mM sodium ascorbate
5 mM Trolox
10 mM sodium azide
In DPBS (Dulbecco's phosphate-buffered saline)
4× Loading buffer (20 ml)
Dissolve 1.6 g of SDS in 6 ml of ddH2O
Add 5 ml of 1 M Tris-HCl pH 6.8
Add 1.23 g of DTT. Dissolve
Add 8 mg of bromophenol blue
Add 8 ml of glycerol
Add ddH2O to 20 ml
Make 1 ml aliquots. Store at -20°C
N2B27 medium (1 L)
475 ml DMEM/F12
475 ml Neurobasal
20 ml B27
10 ml N2
10 ml L-glutamine (200mM)
10 ml non-essential amino acids (100×)
1 ml 0.1 M β-mercaptoethanol
RIPA
50 mM Tris, pH 7.4
150 mM NaCl
1% Triton X-100
0.5% deoxycholate
0.1% SDS
1 mM Trolox
1 mM DTT
Buffer D
1 M KCl
0.1 M Na2CO3
2 M urea
10 mM Tris-HCl, (pH 7.4)
TBS-T
0.1% Tween 20 (v/v) in 1× TBS
Acknowledgments
We thank the technical suggestion from Prof. Alice Ting’s lab. We thank current and former Schekman lab members who provided technical advice in the development of this protocol. We acknowledge the Howard Hughes Medical Institute for funding. We also acknowledge UC Berkeley Tang family scholarship. This protocol has been adapted from Song et al. (2021; Doi: 10.1083/jcb.202101075).
Competing interests
The authors declare no competing interests.
References
- Budnik, V., Ruiz-Canada, C. and Wendler, F. (2016). Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci 17(3): 160-172.
- Christianson, H. C., Svensson, K. J., van Kuppevelt, T. H., Li, J. P. and Belting, M. (2013). Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc Natl Acad Sci U S A 110(43): 17380-17385.
- Chen, C. L. and Perrimon, N. (2017). Proximity-dependent labeling methods for proteomic profiling in living cells. Wiley Interdiscip Rev Dev Biol 6(4).
- De Jong, O. G., Kooijmans, S. A. A., Murphy, D. E., Jiang, L., Evers, M. J. W., Sluijter, J. P. G., Vader, P. and Schiffelers, R. M. (2019). Drug Delivery with Extracellular Vesicles: From Imagination to Innovation. Acc Chem Res 52(7): 1761-1770.
- Hung, V., Udeshi, N. D., Lam, S. S., Loh, K. H., Cox, K. J., Pedram, K., Carr, S. A. and Ting, A. Y. (2016). Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat Protoc 11(3): 456-475.
- Lobingier, B. T., Hüttenhain, R., Eichel, K., Miller, K. B., Ting, A. Y., von Zastrow, M. and Krogan, N. J. (2017). An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells. Cell 169(2): 350-360.e312.
- Raposo, G. and Stoorvogel, W. (2013). Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200(4): 373-383.
- Shurtleff, M. J., Temoche-Diaz, M. M., Karfilis, K. V., Ri, S. and Schekman, R. (2016). Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. Elife 5: e19276.
- Song, L., Tian, X. and Schekman, R. (2021). Extracellular vesicles from neuronal cells promote neural induction of mESCs through cyclinD1. J Cell Biol 220(9): e202101075.
- Temoche-Diaz, M. M., Shurtleff, M. J., Nottingham, R. M., Yao, J., Fadadu, R. P., Lambowitz, A. M. and Schekman, R. (2019). Distinct mechanisms of microRNA sorting into cancer cell-derived extracellular vesicle subtypes. Elife 8: e47544.
- Van Niel, G., D'Angelo, G. and Raposo, G. (2018). Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 19(4): 213-228.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Song, L., Chen, J., Sun, A. and Schekman, R. (2021). APEX-mediated Proximity Labeling of Proteins in Cells Targeted by Extracellular Vesicles. Bio-protocol 11(21): e4213. DOI: 10.21769/BioProtoc.4213.
- Song, L., Tian, X. and Schekman, R. (2021). Extracellular vesicles from neuronal cells promote neural induction of mESCs through cyclinD1. J Cell Biol 220(9): e202101075.
Category
Developmental Biology > Cell growth and fate > Differentiation
Stem Cell > Embryonic stem cell > Cell differentiation
Cell Biology > Cell engineering
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.