- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Measuring Real-time DNA/RNA Nuclease Activity through Fluorescence
Published: Vol 11, Iss 21, Nov 5, 2021 DOI: 10.21769/BioProtoc.4206 Views: 4141
Reviewed by: Khyati Hitesh ShahMukesh MahajanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
DNA and RNA nucleases are wide-ranging enzymes, taking part in broad cellular processes from DNA repair to immune response control. Growing interest in the mechanisms and activities of newly discovered nucleases inspired us to share the detailed protocol of our nuclease assay (Sheppard et al., 2019). This easy and inexpensive method can provide data that enables understanding of the molecular mechanism for novel or tested nucleases, from substrate preference and cofactors involved to catalytic rate of reaction.
Keywords: Nuclease assayBackground
Nucleases are enzymes that act on DNA and RNA by cleaving the phosphodiester bonds between nucleotides. In addition to their crucial role in the DNA repair machinery, they are involved in cell signalling pathways pertinent to DNA damage and immune responses, among others (Sheppard et al., 2018). Their complex roles support several premature ageing-, immune-, and tumour-related processes. All of these can suffer from aberrations in the structural and/or catalytic functions of DNA and RNA nucleases (reviewed in Bartosova et al., 2014; Rigby et al., 2015). The growing interest in understanding the activities of numerous human DNA nucleases that remain contentious [e.g., Mre11 (Paull and Deshpande, 2014) and CTIP (Mozaffari et al., 2021)] and the presence of several uncharacterised proteins with predicted nuclease domains in mammalian genomes led us to design a real-time nuclease assay.
The activity and kinetics of nucleases, DNA polymerases, nickases, RNA:DNA nucleases, or single-strand DNA nucleases can be studied in an uncomplicated and cost-effective manner. The fluorescence signal changes resulting from decreasing amount of intercalated DNA dye can be quickly and safely measured by most plate readers. A wide range of oligonucleotides mimicking the DNA substrate of the tested enzyme can be examined in each experiment simultaneously. We designed and tested an oligomer library of 80mers with different characteristics and substrate potential. The oligonucleotides described allow for the determination of the enzymatic direction of nuclease activity. For example, 3′ or 5′ activity can be tested and compared with oligonucleotides containing biotin- blocked or free 3′ends or 5′ends, in addition to overhangs, gaps, or nicks. This method can also illustrate the importance of cofactors or cations through simple comparison between reactions supplemented or not with the chemical/cation. Using the same principle, a modified protein (phosphorylation, dephosphorylation) or mutated/truncated forms can be easily tested for their nuclease activity. Importantly, the assay is sensitive enough to detect the kinetics of repair enzymes when confronted with DNA mismatches or DNA methylation sites.
Materials and Reagents
Prepare all buffers and solutions using ultrapure, nuclease free-water and analytical grade reagents. Filter with 0.2 µm filter at least once and store at 4°C or -20°C. Always use nuclease-free tubes and cotton-filter tips.
Black bottom plates [black bottom plate 96 well, polypropylene, flat bottom (Chimney well)] are necessary for the fluorescence assay to reduce background and crosstalk, and to absorb light (Greiner Bio-One, catalog number: 655209)
Streptavidin (Pierce, catalog number: PIER21122)
Streptavidin (SA) has a great affinity to biotin triethyleneglycol (BITEG). Oligonucleotides with this modification on 3′, 5′, or both ends are protected from nuclease activity after adding 2 μl (0.02 mg/ml) streptavidin to the reaction mix. Prepare 1 mg/ml in ultrapure water.
Oligonucleotides
Order lyophilised unmodified HPLC-purified oligonucleotide substrates and biotin or BITEG modified HPLC-purified oligonucleotides (Integrated DNA Technologies). Dilute them in 1× Annealing Buffer (Sigma-Aldrich) for DNA substrates (100 μM stock) and siMAXTM dilution buffer (Eurofins; 30 mM HEPES, 100 mM KCl, 1 mM MgCl2, pH = 7.3) for all RNA substrates.
Note: Modified DNA or RNA bases can be ordered in the synthesised oligos. We have done so successfully in the past to measure the effect of base modifications on enzymatic functions.
Oligonucleotides from our library were designed and optimised against secondary structure formation using the ‘Predict a Secondary Structure Web Server’ (https://rna.urmc.rochester.edu/RNAstructureWeb/Servers/Predict1/Predict1.html).
The basic 80bp sequences are as follows:
Forward: GATGGTTTGTTGGTCTATTACTACTTGGAGCTTGTATGATTCGAAACCTTGGAGTACTTGCCTACTTGGAGTGAACTTAG
Reverse: CTAAGTTCACTCCAAGTAGGCAAGTACTCCAAGGTTTCGAATCATACAAGCTCCAAGTAGTAATAGACCAACAAACCATC
For designing all types of library oligomers, the basic oligonucleotide sequence can be shortened from both ends or split into two molecules to produce gaps or nicks in duplex DNA. Single nucleotides can be exchanged to add mismatches, substrate preferences, and so forth (Figures 1 and 2).
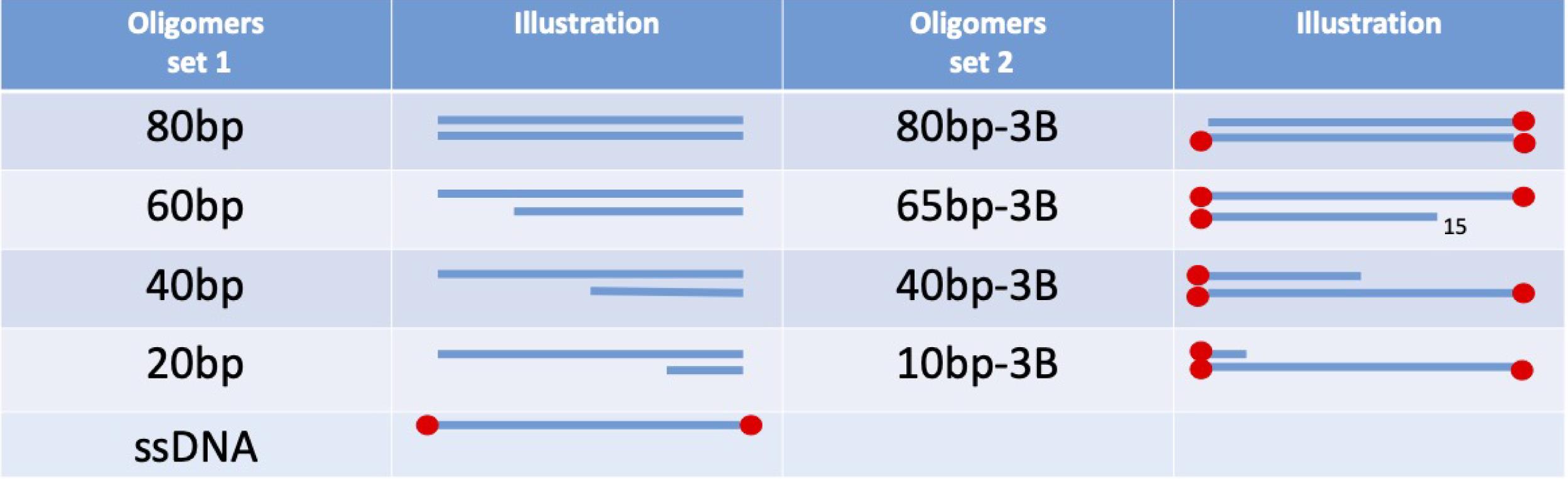
Figure 1. For the calibration curve, two example sets are presented, with and without streptavidin blockade on ends. These need to match the DNA substrates used in the experiment.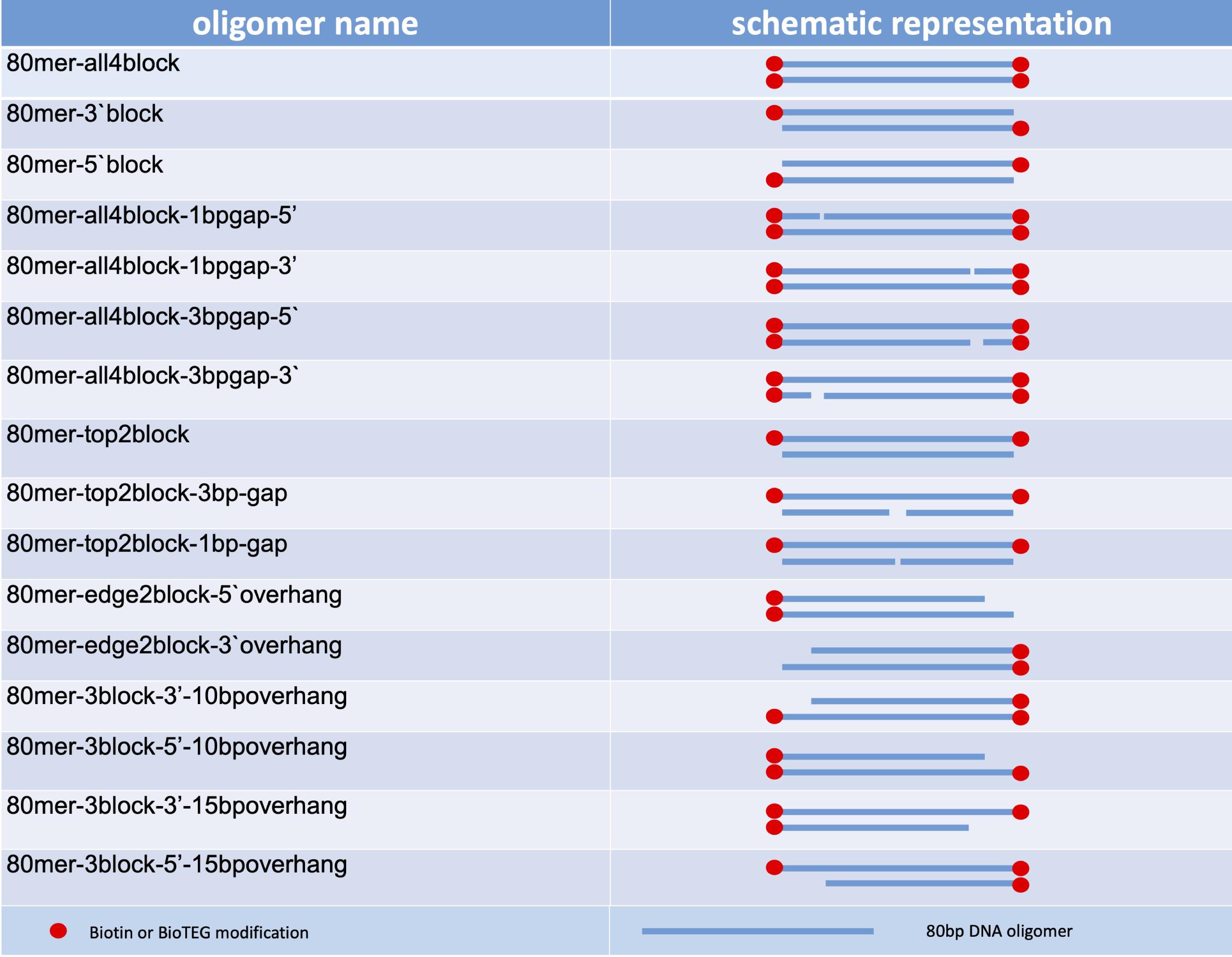
Figure 2. Set of 80-mer oligonucleotide substratesNucleases
Perform the calibration of the method using the commercial nucleases with known activity and preferably acting in a similar mechanism to the predicted activity of the enzyme to be tested. For example, to test the removal of 5′ mononucleotides from duplex DNA, use Exonuclease T7 (T7, New England Biolabs, catalog number: M0263S). For nucleases acting 3′ to 5′, use Exonuclease III (ExoIII, Thermo Scientific, catalog number: EN0191) (Figure 3).
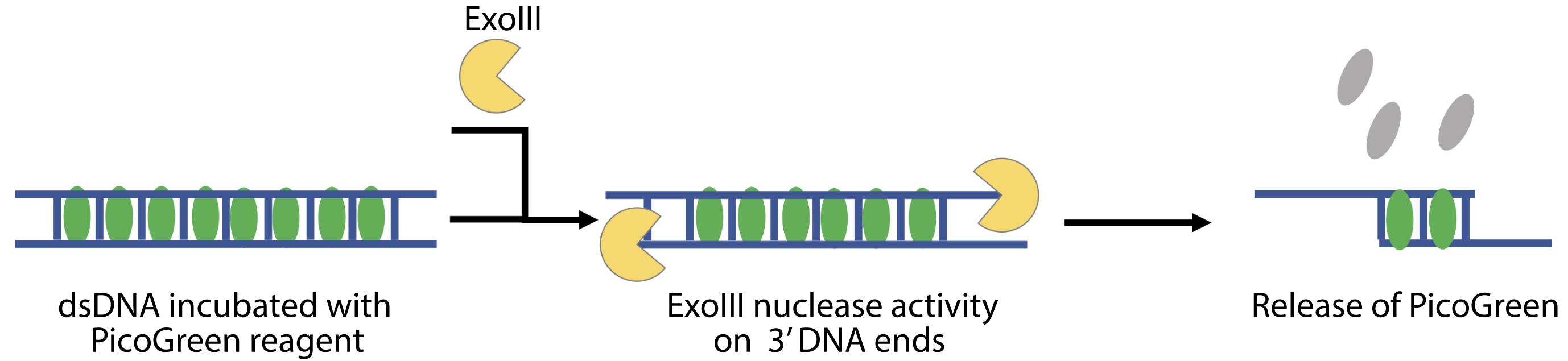
Figure 3. Scheme of PG release from dsDNA after nuclease treatment (ExoIII)1 M Tris, pH 7.5-8.0 (ThermoScientific, catalog number: 15567027)
5 M NaCl, RNase-free (ThermoScientific, catalog number: AM9759)
0.5 M EDTA pH 8.0 (ThermoScientific, catalog number: 15575020)
10× Tango Buffer (ThermoScientific, catalog number: BY5)
CutSmart (New England Biolabs, catalog number: B7204S)
Glycerol (ThermoScientific, catalog number: 15514011)
Quant-iTTM PicoGreenTM dsDNA Assay Kit (Invitrogen, catalog number: P7589)
Quant-iTTM microRNA Assay Kit (Invitrogen, catalog number: Q33140)
Annealing Buffer Composition (1×) (see Recipes)
Storage buffers (see Recipes)
Reaction buffers (see Recipes)
Divalent cations (see Recipes)
PG buffer (see Recipes)
Nucleic acid dyes (see Recipes)
Equipment
Plate reader (Tecan or BMG)
Select the appropriate measurement parameters (temperature, wavelength, number of flashes, and settle time) for your assay.
Preheat the microplate reader to the optimal temperature for enzyme activity (commonly 37°C). To slow down the reaction, set to a lower temperature (e.g., 20°C). Use the wavelength 483-15 nm for excitation (483 nm is the middle excitation peak with a bandwidth of 15 nm; i.e., the excitation range is 475-490 nm) and 530-30 nm for emission (530 nm is the middle emission peak with a bandwidth of 30 nm; i.e., the emission range is 515-545 nm). Increase the number of flashes per well until noise of BLANK wells does not improve further or until measurement time per well becomes unacceptable. Depending on the enzyme, read the samples every 45-60 s for 15 min to 2 h with a shake before each read.
Photobleaching occurring in the samples causes the fluorescence signal to decrease with time. Therefore, run a control curve run in every experiment simultaneously with samples to measure this effect. Longer readings can be inaccurate due to total photobleaching of PicoGreen to the level of the background.
We tested Infinite M Plex (TECAN) and CLARIOstar (BMG Labtech) plate readers. The nuclease assay can also be performed in the qPCR reader, but we found that the plate reader gives consistent results and offers more options, such as shaking the plate before a read.
While the best practices regarding reads per well vary with each enzyme, in general, an optimal duration of reads is approximately 1 h. If using a CLARIOstar, factor the whole time needed to read the entire plate, well by well, which for a full plate took approximately 48 s. So, each well could only be read every 50 s. If the plate reader permits it, try to use bidirectional reading row by row, and add the enzymes in the same order.
Procedure
Plate Preparation
Prepare the plate on ice and protect the samples from light. For every reaction, prepare the mix and run triplicates for the best accuracy. Use of multi-channel pipettes accelerates the set up of the reaction plate but can also lead to air bubbles in wells. Centrifuge the plate to remove them.
Always mark up the plate to aid with pipetting substrate and reaction mixtures and to blank off any empty wells. If using tape to label the rows and columns, peel this off before loading into the plate reader. Take time to plan out experiments and calculate volumes well ahead of time.
Reaction mix per well:
10 µl oligonucleotides 500 nM
10 µl reaction buffer
25 µl ddH2O (or 23 µl if SA)
2 µl of streptavidin (for biotinylated oligos)
50 µl PG
5 µl enzyme
Note: For RNA reactions, a similar procedure could be followed by substituting PG with RNA dye.
Perform the annealing of complementary oligonucleotides in a thermocycler or heat block and use the molar ratio 1:1.
Mix equal volumes of the equimolar oligonucleotides (100 µM each) in a PCR tube.
Incubate for 5 min at 95°C.
Switch off the heat block leaving the samples in to cool down slowly or use the thermocycler (go down 1°C every 30 s).
Keep the annealed duplexes at -20°C for long-term storage.
Dilute the annealed oligos to 500 nM.
Use 10 µl (50 nM) per reaction/well.
For example, to anneal the oligonucleotide duplexes containing the gap, anneal three oligonucleotides using 2.5 µl each, and add 2.5 µl of annealing buffer. The concentration of the end product is 25 µM.
Notes:
Use the reaction buffer appropriate for the enzyme. The wide range of activity buffers, like 10× CutSmart (New England Biolabs, B7204S) or 10× Tango (ThermoScientific, BY5), can facilitate the data analysis (no need for extra controls) and work well with many enzymes. Run a trial experiment to test the effectiveness of different reaction buffers to get the best results.
Use nuclease-free water.
Remember to add 2 µl of streptavidin for BIOTEG or biotinylated oligonucleotides. Incubate for 15 min on ice on a see-saw shaker to enhance binding.
Add PicoGreen reagent (PG) to every well protecting the plate from light. Try to work quickly and cover the rows/columns already supplemented with PG with lid/aluminium foil.
Add the denoted enzymes (or storage buffer as a negative control) to each well.
Work at the bench at room temperature from now on to avoid the risk of enzyme precipitation. Prepare the desired amount of enzyme units/concentration in 5 µl. Take a full box of filter tips and use in the corresponding position in the tip box and plate to avoid pipetting mistakes. This way, if the position on the plate is lost, the tip box could orient the processing. Add 5 µl to each well, trying not to make any bubbles that could affect the fluorescence reads. The reaction starts now, so work quickly to be able to catch the first minutes of the reaction in the pre-heated plate reader. Remember to protect the plate from light.
Notes:
Avoid using multi-channel pipettes as they tend to add bubbles to wells and are less consistent in adding the correct amount of enzyme.
Centrifuge the plate briefly to remove air bubbles. For quick acting enzymes, this may not be advisable as the enzymatic reaction may have already started during that time.
Attempt to titrate out the enzyme. If too much enzyme is present in the solution, the reaction is very fast, causing loss in data read in the early phase of the reaction.
Design of experiment
Plate with test experiment with T7 and ExoIII exonucleases. The samples were prepared in duplicate. The calibration control curve is also included, as illustrated in Figure 4.
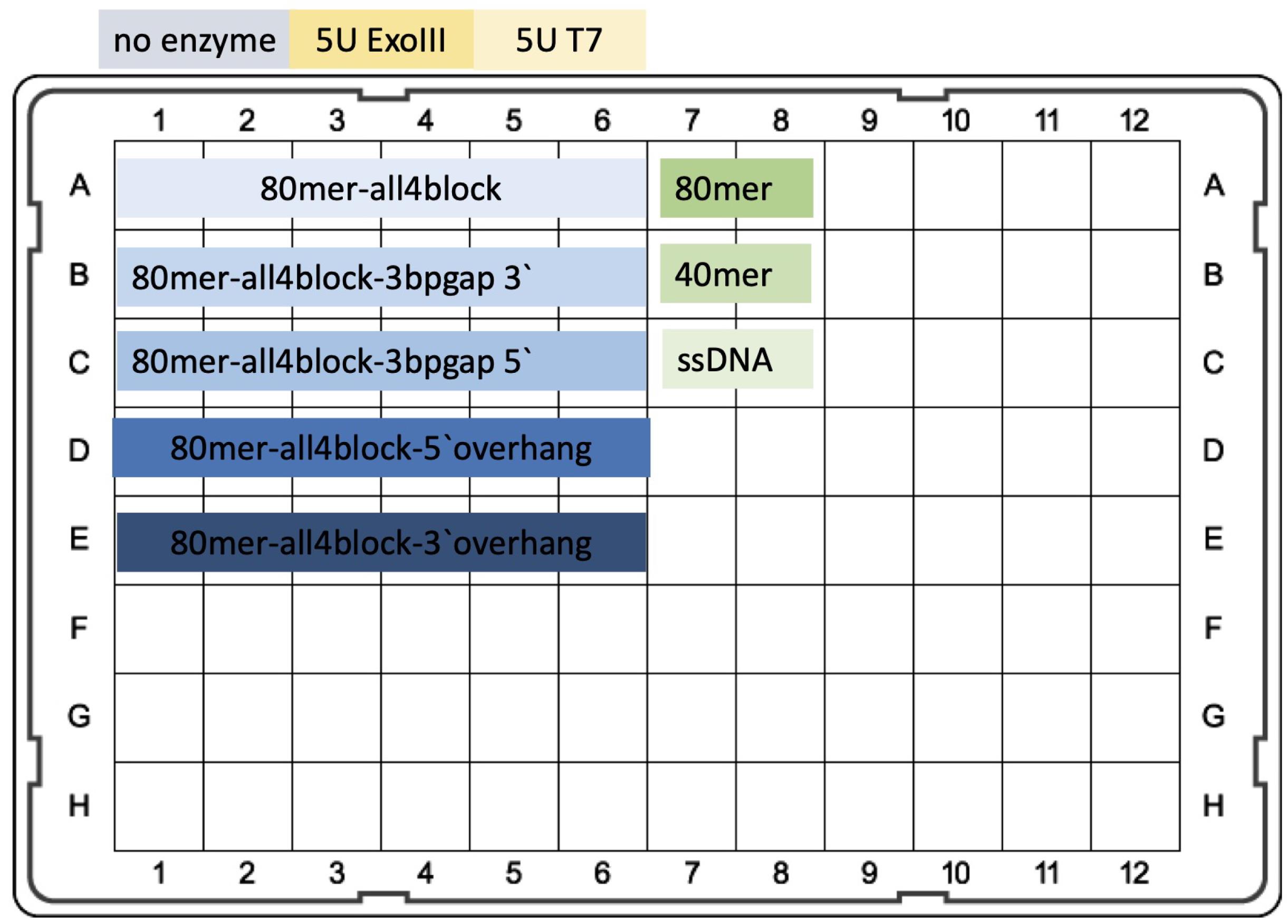
Figure 4. Scheme of the plate for test experiment. A. Row A1-6 contains the negative control oligomer for both tested exonucleases. The correct binding between biotin-modified-oligonucleotides and streptavidin added to the mix protects both 3′ and 5′ ends from digestion. All4block signifies that all four ends on the oligos were blocked by biotin-streptavidin binding. B. Row B1-6 contains oligonucleotides with 3bp gap in proximity, 12bp to 3′ end (which is the substrate for ExoIII acting in the 3′ to 5′ direction). T7 could only digest short 12bp fragments leading to the last nucleotide blocked by Biotin-Streptavidin. C. Row C1-6 contains oligonucleotides with 3bp gap in proximity, 12bp to 5′ end (which is the substrate for T7 exonuclease acting in the 5′ to 3′ direction). ExoIII could only digest short 12bp fragments leading to the last nucleotide blocked by Biotin-Streptavidin. D. Row D1-6 contains oligonucleotides with 15bp overhang on 5′. E. Row E1-6 contains oligonucleotides with 15bp overhang on 3′. In Columns 1 and 2, storage buffer from ExoIII (or T7) was added to the samples. Columns 3 and 4: ExoIII 5U per well. Columns 5 and 6: T7 5U per well. A7 and A8: 80mer_3block (oligos that 80mers with both 3′ ends blocked). B7 and B8: 40mer_3block (oligos that 40mers with both 3′ ends blocked). C7 and C8: ssDNA, 80bp single strain DNA, as a control of totally digested duplex oligomer, minimum value of fluorescence for the experiment.
Data analysis
Create the calibration lines from the oligonucleotides with different nominal lengths: 80, 40, and 0 (single stranded). Obtain the reads from the oligonucleotides as a function of time, possibly two or three replicates per nominal length. Next, at each time point, calculate the linear relationship between the nominal lengths (80, 40, 0) and the raw fluorescence reads. As a result, a time evolution of slopes and intercepts that relate the control signal to the control nominal length is obtained.
Group the data reads of the same oligonucleotide in sets with appropriate negative control, which must be very similar to the tested sample. It could contain no enzyme, or optimally, the inactive form of the tested enzyme. DNA 80-oligomer, which is not the substrate for the enzyme, i.e., 80 mer_all4block, can be the negative control too. At the zero-time point, the raw reads should be similar for all wells.
Correct for the effects of photobleaching and background reads by means of dividing the reads of the specimen by the average reads from the negative control well.
Using the slope and intercept from calibration lines, convert the corrected fluorescence units at each time point to length of duplex DNA. See Supplementary material (Excel file with test experiment and calculations) for a template of data entry and calculations.
The result should reflect the length of oligomers, where 80mer is the maximum size, and single ssDNA shows total dissolved duplex.
Both nucleases were tested on five different oligomers from the library.
The 80mer-all4block duplex is not the substrate for any of the tested nucleases (no nickase activity). Thus, we used the 80mer-all4block as a negative control in calculations. The duplex cannot be degraded during the assay because changes in fluorescence occur only due to photobleaching of the PG reagent.
ExoIII activity nuclease assay
ExoIII, with its 3' to 5' exodeoxyribonuclease activity, releases 5'-mononucleotides from the 3'-end. It acts effectively on 80 mer-3block-5′-15 bp_overhang and on 80mer-all4block-3bpgap-3′.
80mer-all4block-3bpgap-5′ has a gap situated 12bp from the 5′ end of the oligomer. The decrease in fluorescence signal reflects the ability of ExoIII to remove this few bp from the 3′-end starting in the gap.
The lower 80mer_all4block control fluorescence signal compared to 80mer-3block-3′-15bp_overhang confirms that the PG reagent intercalates only with double stranded DNA and not with the ssDNA overhang that is part of the oligo and not a substrate for ExoIII.
The reaction with ExoIII starts quickly, and in this experiment, the substrate oligomers for ExoIII were registered at time point 0 as 60bp and 50bp fragments, respectively (Figure 5A).
T7 activity nuclease assay
T7 exonuclease with its 5′ to 3' activity, non-processively hydrolyses oligomers starting on free 5′-end on both 80mer-3block-3′-15bp and 80mer-all4block-3bpgap-5′. T7 can also remove few bp from the 5′-end of the gap in 80 mer-all4block-3bpgap-5′ (Figure 5B).
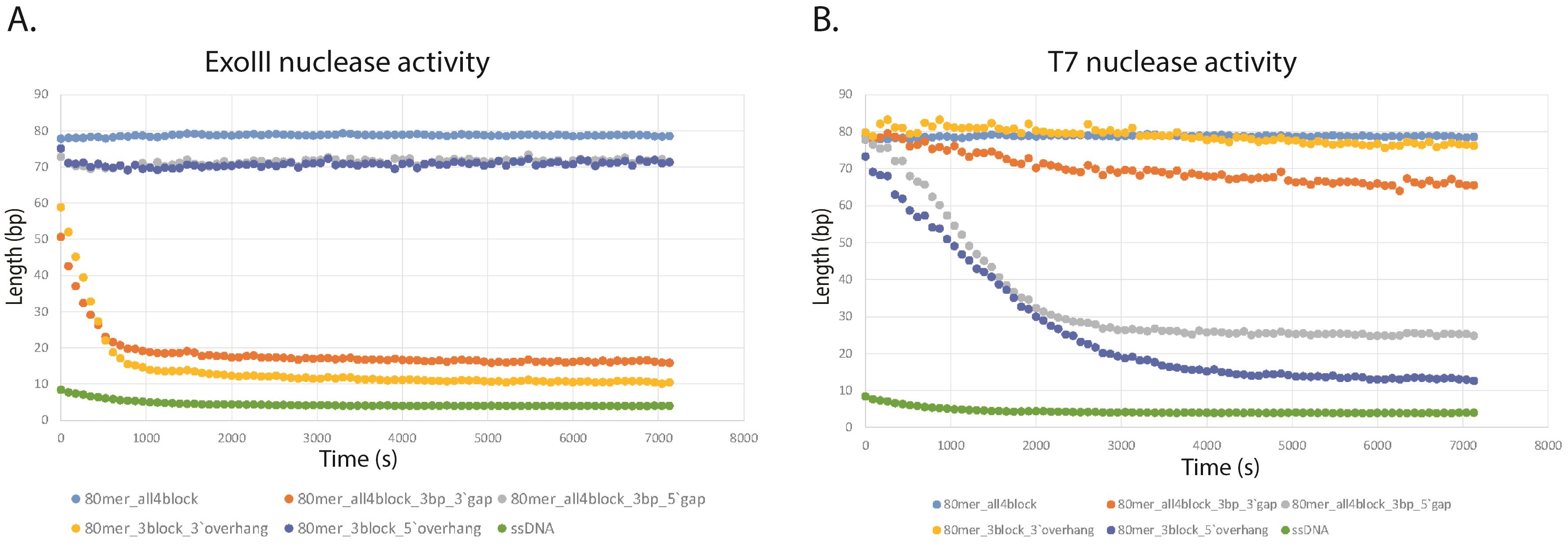
Figure 5. Sample of the nuclease activity results. A. ExoIII nuclease activity, 5U per well. B. T7 nuclease activity, 5U per well. The graphs from the test experiment with ExoIII exonuclease and T7 exonuclease. The data is presented for each enzyme separately. The 80bp oligomer and ssDNA are shown on the graph. The x-axis denotes time in seconds. The y-axis denotes DNA oligo length in base pairs.
Summary: The nuclease assay summarised in this protocol is a powerful and easy toolkit for analysis of the activity of enzymes connected to DNA/RNA. Wide application, simplicity of test preparation, affordable reagents, and accessible equipment makes it a great method to implement in every molecular biology lab. We also postulate that this assay could be used in various variations to measure nucleic acid metabolism through non-nuclease activities.
Recipes
Annealing Buffer Composition (1×)
Make 100 µM stock of the oligonucleotides (ordered from IDT [idtdna.com])
10 mM Tris, pH 7.5-8.0
50 mM NaCl
1 mM EDTA pH 8.0
Storage buffers
Prepare enzymes’ specific storage buffers for both control wells (without any enzyme) and for diluting the enzyme. Do not add glycerol.
- For ExoIII enzyme: 50 mM Tris-HCl (pH 8.0), 50 mM KCl, 1 mM DTT (add fresh prior to use).
- For T7 enzyme: 10 mM Tris-HCl (pH 8.0), 0.1 mM EDTA, 10 mM DTT (add fresh prior to use).
Note: These storage buffers vary depending on the specific enzyme and the manufacturer producing it. They are usually provided by the manufacturer when purchasing specific enzymes.
Reaction buffers
Use recommend using reaction buffers delivered with nucleases. For unknown preference of tested nucleases, try wide range activity buffers, i.e., 10× Tango Buffer and 10× CutSmart.
- 10× Reaction Buffer for ExoIII: 660 mM Tris-HCl (pH 8.0 at 30°C), 6.6 mM MgCl2.
- 10× Reaction Buffer T7: 10× NEBuffer 4 (B7004S): 500 mM potassium acetate, 200 mM Tris acetate, 100 mM magnesium acetate, 10 mM DTT, pH 7.9 at 25°C.
Note: As reaction buffers can make a big difference, research the best one for each enzyme carefully and use the most appropriate one for the specific enzyme being tested.
Divalent cations
Supplement reaction buffer with MgCl2 or MnCl2 when needed. Use both in the first experiments; the information about preferred cation can be determined later.
PG buffer
TE buffer with v/v glycerol for diluting PG reagent
10 mM Tris-HCl
1 mM EDTA, pH 7.5
40% glycerol
Nucleic acid dyes
DNA dyes: The PicoGreen (PG) reagent from Quant-iTTM PicoGreenTM dsDNA Assay Kit was prepared immediately (producer recommendation) before use by making a 1:200 dilution of the PG in TE buffer with v/v glycerol.
Store the PG reagent long term at -20°C; while thawing, wipe off any moisture before opening the tube. Light and moisture both harm the PG reagent. We routinely keep the PG aliquoted and frozen, 5 µl per tube (1 ml of PG Buffer to be added prior to use), in a light proof box.
Note (RNA dyes): For the RNase assays, the dye from the Quant-iTTM microRNA Assay Kit was prepared by diluting the microRNA reagent A into buffer B in a 1/2,000 dilution, as detailed in the protocol.
Acknowledgments
The RC lab is funded by the BBSRC (BB/N017773/2), SNF (CRSK-3_190550), Rosetrees Trust Fund (M713), and UZH Research Priority Program (URPP–Translational Cancer Research). This protocol was adapted from the publication by Sheppard et al. (2019).
Competing interests
The authors declare no competing interests.
References
- Bartosova, Z. and Krejci, L. (2014). Nucleases in homologous recombination as targets for cancer therapy. FEBS Lett 588(15): 2446-2456.
- Mozaffari, N. L., Pagliarulo, F. and Sartori, A. A. (2021). Human CtIP: A 'double agent' in DNA repair and tumorigenesis. Semin Cell Dev Biol 113: 47-56.
- Paull, T. T. and Deshpande, R. A. (2014). The Mre11/Rad50/Nbs1 complex: recent insights into catalytic activities and ATP-driven conformational changes. Exp Cell Res 329(1): 139-147.
- Rigby, R. E. and Rehwinkel, J. (2015). RNA degradation in antiviral immunity and autoimmunity. Trends Immunol 36(3): 179-188.
- Sheppard, E. C., Morrish, R. B., Dillon, M. J., Leyland, R. and Chahwan, R. (2018). Epigenomic modifications mediating antibody maturation. Front Immunol 9: 355.
- Sheppard, E. C., Rogers, S., Harmer, N. J. and Chahwan, R. (2019). A universal fluorescence-based toolkit for real-time quantification of DNA and RNA nuclease activity. Sci Rep 9(1): 8853.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wyrzykowska, P., Rogers, S. and Chahwan, R. (2021). Measuring Real-time DNA/RNA Nuclease Activity through Fluorescence. Bio-protocol 11(21): e4206. DOI: 10.21769/BioProtoc.4206.
Category
Cancer Biology > Genome instability & mutation > Biochemical assays > DNA structure and alterations
Cancer Biology > General technique > Biochemical assays
Biochemistry > Protein > Activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.