- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Inter-species Transplantation of Blastocysts between Medaka and Zebrafish
Published: Vol 11, Iss 18, Sep 20, 2021 DOI: 10.21769/BioProtoc.4166 Views: 3918
Reviewed by: Giusy TornilloAlberto RissoneAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Transplantation of blastocysts from a donor to a host blastula constitutes a powerful experimental tool to tackle major developmental biology questions. The technique is widely implemented in diverse biological models including teleost fish, where it is typically used for intra-species blastula transplantations – i.e., labeled blastocysts into a non-labeled host to follow lineages, or mutant blastocysts into a wild-type host to address autonomous vs. non-autonomous roles of a gene of interest. We have recently implemented a protocol to transplant blastocysts between zebrafish (D. rerio) and medaka (O. latipes), two species in which blastocysts show different developmental dynamics and sizes (Fuhrmann et al., 2020). We present here a detailed protocol on how to overcome the early differences in chorion structure, blastula size, and speed of development to achieve trans-species blastocyst transplantation.
Keywords: Blastomere transplantationBackground
The Chimera is a mythological creature containing parts of different animals – the body of one species, the head of another species, the tail of yet another species, and so on. In genetics, the term is used to define an organism where cells have diverse genetic information – usually a different genetic origin. In the field of developmental biology, chimeras and genetic mosaics have long been used as powerful tools to differentiate cell intrinsic from cell extrinsic processes, as well as to establish lineages (Le Douarin, 1993; Le Douarin and Dupin, 1993; Buckingham and Meilhac, 2011; Kretzschmar and Watt, 2012). The generation of chimeras and mosaics can be done genetically or mechanically, depending on the animal model of choice. For teleost fish, their external development allows the early, mechanical mixing of blastomeres among siblings and/or between a wild type and a mutant embryo. The use of a tracer in donor cells is typically used to identify the foreign cells in the developing host. There are well-established protocols for blastomere transplantation in different species of teleost fish – examples of blastula transplantation in the context of a specific biological question and explanatory videos on the process can be found (Haas and Gilmour, 2006; Rembold et al., 2006; Kemp et al., 2009; Centanin et al., 2011). Each of them takes into account the species-specific morphology of the early embryos and their developmental dynamics. Here, we modify existing protocols and establish a pipeline to generate zebrafish/medaka chimeras (Fuhrmann et al., 2020).
Materials and Reagents
Petri dish, 100 mm diameter (Greiner Bio-One, catalog number: 627102)
12/24 well plate (Corning, catalog numbers: 3512 [12-well]; 3527 [24-well])
Agarose coated dishes (homemade): 10 cm diameter, 6 cm diameter (Greiner Bio-One, catalog numbers: 627102 [10 cm diameter]; 628102 [6 cm diameter]) and 6-well plates (Roth, catalog number: ATCO.01). For the coating, use agarose 1% in ERM (medaka) or E3 (zebrafish). The base of the plastic plate should be covered by agarose; 1-2 mm thick is enough.
Cell saver tips (200 μl) (Biozyme, catalog number: 729051)
Borosilicate glass capillaries, 1.2 mm outer diameter × 0.94 mm internal diameter (Mind that these are different from the injection needles. They should NOT contain an internal filament. HARVARD Apparatus, model: 30-0016 GC120T-10 or GC120T-15)
Razor blade (to cut open the borosilicate needle) (Schreiber Instrumente, catalog number: 11-0240)
For Homemade (Air) transplantation device
Tube connector, Syringe Mouthpiece (Narishige, model: CI-1)
Teflon Tubing (Narishige, model: CT-1)
Handle Probe (WPI, catalog number: 2505)
Needle Holder (2 MM Press Port 1.2 MM, WPI catalog number: MPH412)
Epoxy Potting (Gray, WPI, catalog number: 4886)
1 ml Syringe (BD Plastipak, catalog number: REF300013)
Glass Pasteur pipette (WU Mainz, catalog number: 200760)
Glass Pasteur pipette, opened tip
Glass plates (40 mm diameter; Roth, catalog number: T937.1)
Forceps (Dumont No. 5 or No. 55)
Long tips – to handle medaka embryos after dechorionation (Eppendorf, model: 5242956003, Microloader 20 µl in Racks)
Sandpaper (240/180)
Plastic Pasteur pipette (Biosigma, catalog number: 390512)
Sieve to wash zebrafish embryos
Net to collect medaka females (homemade)
Hook to collect medaka embryos (homemade, see details in Figure 1)
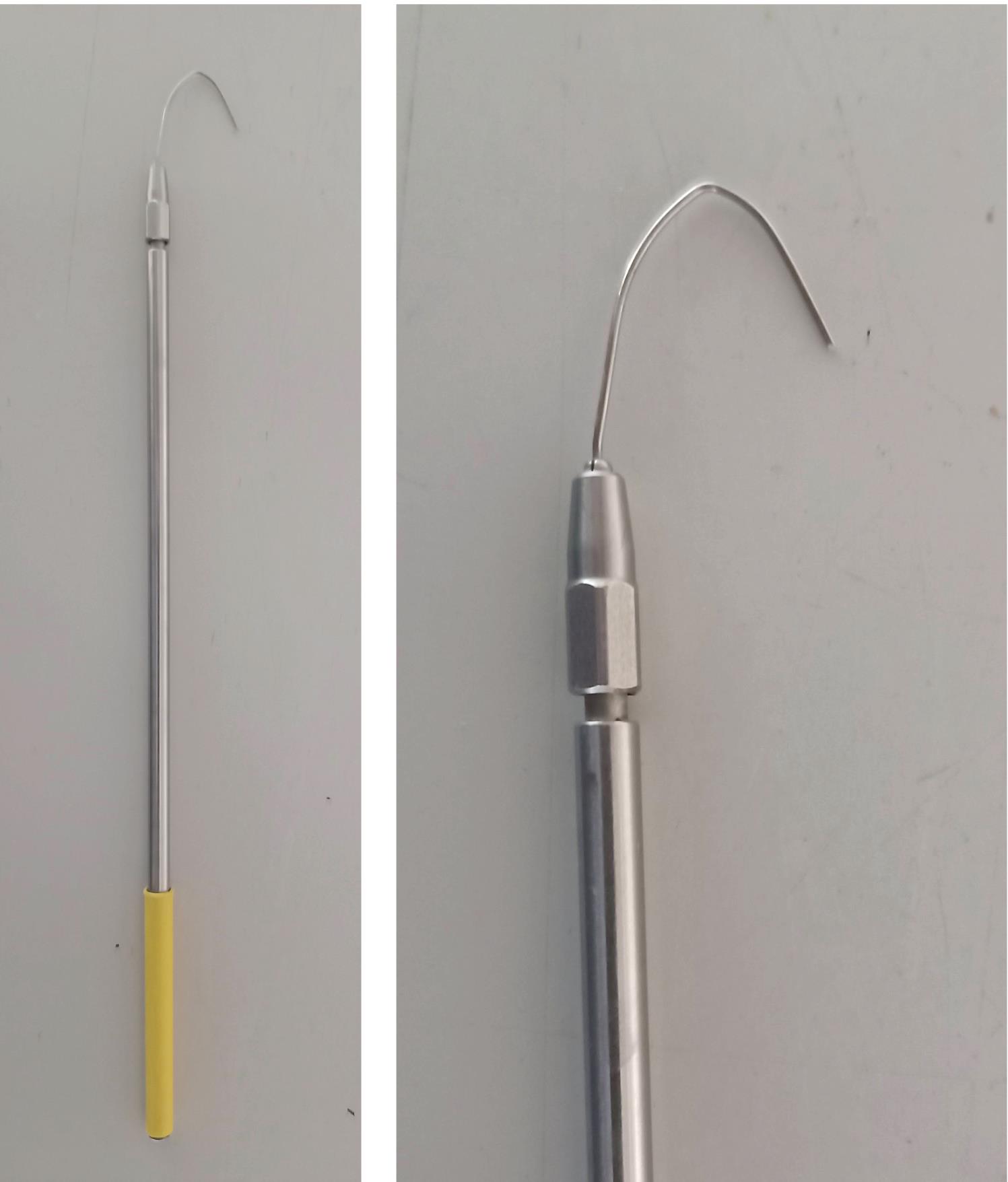
Figure 1. Hook to collect medaka embryos. Inoculation loop holder (Roth, KL97.1), length 21 cm; wire 1.4301: length 2 cm, width 2.5 cm.Glass flasks (100 ml, 500 ml)
100 ml Beakers (Schott, catalog number: 211062402)
Low melting point agarose (Roth, catalog number: 6351.5)
Pronase (Roche, catalog number: 10165921001)
Hatching enzyme (HE) medaka (home-made, can be ordered at NBRP Medaka: https://shigen.nig.ac.jp/medaka/strain/hatchingEnzyme.jsp;jsessionid=8F08DDE7593CDD7D1BFA32E6382E7E0F)
Transplantation mold (we use homemade molds; see Figure 2. Similar commercial molds can be found at: https://www.wpi-europe.com/products/pumps-and-microinjection/oocyte-injection/z-molds.aspx)
Embryo rearing medium for medaka (ERM) (see Recipes)
Zebrafish medium (see Recipes)
20× Tricaine (Sigma-Aldrich, catalog number: A5040-100G) (See Recipes)

Figure 2. Transplantation molds. To create the transplantation plate, molds are inserted top-down in 1.5% Agarose (see section B1). (A) Petri dish filled with 1.5% agarose in water, ca. 60°C. (B-B’) Transplantation molds with a detailed view of the structures that will form the wells. (C) Transplantation molds are placed in the Petri dish containing agarose, with the wells facing the agarose. (D) Transplantation mold stays in the Petri dish until the agarose has formed a stable gel. (E) When the transplantation mold is removed, the agarose gel contains wells in which the embryos will be placed. The transplantation plate is ready then.
Equipment
Zebrafish housing system (Tecniplast ZEBTEC equipped with 3.5 L tanks)
Zebrafish mating boxes (homemade 5 L boxes; see Figure 3. Alternatively, we have used breeding tanks from Tecniplast – 0.8 or 1.7 L, sloped tank)

Figure 3. Zebrafish mating box. Lateral view (top) and top view (bottom) of the zebrafish mating box. This setup accommodates up to five couples.Medaka housing system (Tecniplast ZEBTEC equipped with 3.5 L tanks and Müller & Pflege, 4 L tanks)
Incubator, 23°C (no need of CO2 control) (Binder, model: 9020-0339)
Incubator, 28°C (no need of CO2 control) (Rumed, model: 200330160)
Incubator, 32°C (no need of CO2 control) (Binder, model: 9010-0081)
Micropipette puller (we use a horizontal puller, Sutter Instrument CO, model: P-97)
To pull Harvard Apparatus GC120T – 10 capillaries, we use the following program: Heat 505; Pull 25; Vel 250; Time 10)
Transplantation device (a homemade air system; see Figure 4. We have also successfully used the Eppendorf CellTramR Air/Oil system)
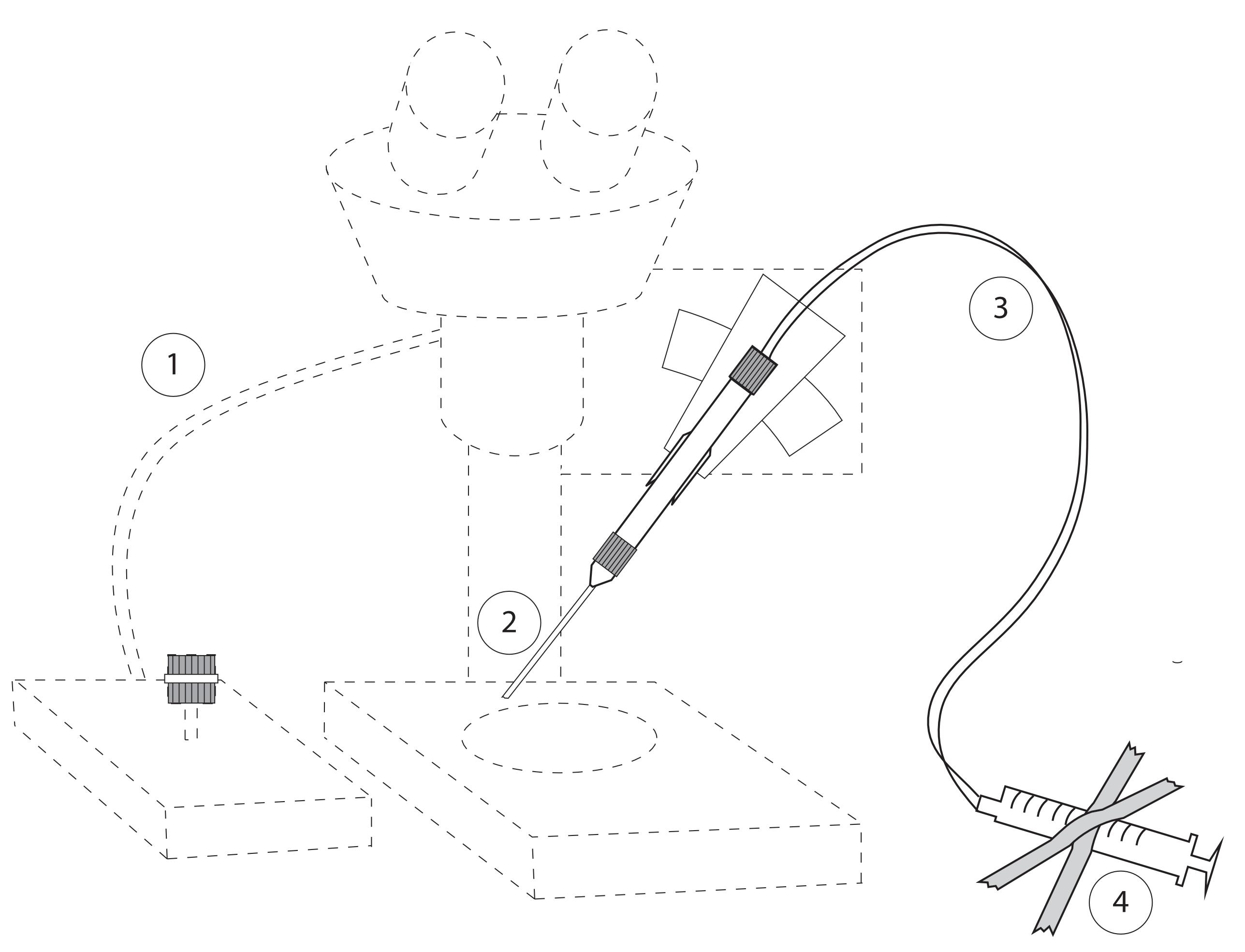
Figure 4. Set up for Transplantations. Main components: (1) Micromanipulator and Stereoscope with light source; (2) transplantation needle inserted into needle holder; (3) tube attached to the needle and a syringe (airtight); (4) syringe (volume 1 ml) attached to desk.Stereoscope with light source (Olympus, model: SZX7)
Micromanipulator (Eppendorf, model: InjectMan NI 2)
Pipette (Gilson 20-200 µl, 200-1,000 µl)
Fluorescence stereomicroscope (Nikon, model: SMZ18, solar light engine from Lumencor)
Microwave
Procedure
Please consider Figure 5 for the order of steps taken to complete the protocol and the respective temperatures settings used.
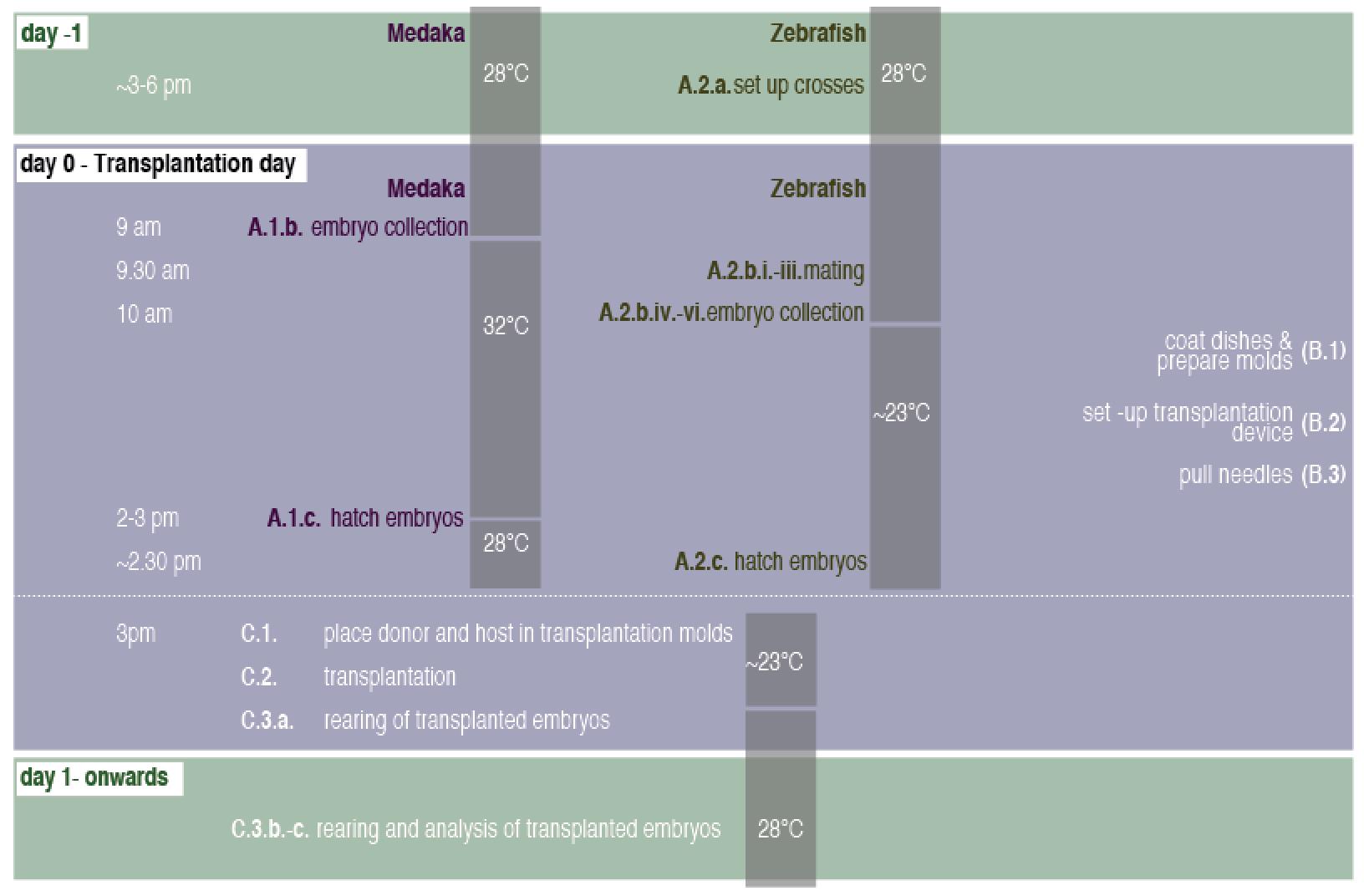
Figure 5. Timeline for inter-species transplantations
Set up fish to produce embryos
Medaka
Medaka crosses
Medaka are kept in their respective breeding group to allow for early breeding on the next day. There is no need to split females and males the day before. Collection of embryos can be done every day for each female, which lays approximately 20 eggs each. Embryos are attached to the mother for some hours after fertilization. Beware that you need to use more medaka females than zebrafish females.
9:00-9:30 am – Embryo collection
On the morning of the experiment (~9:00 am), identify a female harboring embryos and gently catch it with a fishnet (Figure 6).
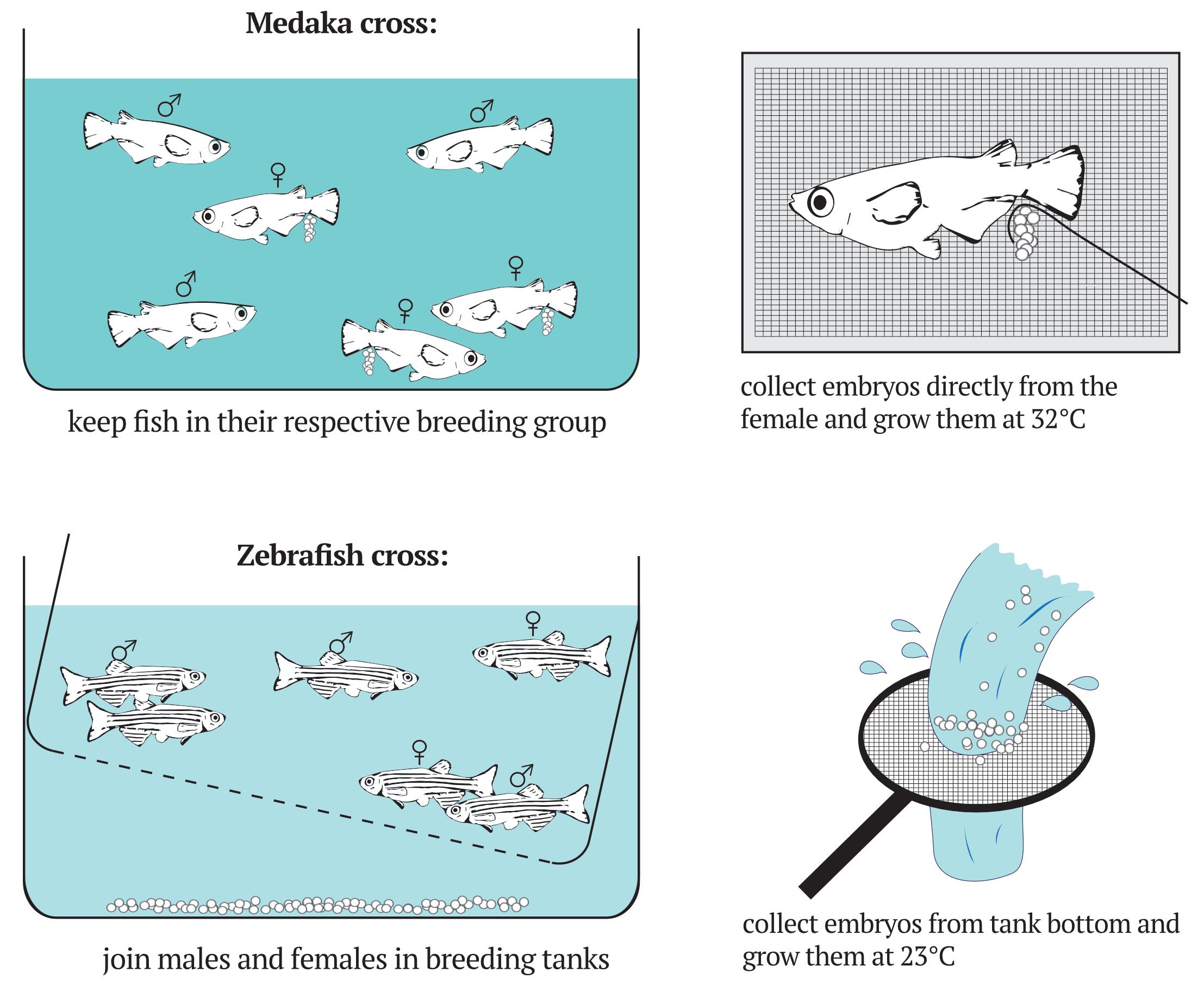
Figure 6. Setup for medaka and zebrafish crosses and embryos collectionCollect the embryos using a thin metal hook (Figure 1), brushing them gently from the female’s belly. Place the female back in the tank.
Transfer the embryos from the net or from the hook into a 10 cm Petri dish filled with ERM.
Place medaka embryos in ERM at 32°C. This helps embryos to develop faster to catch up with zebrafish blastulae (Furutani-Seiki and Wittbrodt, 2004).
2:00-3:00 pm – Hatch medaka embryos
Hatching of medaka embryos is best performed at the 256-512 cell stage (~2 pm). Younger embryos are more fragile; the mortality rate increases if hatched earlier.
Take medaka embryos out of the fish medium and use your fingers to roll them gently on sandpaper (grain 240) to remove hairs from the chorion.
Note: The goal is to create holes in the external part of the chorion to favor the hatching enzyme degrading the chorion from the inside. We use the removal of hairs as a proxy for efficient creation of holes.
Check embryos under the stereomicroscope to monitor hair removal. Repeat the rolling step until all hairs are gone.
Discard dead embryos before moving forward.
Rinse the embryos in ERM medium
Transfer the embryos to a glass tube using a Pasteur pipette and remove all liquid.
Use a plastic Pasteur pipette from which the tip has been cut out
Do not put more than 20 embryos per tube.
Cover embryos with hatching enzyme (HE) and incubate at 28°C for 30 to 60 min maximum.
Use the minimal possible amount of HE, just covering the embryos.
Always cover the tube with parafilm to avoid evaporation of HE.
Incubation time depends on: a) the quality of the HE, especially when using homemade HE, and b) the number of embryos in the tube.
Notes:
1)Increasing the HE-volume to embryo ratio leads to faster hatching.2)ii) if hair removal was inefficient, hatching takes more time or can even fail entirely.Check the progress of the HE reaction under the stereomicroscope every 10-15 min.
When embryos have holes in the chorion, wash them carefully four times with ERM.
Rinse embryos by letting the ERM flow down slowly on one side of the tube.
Be aware that the embryos are extremely fragile when dechorionated.
It is important to use ERM at 28°C to avoid a heat shock.
Transfer the embryos to an agarose-coated Petri dish – follow steps in Figure 7.
Fill the glass tube entirely with ERM.
Place a finger on top, sealing the tube completely - avoid air bubbles.
Invert slowly and place the top of the tube underneath the surface of the medium in the agarose-coated dish.
Release finger and let the embryos sink.
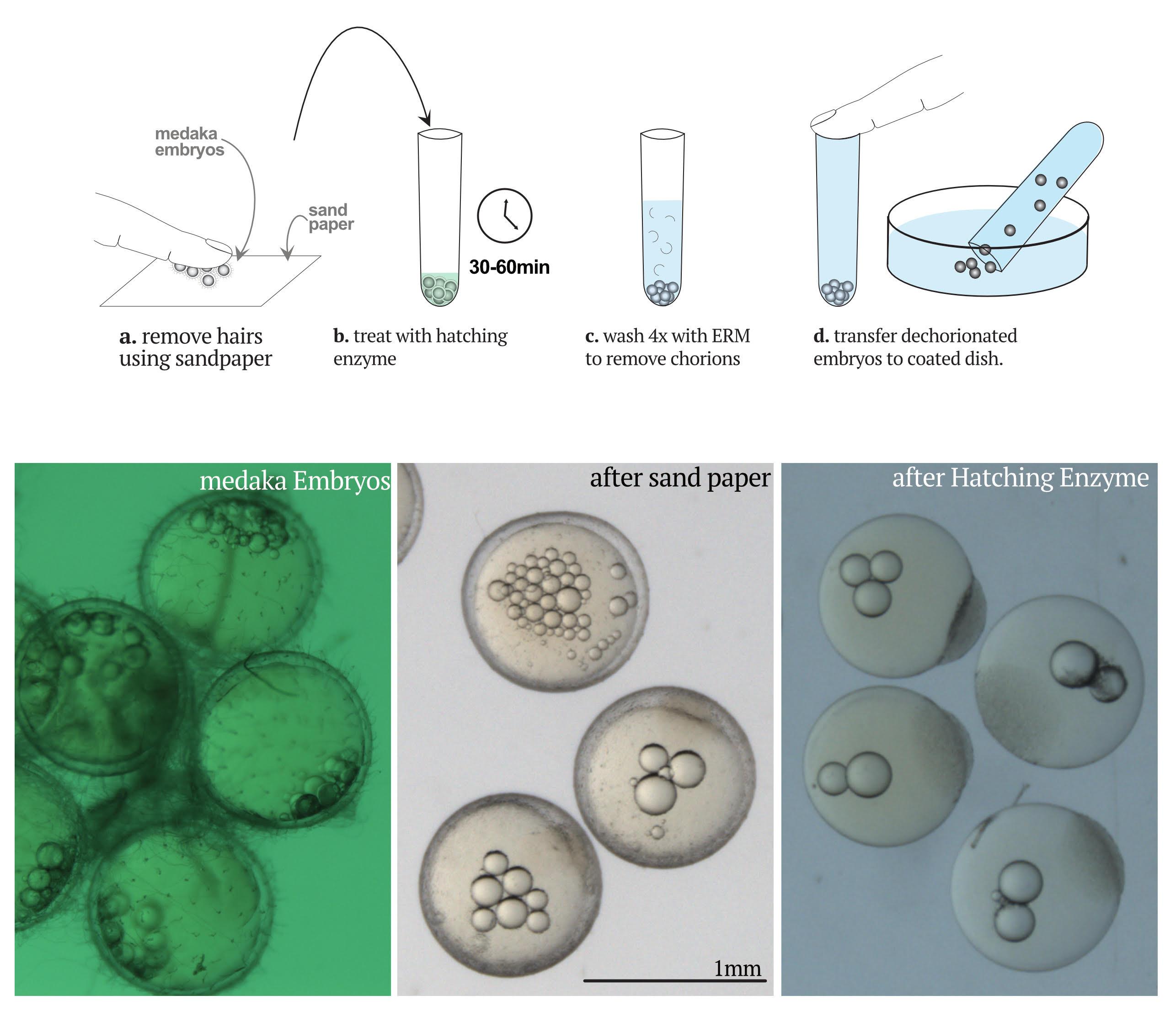
Figure 7. Steps to dechorionate medaka embryos
Zebrafish
3:00-6:00 pm (day -1) Set up zebrafish crosses (Westerfield, 2007)
On the afternoon before the transplantation experiment, split females and males using a large breeding tank.
Fill a breeding tank with 2:1 fishwater:deionized water.
The breeding tank consists of 2 compartments separated by a mesh. Place the males in the bottom compartment and the females in the upper compartment.
One cross consists of 5 males and females; depending on your experiment, you might need to set up more than one cross.
9:30-10:30 (day 0, transplantation day): Embryo collection
On the morning of the experiment (9.30 am – 10:00 am), transfer the males into the upper compartment – they are now together with the females.
Tilt the mesh separator to create a slope with a shallower and a deeper region (Figure 6).
Note: Zebrafish usually breed in shallow waters; you should observe couples mating in the upper extremity.
Leave crosses undisturbed for 20-30 min.
Note: You should observe embryos accumulated at the bottom of the tank afterwards.
Transfer fish into their tank and remove the mesh bottom separator.
Collect eggs by running the breeding tank water through a fine sieve.
Transfer embryos in a 10 cm diameter Petri dish and rinse with zebrafish medium
Keep embryos at room temperature (~23°C).
2:30-3:30 pm – Hatching of zebrafish at early blastulae stage, 258 to 512 cell stage (Westerfield, 2007)
Transfer zebrafish blastulae to 100 ml glass beakers.
Remove the zebrafish medium and add 30 ml of fresh zebrafish medium.
Add 1 ml Pronase (30 mg/ml).
Note: Thaw Pronase and keep it on ice before using it.
Move the beaker in circles every 30 s, checking Pronase activity under the stereoscope (Figure 8).
Embryos without the chorion are the first to accumulate at the center.
Time ranges from 3 to 7 min and depends on the quality of the Pronase and the number of embryos per beaker. Pre-aliquot Pronase to avoid multiple thawing and freezing cycles, which impair its quality and extend the hatching time.
When you see a few embryos without the chorion, it is time to stop the reaction. The chorion of the other embryos will be gone during the washes.
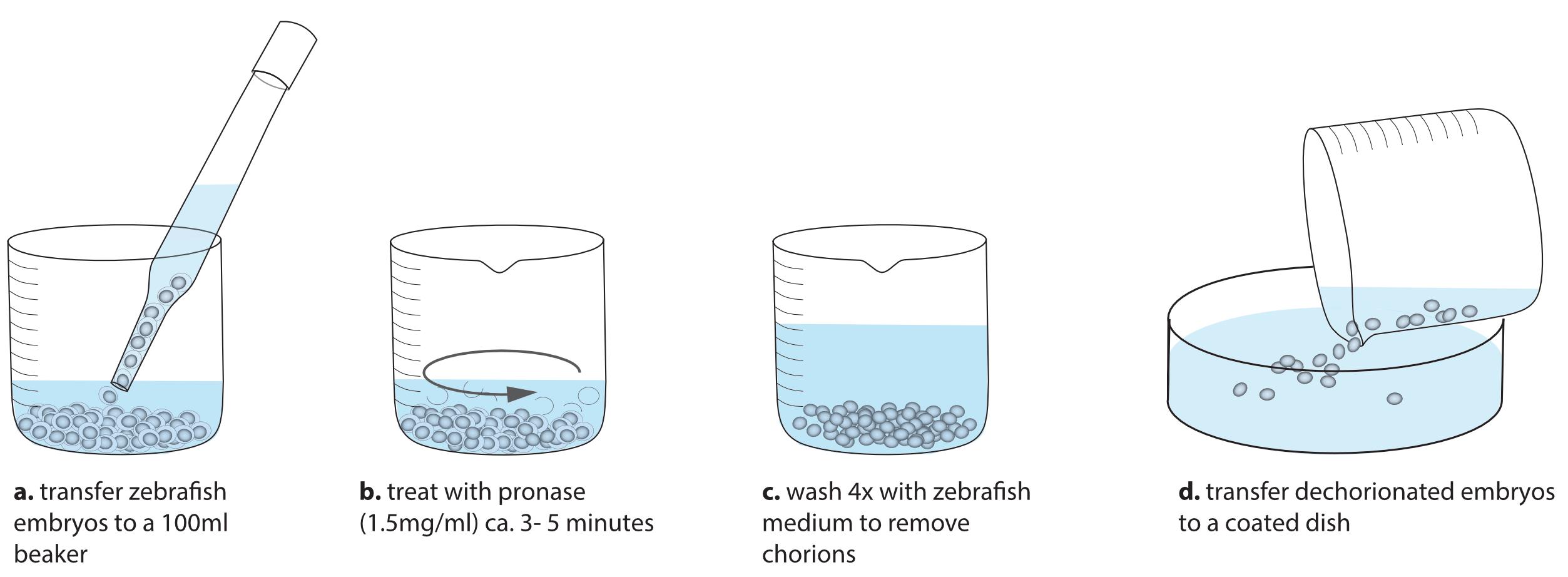
Figure 8. Steps to dechorionated zebrafish embryosDiscard the Pronase solution and wash zebrafish medium at least four times.
Note: Each wash will remove a considerable number of chorions.
Transfer freshly hatched embryos into a coated Petri dish and keep them in zebrafish medium until transplantation.
2:30-4:00 pm Preparing the transplantation setup
Agarose coated dishes and transplantation molds
Coated dishes
Prepare a 1.5% solution of agarose in ddH2O and boil it in a microwave until dissolved.
Agarose solution can be prepared fresh or stored pre-made:
Use 1 L Bottles and prepare 800 ml of agarose solution.
Keep agarose solution at 65°C until used.
If using plastic dishes, coat them with 1.5% agarose.
Glass dishes do not need to be coated.
To coat: pour the Agarose solution into a 10 cm Petri Dish until the bottom is just covered (ca. 20 ml).
Prepare at least one 10 cm Petri Dish for each genotype to keep dechorionated embryos before transplantation.
Prepare a 10 cm Petri Dish or 6-/12-/24-well plates for the transplanted blastulae.
Which of the above is used depends on the experimental design. If donors need to be kept for later screening (i.e., to check genetic background), smaller well plates are useful to keep donors and hosts individually, allowing for the identification of the most relevant chimeras.
When using medaka, few embryos per shared volume is preferential as death of neighboring embryos affects survival rates. Keep one blastula per well in a 24-well or 12-well plate, or approximately three blastulae per well in a 6-well plate.
When using zebrafish (and if donor/host combinations are not tracked), 10 cm Petri dishes give a good survival rate.
Transplantation plates
Pour the Agarose solution into a 10 cm diameter Petri dish until the bottom is covered.
Place the transplantation mold (Figure 2) on top of the agarose.
Wells should face down.
Avoid bubbles.
The transplantation mold should not be covered by agarose.
Once the agarose is firm, the mold can be removed using forceps.
Prepared plates can be stored in the fridge for one day but need to be equilibrated at room temperature before usage.
Prepare transplantation needles
Pull glass capillary with a horizontal needle puller.
Transplantation needles should be long and thin (see Figure 9).
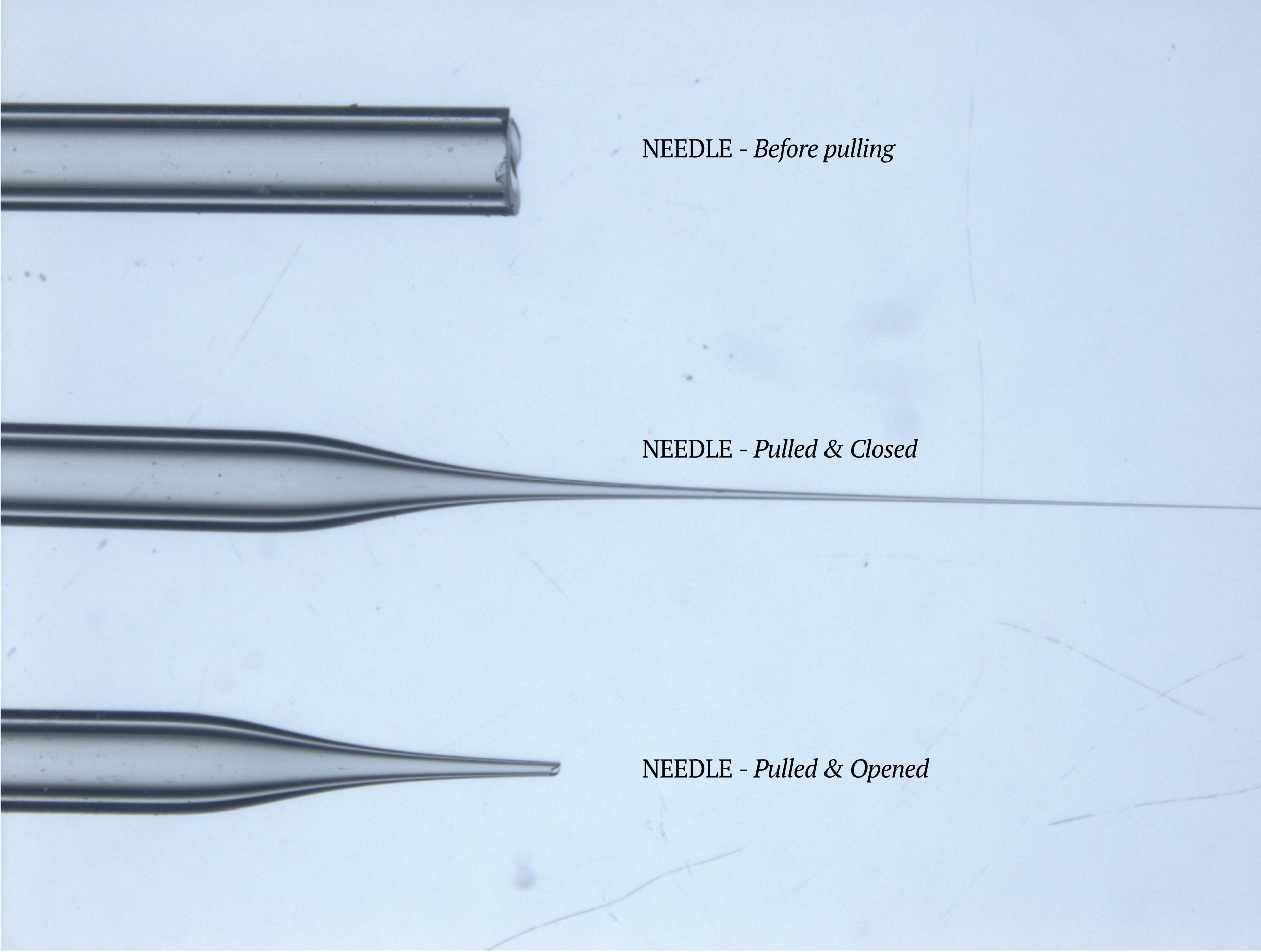
Figure 9. Transplantation needle. Images showing the needle before pulling as from the manufacturer (Materials #5), after pulling with the needle puller (settings described in Equipment #7), and after being opened with a razor blade (B2a).Open needle with a razor blade
The diameter of the needle should be larger than a cell diameter. Since blastula cells have different diameters according to their developmental stage, you will need to adjust the opening of the needle to the stage you are using and the number of cells to be transplanted.
A needle with small diameter (1-1.5× cell width) takes up cells individually and leads to higher dispersal of cells in the needle, which gives a better control of the number of transplanted cells.
A needle with larger diameter allows for the transplantation of more cells with less liquid, preserving the integrity of the donor cells.
The edge of the needle should not be too jagged. However, a slight tilt helps to push through the donor and host blastulae.
A good needle can be reused for multiple transplantations. To store it, be sure to clean it properly by taking ERM/zebrafish medium many times after the last transplantation.
Setting up the micromanipulation device
Use a stereomicroscope with a ~0.7-7:1 magnification and an integrated light source connected to a 3-axis motorized manipulator with a micromanipulation controller and a needle holder (see set up in Figure 4).
Attach a tube to the needle holder and a syringe (volume 1 ml) and seal airtight.
Attach the syringe to the desk using tape.
Note: This will allow you to one-handedly control the syringe and thereby the cells entering and exiting the transplantation needle.
Place transplantation needle in the needle holder.
Place a plate with fish medium in the field of view and submerge the tip of the needle in the water using the micromanipulator.
Move the syringe plunger and check if there is medium entering and exiting the needle.
Note: If the liquid is responding directly and predictably to the movement of the plunger, then you are ready to go.
3:30 pm onwards – Transplantation
Check that the embryos of both species are at blastula stage (~3 pm) (Kimmel et al., 1995; Iwamatsu et al., 2004). Move plates to the microinjection setup to avoid subsequent water movement.
Place donor and host blastulae in alternating columns (Figure 10).
Fill in the transplantation plate with medium for the HOST species.
Transfer zebrafish embryos to individual wells using a glass Pasteur pipette.
Dechorionated zebrafish blastulae have a diameter slightly smaller than that of the glass Pasteur pipette.
Take up a few blastulae from the plate with the pipette and release them individually in the transplantation plate, placing them along a column, and filling one embryo per well.
Donors should be placed in the left-most column; hosts should be placed in the second to fifth column. Since cells from one donor can be transplanted to many hosts, it is convenient to arrange multiple putative hosts per donor.
Accommodate the embryos with the cells facing upwards.
Embryos usually fall into the molds with the yolk facing down, and no further re-positioning of the blastulae is necessary. However, if necessary, take the blastula up with the pipette and retry, or use MicroloaderTM tips as described below.
Transfer medaka blastulae individually and place each next to a mold using cell saver tips.
Clip the end of a plastic MicroloaderTM tip to 2-3 cm.
Use the tip to gently push the blastulae into the molds with the animal pole (blastula cells) facing upwards. Only touch the cells at the animal pole and not the yolk cell with the plastic tip.
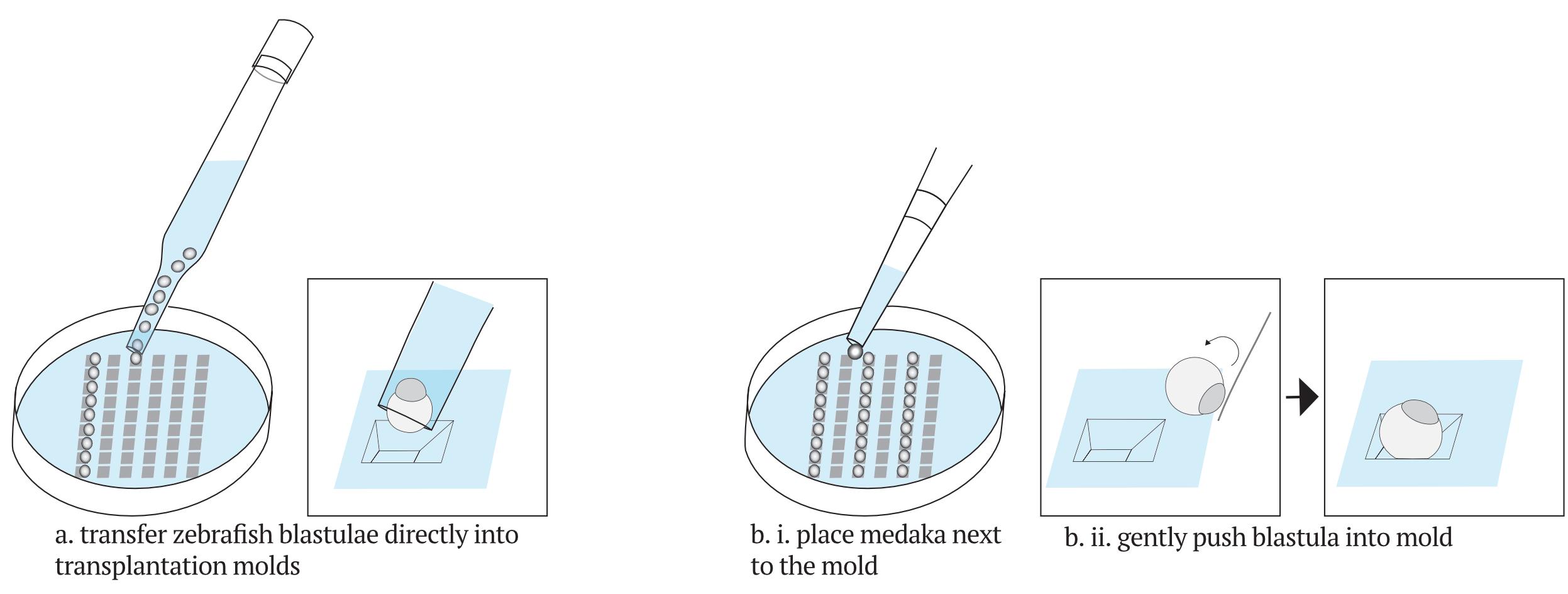
Figure 10. Disposition and orientation of embryos in the transplantation plate. Zebrafish embryos (a) can be handled with a glass Pasteur pipet and accommodated directly into the agarose well. Medaka embryos (b) are bigger and need to be transferred with a plastic Pasteur pipet into the dish and then handled into the wells with a MicroloaderTM tip.Transplantation of blastocysts (Figure 11)
Calibrate the transplantation needle.
Take up a small amount of medium in the transplantation needle by applying a gentle negative pressure through the syringe. Use this to adjust yourself to the response of the water flow inside the needle to the applied pressure.
The surface of the medium inside the thin part of the needle should be visible through the stereomicroscope before attempting to take up cells from the donor, thereby giving direct feedback of the applied flows.
Place the tip of the needle touching the host cells.
Blastula cells can be taken from any position, but caution must be taken for cells close to the yolk cell to avoid sucking up yolk.
In our setup, we use cells closest to the animal pole of the embryo; however, chimeras are also observed when taking up cells in the peripheral regions.
Apply negative pressure to take cells up
Depending on the setup of the transplantation experiment, you can take one or up to a hundred cells from a donor.
When the desired number of cells is reached, apply a gentle opposing force to the syringe to stop the flow.
Remove the needle from the donor blastula and move it to the host blastula.
Place the needle in the desired location of the host and release cells by carefully applying positive pressure.
The position of the cells in the host affects which organs the donor cells will contribute to. In general terms, cells in central positions will be incorporated in anterior structures like the forebrain and retina.
Move the needle to the following host and repeat the procedure until finishing the donor cells.
One blastula can serve as donor for multiple hosts, and the number of cells transplanted can be adjusted to the experimental conditions. Transplanted embryos with up to ~150 donor cells can survive; yet, we usually transplant up to 50 cells.
Clean your needle before going to the next donor blastula.
Allow medium to enter and exit the needle to remove the remaining cells.
Avoid air bubbles in the transplantation dish; this can damage the blastulae.
Repeat the procedure for as many donor blastulae as needed.
After transplantations are completed, remove all dead and non-transplanted blastulae from the transplantation plate.
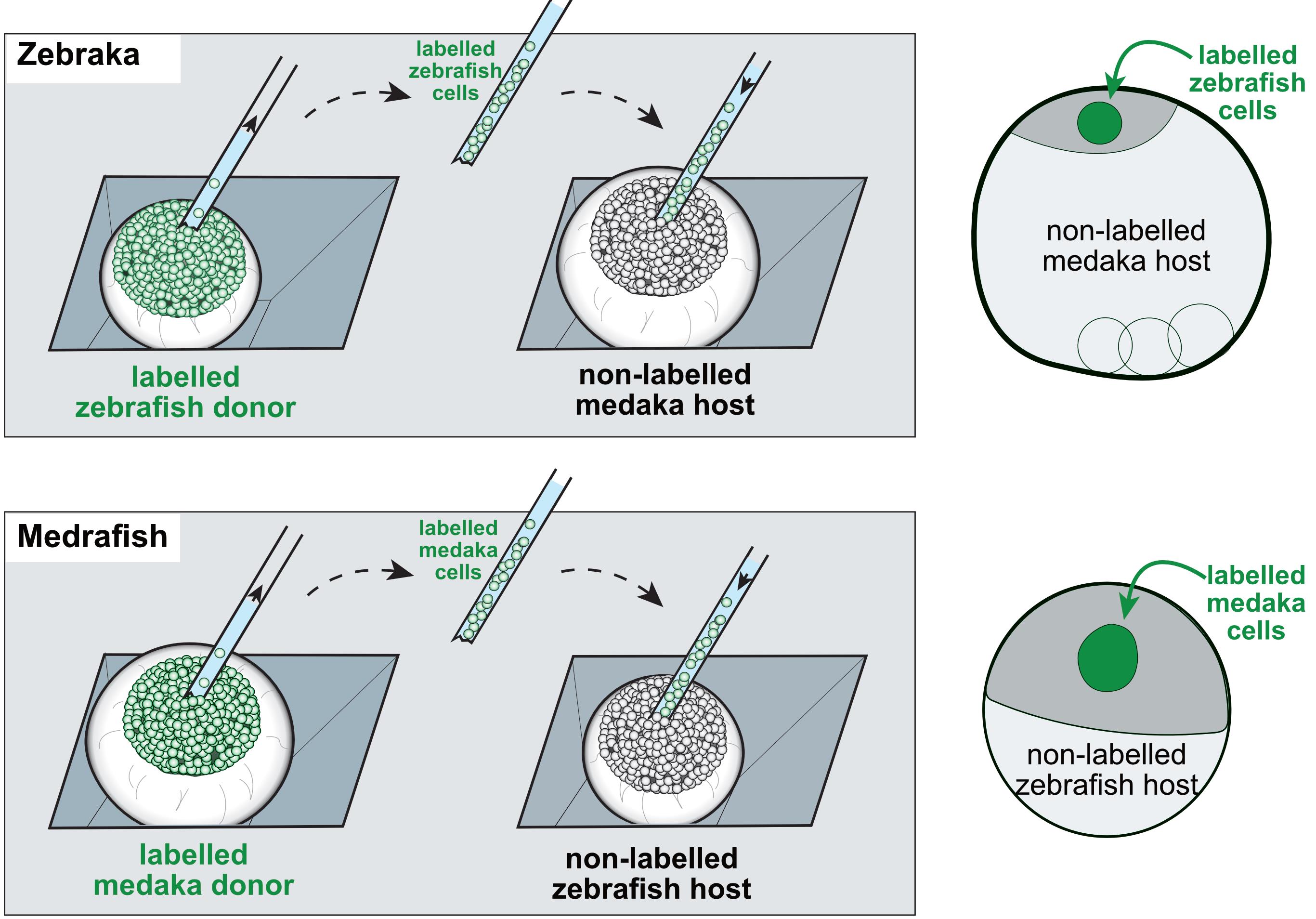
Figure 11. Transplantation of blastomeres between medaka and zebrafishRearing of transplanted embryos
Keep transplanted embryos in the transplantation plate at 28°C overnight.
For Medrafish (zebrafish hosts)
Transfer surviving embryos to a fresh Petri dish on the next day and exchange the zebrafish medium again in the afternoon of the same day.
Replace medium daily and remove dead embryos. We keep embryos up to day 5 of development.
For Zebraka (medaka hosts)
Place surviving embryos individually using cell saver tips in 1.5 ml ERM in a coated 24 well plate (Agarose 1.5%). Alternatively, embryos can be kept in glass dishes (2 cm diameter) or differently sized multi-well dishes.
Exchange ½ the medium daily, avoiding mechanical perturbation during the first 3 days. We keep embryos up to day 9 of development.
Notes
Fish maintenance and mating
Both medaka and zebrafish fish are maintained on a 14 h day – 10 h night cycle at 28°C.
Medaka breeding group:
Each breeding group consists of a single male and two to three females.
Fish are maintained in the same tank; courting behavior starts with the first light (8:00 am in our fish room).
Embryos are collected from each female daily.
Zebrafish breeding group:
Each breeding group consists of five males and five females.
Fish are not fed in the morning of the cross.
Use breeding groups maximum once per week.
Temperature
Medaka embryos tolerate a high temperature range; in our hands, a temperature of 32°C before transplantation (from 2 to 512 cell stage) worked best to speed up development without long-term consequences. Higher temperatures cause mold and increase evaporation but should still be possible in a respective setup.
Zebrafish tolerate only a narrow temperature range, but embryos develop normally at room temperature (~23°C), which delays development sufficiently to achieve synchronization with medaka at the blastula stage.
Handling of dechorionated embryos
Hatched embryos are more fragile than intact embryos. Special care must be taken before the completion of gastrulation as the yolk does not sustain contact with plastic.
Medaka embryos are more fragile than zebrafish embryos due to their larger yolk cell. Mechanical perturbations, such as those induced by water movement, are sufficient to cause ruptures in the yolk and result in the death of the respective embryos. Especially in the first two days after transplantation, the embryos should be perturbed as little as possible.
Dechorionated embryos appear to be more sensitive to the chemical nature of their surroundings. This means that extra care must be taken when handling the fish medium, and dead embryos should be removed from the dish.
If only a few zebrafish embryos are available, dechorionate them with fine forceps (Dupont 55) under a stereomicroscope (Skip step A2c). If done correctly, manual dechorionation is gentler than chemical treatments and improves the survival rate of embryos, but is more time consuming. You cannot hatch medaka embryos with forceps as the chorion is very stiff and the attempt will damage your embryo. However, it is possible to remove a partially opened chorion after an incomplete hatch with HE using forceps.
Imaging
Every transfer to a microscope causes mechanical strain and thus reduces the survival rate and should be done carefully.
Mount embryos for live imaging as follows:
For general observation and imaging in a stereomicroscope, transfer to 3% Methylcellulose (in fish medium).
For confocal live imaging, mount in a 0.6% solution of low melting agarose (should be <40°C before mounting). This is preferential over Methylcellulose as Methylcellulose creates fluorescent background signals.
Once embryos start moving, anesthetize dechorionated live embryos in 1× (1 mg/ml) tricaine in the respective medium (Neiffer et al., 2009; Lischik et al., 2019).
In all stages of development, we check embryos using a fluorescent stereomicroscope if a fluorescent marker is available in the donor. This allows to:
Select for successfully transplanted embryos.
Report the location of most donor cells.
Follow the outgrowth dynamics of larger structures, such as neurons.
Use confocal (live) imaging, for example, to:
Spot smaller clones of donor cells.
Identifying cellular and sub-cellular behaviors.
Visualize donor-host interactions.
Dechorionated zebrafish embryos survive live imaging early on; medaka embryos are more sensitive and survive better at older stages.
Recipes
Embryo rearing medium for medaka (10× ERM)
170 mM NaCl
4 mM KCl
2.7 mM CaCl2·H2O
6.6 mM MgSO4·7H2O
170 mM HEPES
Fill up to 10 L with deionized water.
Zebrafish medium
3 g red sea salt in 100 ml deionized water
20× Tricaine
2 g Ethyl 3-aminobenzoate methanesulfonate
5 g Na2HPO4·2H2O in 500 ml deionized water
Adjust pH 7.0-7.5
Acknowledgments
We are grateful to all members of the Centanin group for their critical input on this protocol and their help collecting pictures. We thank Jochen Wittbrodt for sharing instruments, A. Saraceno, E. Leist and M. Majewski for fish maintenance, and C. Loosli for the preparation of the Hatchling Enzyme. Work in the Centanin Lab is supported by the DFG via the SFB873, project A11. This protocol is an extended adaptation of from Fuhrmann et al. (2020).
Competing interests
The authors declare no competing interests.
References
- Buckingham, M. E. and Meilhac, S. M. (2011). Tracing cells for tracking cell lineage and clonal behavior. Dev Cell 21: 394-409.
- Centanin, L., Hoeckendorf, B. and Wittbrodt, J. (2011). Fate restriction and multipotency in retinal stem cells. Cell Stem Cell 9: 553-562.
- Neiffer, D.L. and Stamper, M. A. (2009). Fish sedation, analgesia, anesthesia, and euthanasia: considerations, methods, and types of drugs. ILAR J. 50: 343-360.
- Haas, P. and Gilmour, D. (2006). Chemokine signaling mediates self-organizing tissue migration in the zebrafish lateral line. Dev Cell 10: 673-680.
- Kemp, H. A., Carmany-Rampey, A. and Moens, C. (2009). Generating Chimeric Zebrafish Embryos by Transplantation. J Vis Exp 29: e1394.
- Kimmel, C. B., Ballard, W. W., Kimmel, S.R., Ullmann, B. and Schilling, T. F. (1995). Stages of embryonic development of the zebrafish. Dev Dyn 203(3): 253-310.
- Kretzschmar, K., and Watt, F. M. (2012). Lineage tracing. Cell 148: 33-45.
- Le Douarin, N. M. (1993). Embryonic neural chimaeras in the study of brain development. Trends Neurosci 16: 64-72.
- Le Douarin, N. M. and Dupin, E. (1993). Cell lineage analysis in neural crest ontogeny. J Neurobiol 24: 146-161.
- Lischik, C. Q., Adelmann, L. and Wittbrodt, J. (2019). Enhanced in vivo-imaging in medaka by optimized anaesthesia, fluorescent protein selection and removal of pigmentation. PLoS One 14(3): e0212956.
- Furutani-Seiki, M. and Wittbrodt, J. (2004). Medaka and zebrafish, an evolutionary twin study. Mech Dev 121: 629-637.
- Rembold, M., Loosli, F., Adams, R. J. and Wittbrodt, J. (2006). Individual cell migration serves as the driving force for optic vesicle evagination. Science 313: 1130-1134.
- Iwamatsu T. (2004). Stages of normal development in the medaka Oryzias latipes. Mech Dev121: 605-618.
- Westerfield, M. (2007). The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio rerio). (5th edition). University of Oregon Press.
- Fuhrmann, J. F., Buono, L., Adelmann, L., Martinez-Morales, J. R. and Centanin, L. (2020). Genetic developmental timing revealed by inter-species transplantations in fish. Development 147(22):dev192500.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Fuhrmann, J. F., Onistschenko, J. and Centanin, L. (2021). Inter-species Transplantation of Blastocysts between Medaka and Zebrafish. Bio-protocol 11(18): e4166. DOI: 10.21769/BioProtoc.4166.
Category
Developmental Biology > Morphogenesis > Organogenesis
Biological Sciences > Biological techniques
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.