- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Fixation and Immunostaining of Endogenous Proteins or Post-translational Modifications in Caenorhabditis elegans
(*contributed equally to this work) Published: Vol 11, Iss 19, Oct 5, 2021 DOI: 10.21769/BioProtoc.4172 Views: 4015
Reviewed by: DURAI SELLEGOUNDERManish ChamoliAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Although the advent of genetically-encoded fluorescent markers, such as the green fluorescent protein (GFP; Chalfie et al., 1994), has enabled convenient visualization of gene expression in vivo, this method is generally not effective for detecting post-translational modifications because they are not translated from DNA sequences. Genetically-encoded, fluorescently-tagged transgene products can also be misleading for observing expression patterns because transgenes may lack endogenous regulatory DNA elements needed for precise regulation of expression that could result in over or under expression. Fluorescently-tagged proteins created by CRISPR genome editing are less prone to defective expression patterns because the loci retain endogenous DNA elements that regulate their transcription (Nance and Frøkjær-Jensen, 2019). However, even CRISPR alleles encoding heritable fluorescently-tagged protein markers can result in defects in function or localization of the gene product if the fluorescent tag obstructs or otherwise interferes with important protein interaction domains or affects the protein structure.
Indirect immunofluorescence is a method for detecting endogenous gene expression or post-translational modifications without the need for transgenesis or genome editing. Here, we present a reliable protocol in which C. elegans nematodes are fixed, preserved, and permeabilized for staining with a primary antibody to bind proteins or post-translational modifications, which are then labeled with a secondary antibody conjugated to a fluorescent dye. Use of this method may be limited by the availability of (or ability to generate) a primary antibody that binds the epitope of interest in fixed animals. Thousands of animals are simultaneously subjected to a series of chemical treatments and washes in a single centrifuge tube, allowing large numbers of identically-treated stained animals to be examined. We have successfully used this protocol (O’Hagan et al., 2011 and 2017; Power et al., 2020) to preserve and detect post-translational modifications of tubulin in C. elegans ciliated sensory neurons and to detect non-modified endogenous protein (Topalidou and Chalfie, 2011).
Keywords: FixationBackground
The ability to detect the presence and localization of endogenous biological molecules or epitopes in organisms is essential to understand their function. Previous methods of fixation and permeabilization of C. elegans for immunofluorescent detection of molecules include both freeze cracking and tube fixations (Finney and Ruvkun, 1990; Miller and Shakes, 1995; Duerr et al., 2006). Here, we present a robust method (see overview in Figure 1), modified from the one described by Finney and Ruvkun (1990).
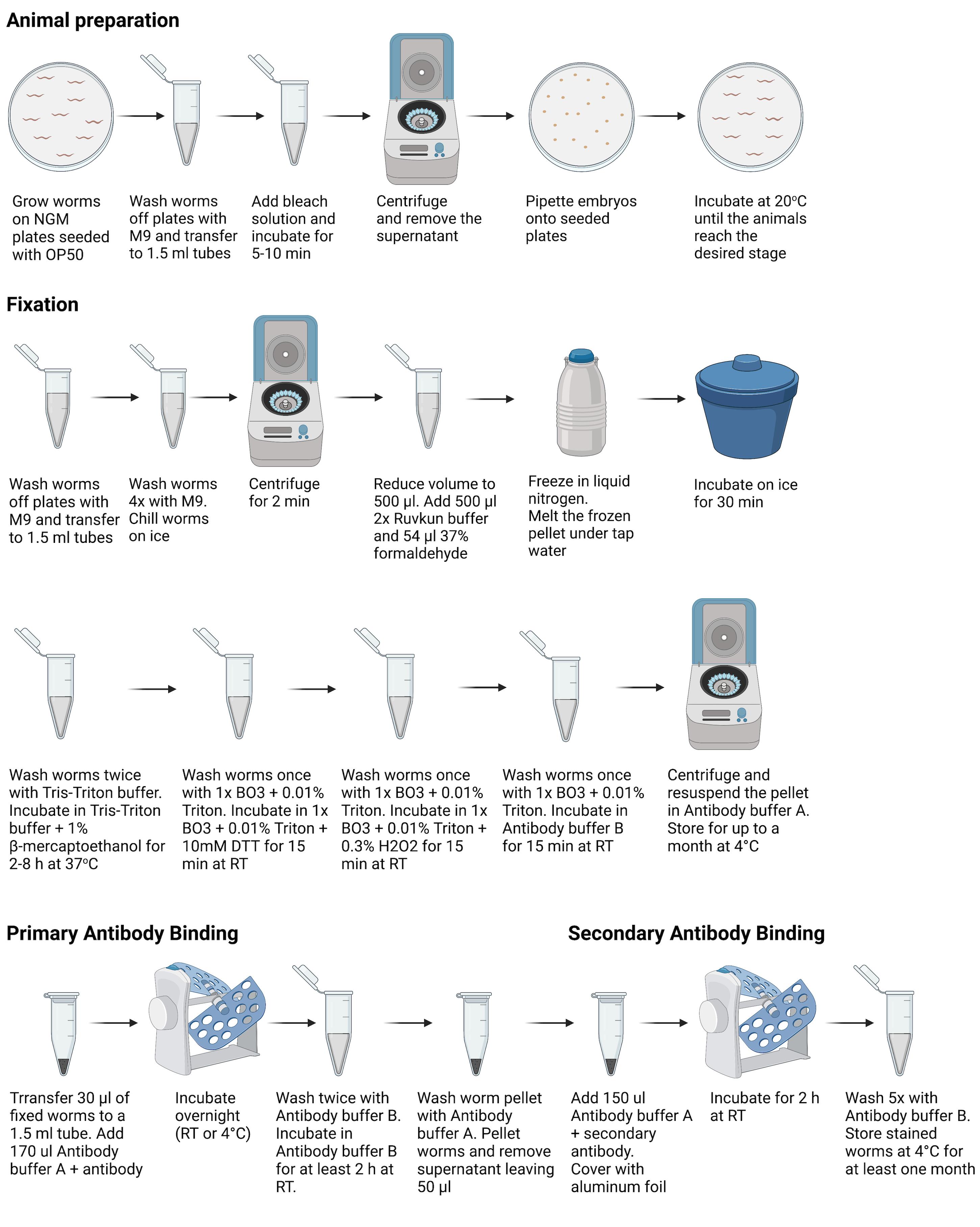
Figure 1. Overview of procedures used for animal preparation, fixation, and primary and secondary antibody binding. Figure 1 was created with Biorender.com.
Materials and Reagents
C. elegans strains of interest (https://cgc.umn.edu/)
OP50 E. coli strain (https://cgc.umn.edu/)
NGM plates seeded with OP50
PBS/phosphate Buffered Saline, 1× Powder, pH 7.4 (Fisher Scientific, catalog number: BP661-10)
Tris Base (Sigma-Aldrich, catalog number: 648311)
EDTA (Sigma-Aldrich, catalog number: E9884)
EGTA (Sigma-Aldrich, Calbiochem, catalog number: 324626)
Spermidine trihydrochloride (Sigma-Aldrich, catalog number: S2501)
PIPES (Sigma-Aldrich, catalog number: P1851)
BME/β-mercaptoethanol (Sigma-Aldrich, catalog number: M6250)
DTT/dithiothreitol (Fisher Scientific, catalog number: FERR0861). Store at 4°C, or in aliquots at -20°C
H2O2/Hydrogen Peroxide 30% with stabilizer (Sigma-Aldrich, catalog number: H1009)
H3BO3/Boric Acid Powder 99.5% (Fisher Scientific, catalog number: 18-609-176)
Formaldehyde Solution, 37% (Sigma-Aldrich, catalog number: 252549). Store at 4°C
Triton X-100 non-ionic detergent (Sigma-Aldrich, catalog number: X100)
BSA (Sigma-Aldrich, catalog number: A2153). Store at 4°C, or in aliquots at -20°C
NaN3/Sodium Azide (Sigma-Aldrich, catalog number: S2002)
GT335 (AdipoGen Life Sciences, catalog number: AG-20B-0020-C100). Store at -20°C
Alexa-fluor 568-conjugated donkey anti-mouse secondary antibody (Fisher Scientific, Invitrogen, catalog number: A10037). Store in the dark at 4°C
Sodium hypochlorite 5.65-6% solution (Fisher Scientific, catalog number: SS290-4) or Clorox bleach (~7.5% sodium hypochlorite)
Potassium hydroxide (KOH) (Fisher Scientific, catalog number: P251-500)
Agarose (Fisher Scientific, catalog number: 16-520-050)
2× Ruvkun Buffer (see Recipes)
Tris-Triton buffer (see Recipes)
20× BO3 buffer (see Recipes)
1× BO3 + 0.01% Triton buffer (see Recipes)
Antibody buffer A (see Recipes)
Antibody buffer B (see Recipes)
M9 solution (see Recipes)
5 M KOH (see Recipes)
Tris-Triton buffer +1% β-mercaptoethanol (see Recipes)
1 M DTT solution (see Recipes)
1× BO3 + 0.01% Triton buffer + 10 mM DTT (see Recipes)
1× BO3 + 0.01% Triton buffer + 0.3% H2O2 (see Recipes)
Liquid 2% agarose (see Recipes)
Equipment
Note: Equipment similar to the items described below is required.
Platform Rocker (Fisher Scientific, Thermo Scientific Vari-Mix, catalog number: 09-047-113Q)
Microcentrifuge (Fisher Scientific, Thermo Scientific Sorvall Legend 21, catalog number: 75-77-2488)
Compound epifluorescence or confocal microscope with 63× (NA 1.4) or 100× (NA 1.4) oil-immersion objectives
Digital microscope camera (Examples of microscope cameras we have used successfully for this protocol include:
Retiga-SRV Fast 1394 digital camera
Photometrics Cascade 512B CCD camera
Hamamatsu C11440-42U ORCA-Flash4.0 LT Digital CMOS camera
Photometrics CoolSNAP HQ2-FW camera
Microscope slides (Fisher Scientific, catalog number: 22-265446)
Carl Zeiss High Performance Coverslips (Fisher Scientific, catalog number: 10474379)
Glass Pasteur pipettes (Fisher Scientific, catalog number: 13-678-20A)
Eyelash glued to a toothpick
Software
ImageJ (NIH, https://imagej.nih.gov/ij/)
Procedure
Optional: Synchronization of animals for fixation
To compare results across control and mutant strains, it is important that animals are age-matched. However, this step may be omitted if the purpose of the experiment is to detect an epitope across all stages of the development of C. elegans nematodes.
Grow C. elegans using standard methods on a 6 cm NGM plate seeded with OP50 E. coli (see wormbook.org for standard growth conditions and instructions for M9 solution, NGM plates, and culture of OP50).
When many gravid hermaphrodites are present, wash worms off the plate(s) by pipetting 0.5 ml M9 solution onto the plate, tilting the plate to wash hermaphrodites off, and pipetting worm suspension into a 1.5 ml centrifuge tube. Add 20 μl of 5 M KOH and 100 μl of 5.65-7.5% sodium hypochlorite (bleach; we use Clorox. Optionally, a 5.65-6% sodium hypochlorite solution works just as well). Incubate for about 5-10 min, occasionally inverting tube to keep worms suspended. This step dissolves the adults but spares the embryos, which are surrounded by a tough eggshell.
Every few minutes, check for disappearance of adults, either by viewing the tube under a dissecting microscope or holding it up to the light and viewing by eye. When few adults are visible in the suspension, spin tube(s) at ~900 × g (~3,000 rpm) in a benchtop centrifuge for ~20 s to pellet embryos (eggs) released by hypochlorite digestion of adults.
Optional: Remove most of the supernatant by pipetting, but leave ~100 μl so that the pellet is undisturbed. Then add 900 μl M9 solution and resuspend to wash, eliminating most of the sodium hypochlorite. Sodium hypochlorite could induce oxidative stress that could alter expression of some genes. Spin tube(s) at ~900 × g (~3,000 rpm) in a benchtop centrifuge for ~20 s to pellet embryos (eggs).
Remove most of the supernatant by pipetting but leave ~100 μl.
Pipette up and down gently to resuspend the pellet of embryos in the 100 μl. (If few worms survive the sodium hypochlorite treatment, consider including one or two washes (Step A4) with M9 solution to remove the sodium hypochlorite).
Pipette drops of the embryo suspension onto fresh NGM plates with lawns of OP50. Place the drops outside the lawn of OP50, and tilt the plates to spread the drop out so that the liquid (which still includes sodium hypochlorite) will more rapidly soak into the NGM plate and/or evaporate. (If embryos continue to soak in the hypochlorite mix too long, they will become non-viable). Try adding approximately 100-200 embryos per plate to each of five plates. If you don’t have enough embryos, scale up the number of starting plates that you synchronize by bleaching.
Incubate at 20°C for approximately three days to reach the first day of adulthood. Temperature or time can be adjusted depending on differences in aging rate and reproduction of your worm strain to provide a loosely synchronized population of young adults or your desired larval stage.
Note: This method isolates only embryos that are contained in the uterus of adult hermaphrodites. Embryonic development is occurring inside the adult hermaphrodite uterus, such that embryos could be several hours apart in terms of development. Therefore, embryos will be only loosely developmentally synchronized using this method. For tighter synchronization, other methods must be used.
Fixation
Collect worms from 3-5 non-starved, non-contaminated plates by washing them off with M9 and transfer them to 1.5 ml centrifuge tubes. Many worms from several plates are needed because some worms are lost at each subsequent step. Because worms stick to the inside of plastic pipette tips, using sterile glass Pasteur pipettes to transfer worms in this step can help prevent the loss of worms.
Wash four times over ~60 min by resuspending in 1 ml M9 so that the bacteria in the gut are excreted. Between washes, pellet worms by centrifuging at ~900 × g (~3,000 rpm) for 2 min before carefully withdrawing most of the supernatant using a pipette. Be sure to leave the pellet of worms untouched.
Chill worms on ice and centrifuge at ~900 × g (~3,000 rpm) for 2 min. Remove supernatant to reduce volume to 500 μl, then add 500 μl ice-cold 2× Ruvkun buffer to a final concentration of 1×. Add 54 μl 37% formaldehyde (final concentration ~2%). Fixation by formaldehyde may negatively affect some epitopes, and its concentration (within a range of ~1-4%) may need to be adjusted by trial and error. Mix by inverting the tubes or pipetting.
Freeze rapidly in liquid nitrogen or dry ice/ethanol. This step may help crack or permeabilize the tough worm cuticle. Frozen samples can be stored at -80°C.
Melt the frozen worm pellet under tap water.
Incubate on ice, inverting the tube occasionally, for 30 min.
Wash worms twice in Tris-Triton buffer by centrifuging at ~900 × g (~3,000 rpm) for 2 min, carefully withdrawing supernatant, and resuspending in 1,000 μl Tris-Triton buffer. Triton detergent helps prevent worms from sticking to the pipettes, tubes, and one another.
Pellet worms by centrifuging at ~400 × g (~2,000 rpm) for 2 min. After this step, the animals should be considered fragile. From this point on, when pelleting worms, centrifuge at no more than ~400 × g (~2,000 rpm). Remove supernatant, carefully avoiding pellet, and resuspend the worms in 500 μl Tris-Triton buffer +1% β-mercaptoethanol. Incubate for 2-8 h with gentle agitation on a rocker platform in a 37°C incubator. β-mercaptoethanol can reduce disulfide bonds in proteins, which may improve permeability of the cuticle. The incubation with β-mercaptoethanol at high temperature may also help to denature enzymes like DNases, proteases, and peroxidases that could damage epitopes.
Wash the worms once by pelleting in the centrifuge, carefully removing supernatant, and resuspending with 1,000 μl 1× BO3 + 0.01% Triton buffer. The BO3 buffer has a basic pH that benefits the redox reactions in the subsequent steps.
Pellet worms, remove supernatant leaving pellet undisturbed, and resuspend the worms in 500 μl of 1× BO3 + 0.01% Triton buffer + 10 mM DTT. Incubate for 15 min with gentle agitation on the rocker platform at room temperature. DTT also may improve cuticle permeability by further reducing disulfide bonds.
Wash the worms once by pelleting in the centrifuge, carefully removing supernatant, and resuspending with 1,000 μl of 1× BO3 + 0.01% Triton buffer.
Pellet worms, remove supernatant leaving the pellet undisturbed, and resuspend the worms in 500 μl of 1× BO3 + 0.01% Triton buffer + 0.3% H2O2. Incubate for 15 min with gentle agitation at room temperature. H2O2 may oxidize sulfhydryl groups to prevent disulfide bonds from reforming.
Wash the worms once by pelleting in the centrifuge, carefully removing supernatant, and resuspending with 1,000 μl of 1× BO3 + 0.01% Triton buffer.
Pellet by centrifugation, remove supernatant, and resuspend with 1,000 μl Antibody buffer B. Allow to wash for at least 15 min with gentle agitation at room temperature to remove any residual BO3 buffer or H2O2. Pellet by spinning at ~400 × g (~2,000 rpm) for 2 min and resuspend in ~100-300 μl of Antibody buffer A, depending on how many worms are present. Worms are now fixed and can be safely stored for up to a month at 4°C in Antibody buffer A.
Primary Antibody Binding
Resuspend fixed worms by gently inverting the tube, then transfer 30 μl of fixed worms to a fresh 1.5 ml tube using a pipette tip that has been cut to enlarge the opening (this may prevent damage to the fragile fixed worms). Visually confirm that many worms are present and, if necessary, adjust volume. There should be hundreds of worms.
Add 170 μl Antibody buffer A with the appropriate amount of the desired primary antibody. For example, we often use GT335 (a monoclonal antibody that binds the branch point glutamate of post-translationally polyglutamylated proteins, such as tubulins) at a dilution of 1:450. If we had three C. elegans strains to compare, we would dilute 1.33 μl GT335 into 510 μl Antibody buffer A, and then add 170 μl of the dilution to the 30 μl worm suspension for the final dilution of 1:450 in a final volume of 200 μl. Incubate overnight, either at room temperature or at 4 °C in a cold room, depending on the antibody documentation. (Incubation time can be reduced, but a minimum of 2 h is recommended to allow diffusion through cuticle and tissues.) Dilution and incubation temperature may need to be determined empirically. Especially when trying a new antibody, it is also worthwhile to include a control in which no primary antibody is added. Use a rocker to provide gentle agitation.
Wash the worms twice with Antibody buffer B by pelleting in the centrifuge, carefully removing all but the last ~50-100 μl of supernatant, and resuspending in 500 μl Antibody buffer B. Then pellet worms, remove supernatant, and resuspend worms for a third time in at least 500 μl Antibody buffer B and incubate for at least 2 h to overnight with gentle rocking at room temperature to eliminate all unbound primary antibody.
Wash the worms once by pelleting in the centrifuge, carefully removing all but the last ~50-100 μl of supernatant, and resuspending in 500 μl Antibody buffer A. Pellet worms and remove supernatant leaving approximately the last 50 μl.
Secondary Antibody Binding
Add 150 μl of the secondary antibody diluted in Antibody buffer A for a final volume of 200 μl. We typically use Alexa-fluor 568-conjugated donkey anti-mouse secondary antibody at a dilution of 1:2,000 or 1:2,500 in the final volume of 200 μl of worm suspension. Enclose the tubes in a cardboard freezer box or cover with aluminum foil to protect from light, and incubate at room temperature for 2 h on the rocker.
Wash worms five times over several hours by pelleting in the centrifuge, carefully removing all but the last ~50-100 μl of supernatant, resuspending in 500 μl Antibody buffer B, and gently rocking to remove unbound secondary antibody. If excessive non-specific secondary antibody staining is observed, continue washing worms with Antibody buffer B for several more hours and/or leave on rocker in Antibody buffer B overnight. Fixed stained worms can be stored for a month or more at 4°C.
Make an agarose pad by adding a drop of liquid 2% agarose to a slide and immediately placing another slide on top to create a flat thin layer. Remove the top slide and mount worms for imaging by using a pipette tip that has been cut to slightly enlarge the opening to transfer a drop of 3-5 μl onto the agarose pad. Position gently using an eyelash glued to a toothpick and apply a cover slip before imaging. Use the eyelash to manually clear away any debris or precipitates that might remain from the fixation or staining process. If few stained worms are transferred to the slide, allow worms to settle to the bottom of the tube and either reduce the volume in the tube or draw worms up directly from the pellet rather than resuspending, before attempting to mount worms again.
Data analysis
Because Alexa-fluor 568 fluoresces red, we typically acquire two-color images to capture red immunofluorescence in transgenic animals expressing green fluorescent protein markers. GFP fluorescence appears dimmer after the fixation and staining treatment than in live animals but can often be used to provide visual landmarks to interpret the tissues, cells, or subcellular structures in which red immunofluorescence is detected. We acquire epifluorescence or confocal data by optical sectioning using a monochrome CCD or CMOS camera and observe color channels individually or in combination in z-projections created using ImageJ software.
Results are generally consistent across trials, but some variation is to be expected in the intensity of staining of worms (Figure 2). To confidently report on the tissues, cells, or subcellular structures in which immunofluorescence is detected, it is important to see consistent staining in multiple animals. We typically first examine many animals microscopically and capture images of at least 10 animals for quantitative analysis (presence or absence of staining, area/region of structures stained, or fluorescence intensity of staining). We have used image analysis of animals subjected to this antibody staining method to detect differences in glutamylation across genotypes (O’ Hagan et al., 2011), and others have detected differences based on environmental regulation (Kimura et al., 2018).
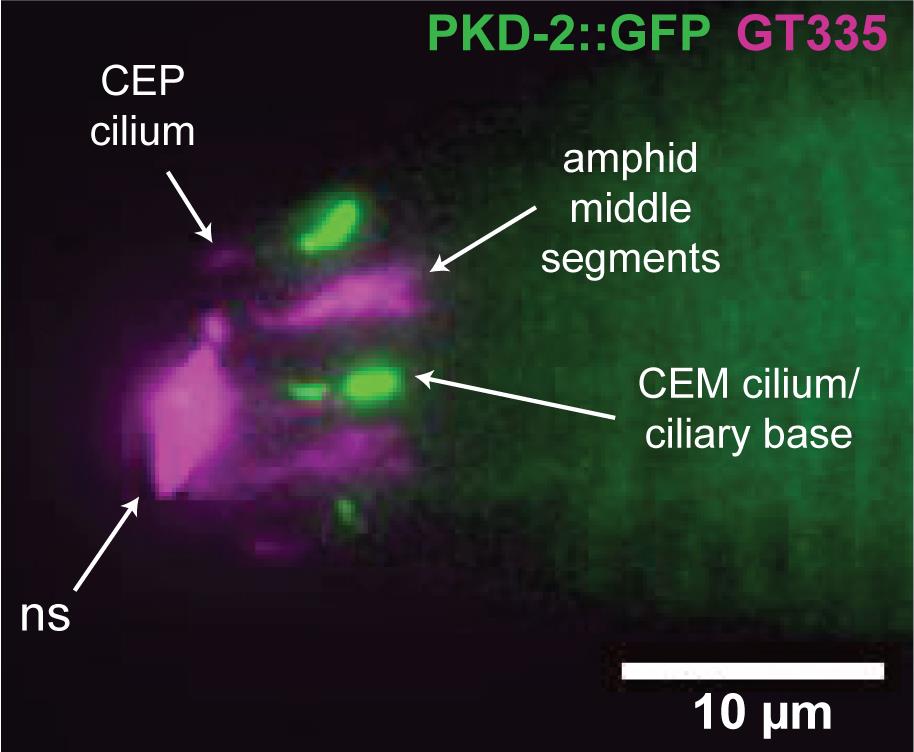
Figure 2. Representative image of staining with GT335 primary antibody and Alexa-fluor 568-conjugated secondary antibody. GT335 detects post-translational glutamylation by labeling the branch point glutamate (Wolf et al., 1992). In this picture of a C. elegans adult male nose, indirect immunofluorescence (magenta) of the GT335 monoclonal primary antibody, labeled with Alexa-fluor 568-conjugated secondary antibody, labels the ciliary middle segments of the amphid channel neurons, as well as CEP (cephalic) cilia. PKD-2::GFP (green) labels cilia of the male-specific CEM (cephalic male) sensory neurons in the nose. This epifluorescence image was captured using a Photometrics Cascade 512B CCD and Zeiss Axioplan2 microscope with 100× (NA 1.4) oil-immersion objective. ns indicates non-specific staining – a clump of debris and fluorescent secondary antibody was stuck to the buccal opening. Inconsistent or variable staining of a structure sometimes indicates that it may be non-specific. The buccal cavity is a common location to observe non-specific staining. Depending on the secondary antibody, non-specific staining is sometimes observed on annuli or other cuticular structures, and may be seen in controls in which the primary antibody is omitted. If available, use genetic mutants that lack the epitope or protein of interest to validate the antibodies used in the experiment.
Recipes
2× Ruvkun Buffer
160 mM KCl
40 mM NaCl
20 mM EGTA
10 mM spermidine-HCl
30 mM PIPES buffer, pH 7.4
50% methanol
Store at 4°C
PIPES buffer pH 7.4 is typically made as a 1 M stock by adding 302.37 g of PIPES powder to 600 ml ddH2O and bringing to pH 7.4 by adding approximately 153 ml of 10 N NaOH before bringing to volume by adding ddH2O. PIPES will not readily dissolve until pH is above 6.5.
Tris-Triton buffer
100 mM Tris-HCl, pH 7.4
1% Triton X-100
1 mM EDTA
Tris-HCl pH 7.4 solution is typically made as a 1 M stock by diluting Tris base in ddH2O and bringing the pH to 7.4 by slowly adding HCl.
20× BO3 buffer
1 M H3BO3
0.5 M NaOH
Dilute to 1× with ddH2O
1× BO3 + 0.01% Triton buffer
Dilute Tris-Triton buffer 1:100 in 1× BO3 buffer
Antibody buffer A
1× PBS
1% BSA
0.5% Triton X-100
0.05% sodium azide
1 mM EDTA
Store at 4°C
Antibody Buffer B
1× PBS
0.1% BSA
0.5% Triton X-100
0.05% sodium azide
1 mM EDTA (The same as Antibody buffer A, but with only 0.1% BSA)
Store at 4°C
Note: Antibody dilutions should be made freshly from refrigerated or frozen aliquots to minimize freeze-thaw cycles.
M9 Buffer
3 g KH2PO4
6 g Na2HPO4
5 g NaCl
1 ml 1 M MgSO4
ddH2O to 1 L
Sterilize by autoclaving
5 M KOH
Add 28.05 g KOH pellets to approximately 75 ml ddH2O.
Allow to dissolve, with occasional shaking. Dissolving KOH generates heat, so do not cap tightly. Then bring to 100 ml with ddH2O.
Tris-Triton buffer +1% β-mercaptoethanol
Working in a fume hood, add 100 µl β-mercaptoethanol to a 15 ml conical tube.
Add Tris-Triton buffer to a final volume of 10 ml.
1 M DTT solution
Working in a fume hood, dissolve 1.55 g of DTT powder in 10 ml ddH2O.
Distribute into 1 ml aliquots and store at -2°C.
1× BO3 + 0.01% Triton buffer + 10 mM DTT
Working in a fume hood, add 100 µl 1 M DTT solution to a 15 ml conical tube.
Add 1× BO3 + 0.01% Triton buffer to a final volume of 10 ml.
1× BO3 + 0.01% Triton buffer + 0.3% H2O2
Working in a fume hood, add 100 µl Hydrogen Peroxide 30% solution to a 15 ml conical tube.
Add 1× BO3 + 0.01% Triton buffer to a final volume of 10 ml.
Liquid 2% agarose
Add 2 g agarose to 100 ml ddH2O.
Heat in microwave until agarose is fully dissolved.
Use while still liquid.
Acknowledgments
This work was funded by the New Jersey Commission for Spinal Cord Research (NJCSCR) CSCR15IRG014. This method was used in our recent research publication, Power et al. (2020), as well as in previous publications (Topalidou and Chalfie, 2011; O’ Hagan et al., 2011 and 2017). This method is minimally modified from the Finney and Ruvkun method (Finney and Ruvkun, 1990).
Competing interests
The authors declare no competing interests.
Ethics
Use of C. elegans does not require institutional approval.
References
- Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W. and Prasher, D. C. (1994). Green fluorescent protein as a marker for gene expression. Science 263: 802-805.
- Duerr, J. S. (2006). Immunohistochemistry. WormBook 19:1-61.
- Finney, M. and G. Ruvkun, (1990). The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell 63(5): 895-905.
- Kimura, Y., Tsutsumi, K., Konno, A., Ikegami, K., Hameed, S., Kaneko, T., Kaplan, O. I., Teramoto, T., Fujiwara, M., Ishihara, T., et al. (2018). Environmental responsiveness of tubulin glutamylation in sensory cilia is regulated by the p38 MAPK pathway. Sci Rep 8(1): 8392.
- Miller, D. M. and Shakes, D. C. (1995). Immunofluorescence microscopy. Methods Cell Biol 48: 365-394.
- Nance, J. and Frøkjær-Jensen, C. (2019). The Caenorhabditis elegans Transgenic Toolbox. Genetics 212(4): 959-990.
- O'Hagan, R., Silva, M., Nguyen, K. C. Q., Zhang, W., Bellotti, S., Ramadan, Y. H., Hall, D. H. and Barr, M. M. (2017). Glutamylation Regulates Transport, Specializes Function, and Sculpts the Structure of Cilia. Curr Biol 27(22): 3430-3441 e3436.
- O'Hagan, R., Piasecki, B. P., Silva, M., Phirke, P., Nguyen, C. Q., Hall, D. H., Swoboda, P. and Barr, M. M. (2011). The Tubulin Deglutamylase CCPP-1 Regulates the Function and Stability of Sensory Cilia in C. elegans. Curr Biol 21(20): 1685-1694.
- Power, K.M., Akella, J.S., Gu, A., Walsh, J. D., Bellotti, S., Morash, M., Zhang, W., Ramadan, Y.H., Ross, N., Golden, A., Smith, H. E., Barr, M. M. and O’Hagan, R. (2020). Mutation of NEKL-4/NEK10 and TTLL genes suppress neuronal ciliary degeneration caused by loss of CCPP-1 deglutamylase function. PLOS Genetics 16(10): e1009052.
- Topalidou, I. and Chalfie, M. (2011). Shared gene expression in distinct neurons expressing common selector genes. Proc Natl Acad Sci U S A 108 (48): 19258-19263.
- Wolff, A., de Nechaud, B., Chillet, D., Mazarguil, H., Desbruyeres, E., Audebert, S., Edde, B., Gros, F. and Denoulet, P. (1992). Distribution of glutamylated alpha and β-tubulin in mouse tissues using a specific monoclonal antibody, GT335. Eur J Cell Biol 59: 425-432.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
O'Hagan, R. and Topalidou, I. (2021). Fixation and Immunostaining of Endogenous Proteins or Post-translational Modifications in Caenorhabditis elegans. Bio-protocol 11(19): e4172. DOI: 10.21769/BioProtoc.4172.
Category
Cell Biology > Cell staining > Protein
Immunology > Immune cell staining
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.