- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation and Quantification of Mouse γδT-cells in vitro and in vivo

(*contributed equally to this work) Published: Vol 11, Iss 17, Sep 5, 2021 DOI: 10.21769/BioProtoc.4148 Views: 5024
Reviewed by: Luis Alberto Sánchez VargasDarshika Udawatterozan romel attili
Original research article
The authors used this protocol in:

Dec 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The skin plays an important role in protecting the body from pathogens and chemicals in the external environment. Upon injury, a healing program is rapidly initiated and involves extensive intercellular communication to restore tissue homeostasis. The deregulation of this crosstalk can lead to abnormal healing processes and is the foundation of many skin diseases. A relatively overlooked cell type that nevertheless plays critical roles in skin homeostasis, wound repair, and disease is the dendritic epidermal T cells (DETCs), which are also called γδT-cells. Given their varied roles in both physiological and pathological scenarios, interest in the regulation and function of DETCs has substantially increased. Moreover, their ability to regulate other immune cells has garnered substantial attention for their potential role as immunomodulators and in immunotherapies. In this article, we describe a protocol to isolate and culture DETCs and analyse them in vivo within the skin. These approaches will facilitate the investigation of their crosstalk with other cutaneous cells and the mechanisms by which they influence the status of the skin.
Graphic abstract:
Overall workflow to analyse DETCs in vitro and in vivo.
Background
The skin is composed of multiple cell types, including keratinocytes, fibroblasts, and various immune cells, that work together to provide a physical and immune barrier against the external environment. One of the skin resident immune cells, dendritic epidermal T cells (DETCs), has been shown to play important roles in tissue homeostasis, repair, and pathophysiology. In the epidermis, DETCs exhibit a dendritic morphology that allows each cell to stay in physical contact with other epidermal cells such as keratinocytes and Langerhans cells (Jameson et al., 2002; Jameson and Havran, 2007). This intercellular communication between keratinocytes and Langerhans cells is mediated by factors that activate DETCs, which in turn modulate tissue homeostasis. For example, during wound healing, damaged and stressed keratinocytes express various antigens that are recognized by DETCs, resulting in their activation and release of various cytokines such as keratinocyte growth factor-1 and 2, insulin growth factor-1, and interleukin-2 (Gustafsson et al., 2020). In addition, DETCs also secrete fibroblast growth factor 9, which mediates hair neogenesis during wound healing (Gay et al., 2013). We recently uncovered a novel role of skin resident DETCs in regulating hair follicle stem cell activity in wounded skin (Lee et al., 2017). In addition, DETCs have also been shown to play an important role in maintaining the epidermis in unwounded skin as mice lacking DETCs exhibit higher levels of epidermal apoptosis (Sharp et al., 2005). In this protocol, we provide a detailed method for the isolation and culture of DETCs to investigate the effect of various soluble factors and cell-cell contacts on DETC activation and the downstream consequence on other cutaneous cells. We also describe a method for the in vivo analysis of DETCs, which will help in understanding the intercellular communication of DETCs at a tissue level in both physiological and pathological conditions.
Materials and Reagents
10 ml serological pipette (Stem Cell Technologies, catalog number: 38004)
70 µm cell strainer (Corning, catalog number: 431751)
10 cm Petri dish (Eppendorf, catalog number: 30702118)
FACS tubes (Stem Cell Technologies, catalog number: 38007)
50 ml Falcon tube (Thermo Fisher, catalog number: 10788561)
15 ml Falcon tubes (Stem Cell Technologies, catalog number: 05860)
Ultra-low attachment culture plates (Corning, catalog number: CLS3471-24EA)
96-well dish (Eppendorf, catalog number: EP0030730011-80EA)
Kimwipes (Kimberly-Clark Kimtech Science, catalog number: 34155)
C57BL6 mice (Jackson Laboratories)
Ethanol (Sigma-Aldrich, catalog number: T4049 02870)
Fetal Bovine Serum (Gibco, catalog number: 10270106)
Phosphate Buffer Solution (PBS) (CSH Protocols, http://cshprotocols.cshlp.org/content/2006/1/pdb.rec8247)
Trypsin solution (Sigma-Aldrich, catalog number: T4049)
E media without calcium (Nowak and Fuchs, 2009)
7-Aminoactinomycin D (7-AAD) (Thermo Fisher Scientific, catalog number: A1310)
FITC isotype control (Thermo Fisher Scientific, catalog number: GM4992)
PE-Cy7 isotype control (Thermo Fisher Scientific, catalog number: 25-4714 - 80)
PE isotype control (Thermo Fisher Scientific, catalog number: 12-4714-42)
FITC anti-Vγ3 TCR (Thermo Fisher Scientific, catalog number: MHGD01)
PE-Cy7 anti-CD3ϵ (Thermo Fisher Scientific, catalog number: 25-0038-42)
PE anti-γδTCR (Thermo Fisher Scientific, catalog number: 12-9959-42)
Concanavalin A (Sigma-Aldrich, catalog number: C5275)
Glutamine (Sigma-Aldrich, catalog number: G8540)
HEPES (Sigma-Aldrich, catalog number: H3375-25G)
Sodium pyruvate (Himedia, catalog number: PCT0503)
NEAA (Merck, catalog number: 7145 M7145)
Penicillin-Streptomycin (Merck, catalog number: P4333)
β-Mercaptoethanol (ME) (Sigma-Aldrich, catalog number: M6250)
Recombinant human IL-2 (Promo Cell, catalog number: 61241)
Gentamicin (Thermo Fisher Scientific, catalog number: 15750078)
RPMI-1640 medium (Thermo Fisher Scientific, catalog number: 11875101)
Mouse IL-17 Quantikine ELISA Kit (R&D Systems, catalog number: M1700)
FGF7 (Sigma-Aldrich, catalog number: RAB0188)
TNFα (Thermo Fisher Scientific, catalog number: KHC3011)
IFN-γ (Thermo Fisher Scientific, catalog number: RAB0223)
IL-23 (Thermo Fisher Scientific, catalog number: PHC9321)
Recombinant IL-1beta (10 ng/ml) (R&D Systems, catalog number: 201-LB-005/CF)
DMEM/F-12, powder (Gibco, catalog number: 12500062)
Sodium bicarbonate (Gibco, catalog number: S5761)
Cholera toxin (Sigma-Aldrich, catalog number: C8052-.5MG)
Hydrocortisone (Sigma-Aldrich, catalog number: H0888)
Autoclaved Milli-Q distilled water
Hydrochloric acid (Sigma-Aldrich, catalog number: 320331-500ML)
Anti-CD3 (1 μg/ml) (Abcam, catalog number: 5690)
Trypan blue (Thermo Fisher Scientific, catalog number: 15250061)
Anti-JAML (Abcam, catalog number: 67843)
WST-1 reagent (Merck, catalog number 5015944001)
MTT reagent (Merck, catalog number: CT01-5)
OCT medium (Thermo Scientific, catalog number: 23-730-571)
16% paraformaldehyde (Fisher Scientific, catalog number: 50-980-487)
Triton X-100 (Thermo Scientific, catalog number: PI28313)
Keratin 5 (Abcam, catalog number: ab52635)
Anti-Ki67 (Abcam, catalog number: ab16667)
Anti-γδTCR antibody (eBioscience, catalog number: 12-5711-82)
Goat anti-Rabbit Alexa Fluor 488 (Molecular Probes, catalog number: A-11008)
Goat anti-chicken Alexa Fluor 647 (Molecular Probes, catalog number: A-21449)
DAPI (Abcam, catalog number: ab228549)
Vectashield (Vector Laboratories, catalog number: H-1500)
MOWIOL 4-88 Reagent (Sigma-Aldrich, catalog number: 475904-100GM-M)
Tris (Sigma-Aldrich, catalog number: 10708976001)
Glycerol (Sigma-Aldrich, catalog number: G5516)
Superfrost Plus slides (VWR, catalog number: 48311-703)
PBS with 2× antibiotics (see Recipes)
E media without calcium (see Recipes)
FACS Staining Buffer (see Recipes)
RPMI media (see Recipes)
0.2% Triton X-100 (see Recipes)
Blocking buffer for permeabilization (see Recipes)
4% Paraformaldehyde (PFA) (see Recipes)
70% Ethanol (see Recipes)
Mowiol (see Recipes)
Equipment
Fine forceps (Fisher Scientific, catalog number: NC9924848)
Scissors (Fisher Scientific, catalog number: 08-951-20)
Cell culture incubator (Eppendorf, model: EppendorfTM GalaxyTM 170)
Aspirator (Sigma-Aldrich, catalog number: BMSV0020-1EA)
FACS Aria (BD, model: FACSAriaTM III sorter)
Centrifuge (Eppendorf, model: 5702)
Spectrophotometer (Thermo ScientificTM GENESYSTM 20 Visible Spectrophotometer)
Cell culture incubator (Eppendorf, model: EppendorfTM GalaxyTM 170 S)
Cell counter (Thermo Fisher Scientific, model: Countess3)
7-17 DETC cell line (Boismenu and Havran, 1994)
Cryostat (Leica, model: CM1950)
-80°C freezer (Thermo Scientific, Forma Ultra-Low Temperature Upright DD Freezer)
Hydrophobic pen (Merck, catalog number: Z377821-1EA)
Humidifying chamber
Compound Binocular Microscope (Celestron Labs, model: CB2000CF)
Fluorescent microscope (Olympus, model: IX73), Confocal microscope FV 3000 5 laser (IEC60825-1:2007)
Software
ImageJ version 1.46 software
GraphPad Prism 6
Procedure
Isolation, maintenance, and proliferation of DETCs
Given the important roles of DETCs, methods to investigate the regulation and function of these cells are required. This is facilitated by the ability to isolate and establish primary cultures of DETCs and reconstitute their intercellular crosstalk with different cells in vitro.
Isolation and culture of DETCs
Isolation of DETCs from the skin via FACS (Kashem and Kaplan, 2018)
Euthanize C57BL6 pups of postnatal days 0 to 5 via decapitation or a method approved by the Institutional Animal Ethics Committee. Three to four pups are required for a 3.5 cm dish of cultured DETCs.
Clean the surface of the pup using 70% ethanol to decrease the chances of microbial contamination.
Remove the limbs and tail of each pup as close to the core body as possible using sharp scissors. Insert the scissors through the hole made by the removal of the tail and cut the skin along the dorsal midline of the body all the way to the neck.
Using forceps, grasp the skin and peel the whole skin off of the body, taking care not to tear the skin into pieces. Rinse the peeled skin by placing it in a tube with 10 ml of sterile PBS containing 2× antibiotics (see Recipe 1) for 10 min. Then remove excess PBS by blotting the skin on a tissue.
In a new 10 cm Petri dish containing 10 ml of 0.25% trypsin, place the skin with the dermis side down making contact with the solution. Avoid submerging the epidermis in the trypsin solution to prevent over-digestion of the epidermis. Spread out the curled edges using fine forceps to maximize contact of the entire dermis with the trypsin solution.
Incubate the skin in trypsin for one hour at 37°C or overnight at 4°C.
Note: Steps A7 to A9 are performed at room temperature.
Separate the epidermis from the dermis using fine forceps. The separated epidermis will appear as a thin opaque sheet. The dermis can be used to isolate dermal cells such as fibroblasts (Kashem and Kaplan, 2018). To view a detailed protocol and video of epidermal cells isolation, refer to Li et al. (2017). Cut the epidermis into small pieces with scissors. Transfer the epidermis and trypsin mixture to a new 50 ml tube.
Using a 10 ml serological pipette, repeatedly pipette the mixture of epidermis and trypsin up and down to facilitate the dissociation of the tissue into individual cells. The serological pipette might get blocked due to clumps of tissue. Tap the pipette to remove the clumps from the pipette.
Keep on pipetting until it becomes easy to pipette up and down without tapping. Pipette the solution around 10 to 15 times slowly to properly remove the clumps.
Pass the cell suspension through a 70 µm cell strainer into a new 50 ml tube. Also, pass 5 ml of E media without calcium (see Recipe 2) through the cell strainer to remove any cells that are trapped in the strainer, which would also inactivate the trypsin.
Centrifuge the cell suspension at 250 × g for 10 min in a swing bucket rotor at 4°C.
Remove the supernatant carefully using an aspirator. Wash the cells once with PBS by resuspending the cell pellet in 5 ml of PBS and centrifuging at 250 × g for 5 min each at 4°C.
Resuspend the cells in 1 ml of staining buffer (see Recipe 3) for FACS sorting.
Note: For Staining Buffer, use chelated fetal bovine serum since the presence of calcium might lead to the formation of cell clumps.
Count the number of viable cells using trypan blue staining (Figure 1).
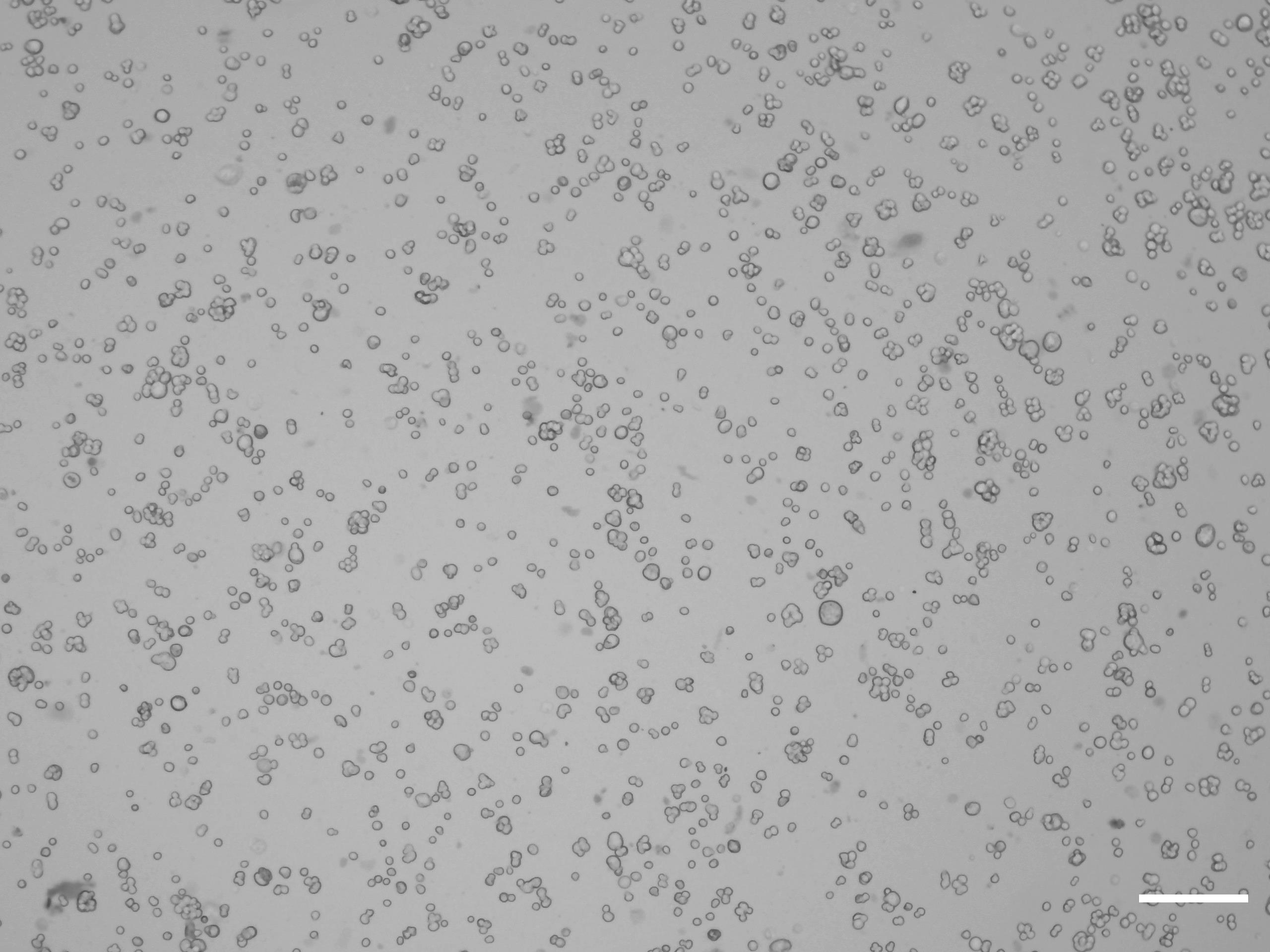
Figure 1. Preparation of cells for counting. Bright-field image of the cell suspension under the microscope for automated counting. Scale bar: 50 μm.To prevent the nonspecific binding of antibodies, incubate the cells with anti CD16/CD32 (1 µg/million cells) for 5 min at 4°C. CD16/CD32 incubation prevents nonspecific binding of immunoglobulins to FcγIII, FcγII, and possibly FcγI receptors.
From this epidermal cell suspension, make five aliquots of 50 µl each in five different FACS tubes. Add 50 µl of staining buffer to each 50 µl aliquot to match the volumes listed in Table 1 and label samples as indicated.
Note: At least 100,000 cells are required to calibrate the flow cytometer.
Table 1. Summary of control and analyte samples required for FACS analysis. To calibrate the flow cytometer, various controls are required: unstained control (#1) and single stained and isotype controls (#2-5). For samples that are to be used to isolate DETCs (#6/analyte sample), cells are stained with all antibodies and resuspended in a staining buffer with 5 µl of 7AAD (Fluorochrome used to stain non-viable cells). Samples used for isolating DETCs might need to be diluted such that the event rate on the FACS machine is between 1,000-4,000 event/s (it is ideal to start with a higher concentration and dilute it later based on the event rate).
After incubating the cells for 30 min in the dark, pellet the cells by centrifuging the tubes in a swing bucket rotor at 250 × g for 5 min at 4°C. Remove the supernatant carefully with an aspirator. Wash the cell pellet three times by resuspending it in 1 ml of staining buffer and centrifuging at 250 × g for 5 min at 4°C.
DETCs can then be isolated using FACS, as explained in detail in Badarinath et al. (2019) and Nielsen et al. (2014).
Other than primary DETCs, there is also a 7-17 DETC cell line, which can be used to study the effects of various factors on DETCs and their interactions with other cutaneous cells. The 7-17 cell line was originally established from FACS-purified DETC from AKR mice and expanded by repeated stimulation with concanavalin A (1 µg/ml) supplemented with rIL-2 (Edelbaum et al., 1995; Nielsen et al., 2014).
Notes:
Preparation of samples for flow cytometry and usage of the machine are complex processes and beyond the scope of this chapter. Before planning the FACS experiment, one should be familiar with the general background and theory of flow cytometry (Shapiro, 2003).
For more details on the gating strategy while sorting, please refer to Wohn et al. (2014) and Havran et al. (1989).
Culture of primary DETCs and 7-17 DETC cell line
Both primary DETCs isolated by FACS and 7-17 DETC stable cell lines are cultured in RPMI 1640 media at 37°C, 5% CO2. The RPMI medium is supplemented with 10% FBS, 2 mM glutamine, 500 µl of Penicillin-Streptomycin (100×), 50 µM β-mercaptoethanol, 25 mM HEPES, 1 mM Na pyruvate, 100 µM nonessential amino acids, and 20 U/ml recombinant human IL-2 (Sharp et al., 2005; Nielsen et al., 2014).
Primary DETCs and DETC cell lines can be cultured as suspension cultures by using ultra-low attachment culture plates. Every two days, remove half of the media by tilting the dish gently, allowing the cells to settle down and replenish with fresh media.
Once cells become 70% confluent, passage them by collecting the total cell suspension into a 15 ml tube and centrifuge the cells at 300 × g for 5 min at room temperature. Remove the supernatant gently and resuspend the cells in 1 ml of fresh media. From this, add 200 µl to a new 10 cm dish containing 10 ml of fresh growth media and culture them in the same way as described above.
In vitro cultures of DETCs are a useful platform to study their interactions with different cell types and the effect of various soluble factors. For example, DETCs can be activated by either cytokines secreted from neighbouring cells or by direct cell-cell interactions. It has also been observed that DETCs are activated by co-culturing them with hair follicle stem cells (Badarinath et al., 2019).
Note: While culturing different cell types in co-culturing experiments, we should always be careful about the growth conditions of different cell types as inappropriate conditions for culturing any of the cell types can affect the cells in various ways such as stress, proliferation, and apoptosis.
We can study the effect of various soluble secreted factors on the activation of DETCs. One method utilizes conditioned media from skin explants in which researchers examined the effect of IL-1α secreted from wounded keratinocytes on the activation of DETCs and is explained in detail in Lee et al. (2017) and Badarinath et al. (2019). Controls required for this experiment are DETCs treated with recombinant IL-1α and isolated DETCs from IL-1 receptor KO animals treated with IL-1α. The isolated cells can be utilized in experiments to determine the soluble and intercellular signals that affect both DETC behaviour (such as proliferation and activation) as well as its impact on neighbouring cells in the skin.
Notes:
For conditioning, use the same medium in which the subsequent culture experiments will be performed. Avoid serum in media for these experiments as serum will have its own effect on the cells.
As an alternative control for DETC activation, these cells can be treated with recombinant IL-23 (10 ng/ml), recombinant IL-1beta (10 ng/ml), anti-CD3 (1 μg/ml), or combinations of these and scored for activation by measuring the expression of IL-17, FGF7, TNFα, and IFN-γ by ELISA or transcript levels as mentioned above.
Proliferation assays for activated DETCs in vitro
Elevated proliferation is one of the hallmarks of activated DETCs and can thus be used as a readout for activation. In addition to proliferation, other biomarkers of activated DETCs include the expression and secretion of various cytokines such as IL-17, FGF-7, TNFα, and IFN-γ. These secreted cytokines can be detected after 48 h using an ELISA kit (Nielsen et al., 2014) or are evident at the transcript levels after 24 h of treatment (Lee et al., 2017).
Effect of secreted factors on DETCs
For proliferation assays, culture DETCs as explained earlier. When cells are 70% confluent, collect the cells in a 15 ml Falcon tube and centrifuge at 300 × g for 5 min at room temperature.
Dilute the control and test conditioned media 1:3 with fresh RPMI media.
Note: Preparation of conditioned media is explained in detail in Badarinath et al. (2019).
Resuspend the cells in 100 µl of respective conditioned media and, from those, plate 30,000 cells in each well of a 96-well dish.
Incubate the cells in 100 µl of conditioned media for 24 to 48 h at 37°C.
After incubation, remove the media containing DETCs and quantify proliferation at different time intervals such as 24, 48, and 72 h.
There are various assays available by which you can count the number of cells for proliferation assays after treatment with various stimuli such as the MTT cell proliferation assay, WST-1 cell proliferation assay, and trypan blue cell counting.
Effect of various factors secreted by DETCs on hair follicle stem cells
Note: Isolate primary hair follicle stem cells as previously described in Nowak and Fuchs (2009).
Treat DETCs with conditioned media from control or test skin/epidermal explants animal for 16-24 h.
Collect the suspension culture in a 15 ml Falcon tube and centrifuge at 300 × g for 5 min. Collect the supernatant and discard the cell pellet.
Dilute the conditioned media 1:5 with fresh E-media.
Incubate hair follicle stem cells with diluted conditioned media for 24-48 h at 37°C, 7% CO2.
Count the cells at different time intervals over the 24-48 h time period.
In vivo analysis of DETC activation
The tissue microenvironment strongly dictates the regulation and function of DETCs. Under homeostatic conditions, DETCs have a distinctive dendritic morphology, but after injury or stress, DETCs proximal to the wound site acquire a rounded morphology and transiently lose their dendrites. In addition to morphological changes, another marker of DETC activation is an increased proliferative index. Upon activation, DETCs release certain cytokines that play a significant role in maintaining the protective physical and immune barrier of the murine skin. A variation in DETC function can aggravate skin-related autoimmune diseases, impede tumour eradication, or disrupt proper wound healing (Cruz et al., 2018). Hence, in vivo analysis of DETCs advances our understanding of the function of these cells in both physiological conditions such as wound healing and a variety of pathological scenarios including fibrosis, inflammatory diseases, and carcinomas.
Data analysis
Sectioning of mouse skin
To embed skin in OCT medium, follow the protocol described in Gund et al. (2021).
Section the frozen blocks as described in Fischer et al. (2008).
Store the sections on the charged slides at -80°C.
Notes:
Collect the skin from the same region of the mice to compare the DETCs between the control and the test animal. It is known that there is heterogeneity of DETCs in different regions of the mouse skin.
Since DETCs are dendritic in morphology when inactive, it is better to have thicker sections to visualize the morphology and quantify dendrites. Hence, take sections of ≥10 µm thickness.
Immunofluorescence assay for γδTCR and Ki67
Remove the frozen slides with skin sections from the -80°C freezer and thaw them at room temperature for at least a minute (but not longer than 5 min).
Using a kimwipe, carefully remove the condensation around the skin tissues. Place the slides in a humidifying chamber (Gund et al., 2021).
Make a hydrophobic barrier around the tissue section using a hydrophobic pen. To fix the sections, add 50-100 µl of 4% PFA per tissue section for 10 min at room temperature. Make sure the tissue sections are completely covered with PFA.
Notes:
This allows you to minimise the volume of buffers and antibodies being used and gives the ability to differentially stain multiple skin sections on one slide.
Make sure to read the antibody datasheet to utilize the appropriate fixative. This protocol is described for the antibodies mentioned in the reagents section.
Do not exceed a fixation time of more than 10 min for tissues less than 10 µm thick as over-fixing leads to excess cross-linking of antigens and can produce false negative results.
Thicker sections must be fixed for a longer time – An overnight incubation at 4°C is recommended.
Aspirate the 4% PFA after 10 min of incubation. Wash the sections thoroughly with 50-100 µl of 1× PBS for 5 min each three times.
Note: Aspiration can be avoided if the sections are loosely attached to the slide. Instead, one can remove the fixative/buffer using a pipette.
Add enough blocking buffer to cover the tissue sections to block nonspecific interactions for 1 h at room temperature.
Note: Freeze thawing of the section causes permeabilization of the plasma membrane and the integrity of the membrane proteins is maintained. Hence, additional permeabilization steps with any harsh detergents such as Tween-20 or Triton-X should be avoided as this can disrupt membrane proteins, especially if left for too long.
Incubate the sections with primary antibody (γδTCR to mark DETCs and Ki67 to mark the proliferating cells) diluted in the blocking buffer (refer to table 2 for dilutions). Add 50-100 µl of the diluted primary antibody to each section and incubate overnight at 4°C in the humidifying chamber.
Note: Make sure that the hydrophobic barrier is intact. If not, mark the boundaries again with a hydrophobic pen around the sections before adding the primary antibody.
Aspirate the primary antibody and wash the sections with 1× PBS for 5 min each three times.
Add 50-100 µl of secondary antibody (refer to Table 2 for the antibody dilutions) diluted in the blocking buffer onto the sections and incubate for 20 min at room temperature in the humidifying chamber.
Note: γδTCR antibody used in this protocol is a conjugated antibody and does not require a secondary antibody.
Aspirate the secondary antibody and wash the sections with 1× PBS for 5 min each three times.
Mount the sections with Vectashield mounting media.
Note: Sections can be mounted using alternative mounting media such as Mowiol or 80% glycerol.
Image the slides under a fluorescent microscope.
Note: Though the signal is optimal if imaged immediately after staining, the stained sections can be stored at 4°C before imaging the slides.
Alternative method: If using other antibodies along with γδTCR that require additional permeabilization of the tissue, follow the procedure below:
For staining of γδTCR, follow the protocol in section B up to step 8.
Repeat steps B1 to B5 on the sections.
Add 50 µl of permeabilization buffer (0.25% Triton X-100 in PBS) and incubate for 15 min at room temperature.
Note: The concentration of Triton X-100 used for permeabilization should be determined for each antigen of interest.
Block the sections with a blocking buffer containing goat serum and 0.1% Triton X-100 for 1 h at room temperature.
Note: Blocking buffer should contain heat-inactivated normal serum from the same species as the host of the secondary antibody.
Incubate the sections with primary antibody diluted in the blocking buffer for 2 h at room temperature.
Follow steps B8 to B12 from section B of the protocol.
Table 2. Summary of various primary and secondary antibodies used in section B along with their respective dilutions
Imaging acquisition and analysis
Image the stained sections under a fluorescent microscope. To quantify the number of proliferating DETCs, lower magnification images of 10× and 20× will suffice. To visualize the morphological differences between the inactive and active DETCs, higher magnification (>40×) images are required.
Note: z-stack images are recommended to fully visualize the dendrites of DETCs that spread throughout the epidermis in multiple planes.
Under homeostatic conditions, DETCs are generally inactive and restricted to the basal layer of the epidermis and the upper region of the hair follicle (Lee et al., 2009). As their name indicates, they possess a dendritic morphology. On the other hand, conditions in which DETCs are activated, such as a wound and mouse models of atopic dermatitis (Lee et al., 2009 and 2017) and squamous cell carcinoma (Du et al., 2010; De Craene et al., 2014), the cells lose their dendritic extensions and appear more rounded in morphology (Lee et al., 2009; Du et al., 2010). It has been previously reported that caspase 8 cKO mice, a model for atopic dermatitis (Lee et al., 2009; Du et al., 2010), exhibits activated DETCs (Lee et al., 2017). In this protocol, we demonstrate that this activation of DETCs (both morphological changes and increased proliferation) is conserved across mouse models with a strong inflammatory phenotype. For instance, we have observed that a mouse model for cutaneous squamous cell carcinoma (K14 Snail transgenic mice) (Du et al., 2010) also exhibits activation of DETCs as early as the neonatal stage (Figure 2 and Figure 3).
Figure 2. Visualization of activated DETCs in WT and Snail transgenic skin. Immunofluorescence assay for Ki67 (green) to depict proliferation, γδTCR (red) to mark DETCs and K5 (blue) to mark the basal layer of epidermis on WT and SnTg skin section to observe the activation of DETCs. Scale bars: 50 µm.
Figure 3. Morphological changes upon activation. Round morphology of DETCs marked by γδTCR (red) in Snail transgenic skin shows signs of activation compared to the dendritic morphology of DETCs in WT skin. Scale bars: 25 µm.When activated, DETCs proliferate and hence are Ki67 positive. Therefore, to quantify the number of proliferating DETCs, we counted the number of γδTCR positive cells that are positive for Ki67 across the epidermis of postnatal day 7 – WT and K14 Snail transgenic mice. The number of DETCs across the epidermis of WT and Snail transgenic skin is variable. Hence, to ensure consistency in quantification among different backgrounds, we counted all the γδTCR positive cells per 1 mm skin section.
The percentage of DETCs that are Ki67 positive and negative can be quantified by using ImageJ as follows: Open the merged image of γδTCR and Ki67 on ImageJ software > Main menu > Plugin > Cell counter > Initialize > Type 1. Click on the cells that are positive for both γδTCR and Ki67. This will give the number of DETCs that are proliferating. To count the number of DETCs that are negative for Ki67, go to ImageJ software > Main menu > Plugin > Cell counter > Initialize > Type 2. The percentage of proliferating and non-proliferating DETCs can be calculated using the following formulae:

The proportions of DETCs that are Ki67 positive and negative in WT and K14 Snail transgenic skin are as shown in Figure 4.
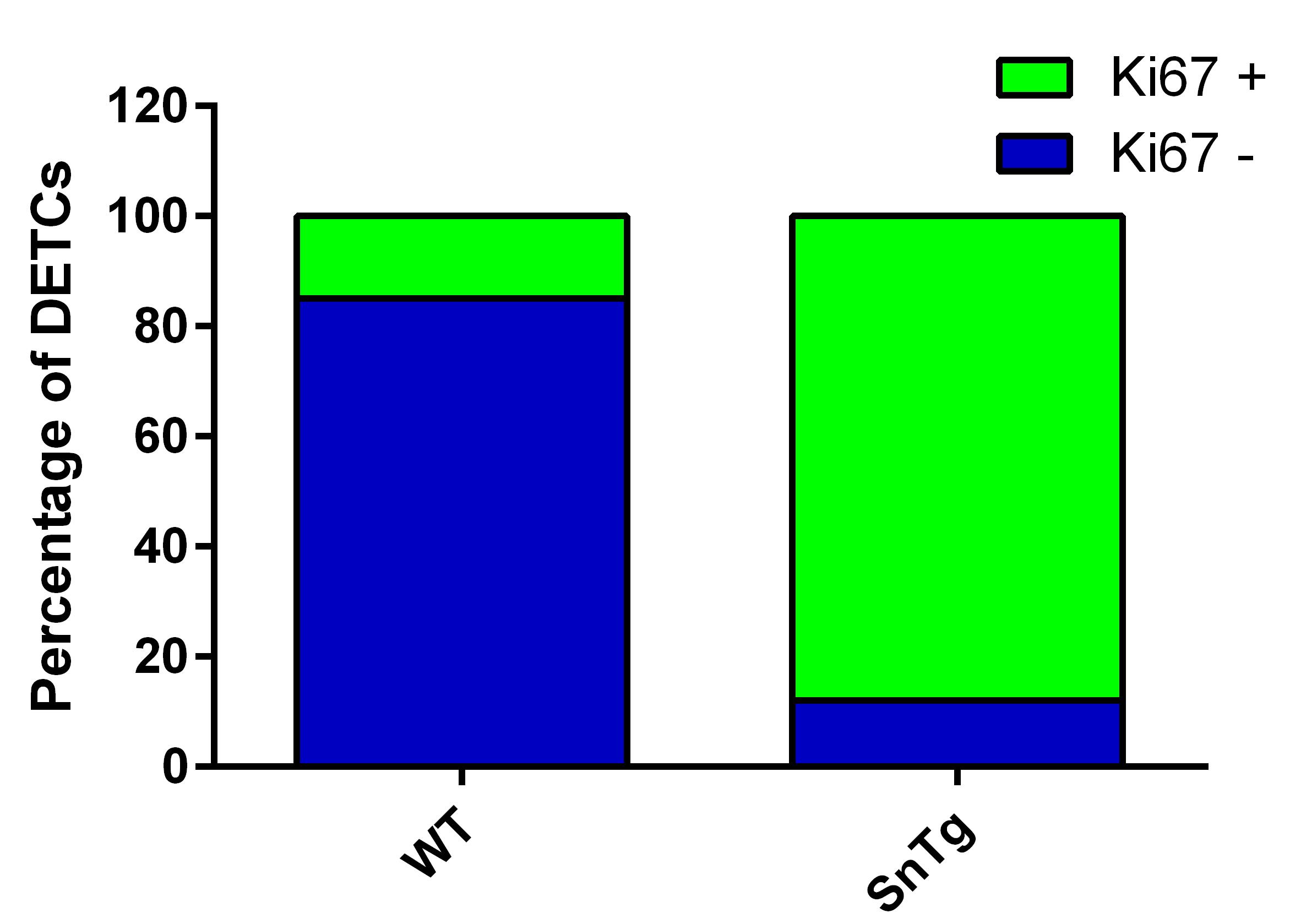
Figure 4. Quantification of activated DETCs in WT and SnTg skin. Percentage plot depicting the proportion of proliferating DETCs, which suggests that Snail transgenic skin has more proliferating DETCs than WT skin.To quantify the circularity of the DETCs in WT and SnTg epidermis, ImageJ software is used. Taking the image of just the DETCs (γδTCR positive cells) > Mark the outline using the polygon selection tool on ImageJ for each DETC > Analyze > Set measurement > Area and shape description (select “Shape descriptor” parameter to measure and display) > Analyze > Measure or Ctrl+M. This will display a table with measurements. Perform this step for all DETCs in WT and SnTg skin sections. Circularity value of 1 indicates that a cell is a perfect circle, and values that approach 0 indicate that the cell is elongated. Here in this protocol, we have used a threshold circularity value of ≥ 0.5 to call a DETC active. A graph plotted using these measurements (Figure 5) shows that the DETCs in K14 Snail transgenic skin are more circular, indicating activation compared to that in the WT skin.
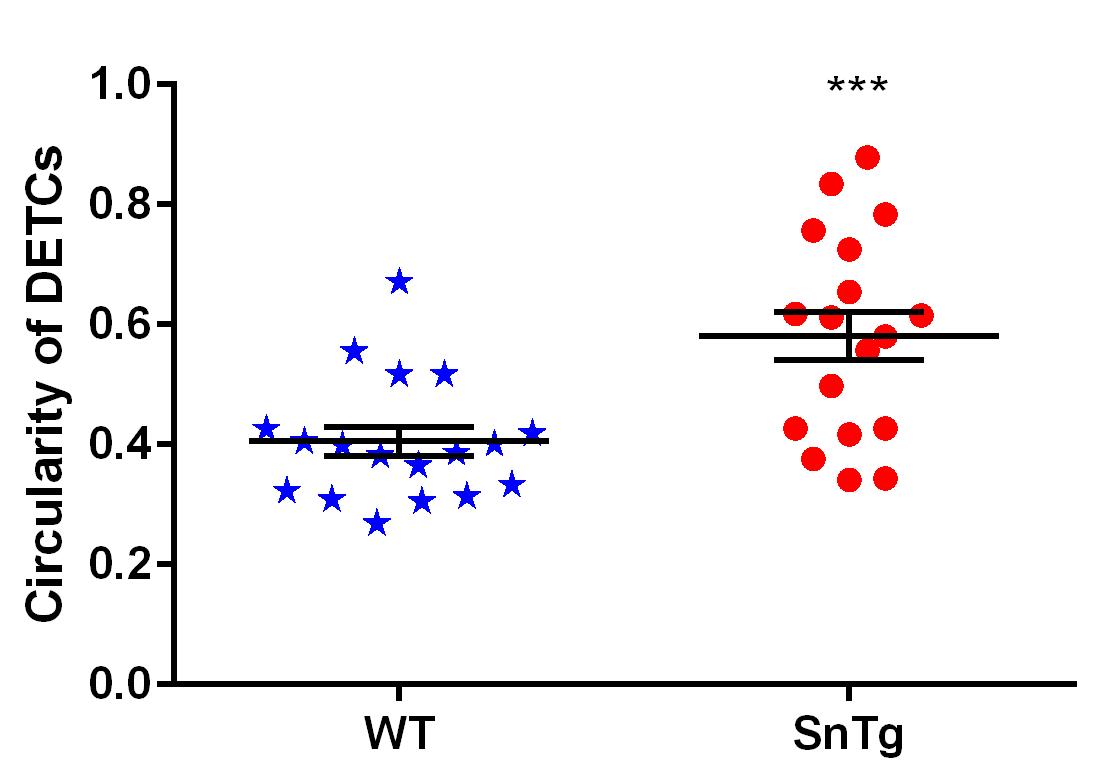
Figure 5. Quantification of morphological changes observed in DETCs. Activated DETCs show morphological changes from a dendritic to circular shape, which were quantified using ImageJ software. Scatter plot shows that DETCs in Snail transgenic epidermis are more circular (circularity value ≥ 0.5) compared to DETCs in WT epidermis. Data represent the values ± SEM, ***P < 0.0001, based on the Student’s t-test.
Recipes
PBS with 2× antibiotics
Add 2 ml of Penicillin-Streptomycin (100×) to 10 ml of 10× PBS and bring volume up to 100 ml with ddH2O.
E media without calcium
Prepare E Media without Calcium for epidermal keratinocytes as described in Nowak and Fuchs (2009).
FACS Staining Buffer
2 ml of chelated Fetal Bovine Serum
1 ml of Penicillin-Streptomycin (100×)
100 µl of 50 mg/ml Gentamicin
Add to 5 ml of 10× PBS
Bring volume up to 50 ml with ddH2O
RPMI media
10% FBS
2 mM glutamine
500 µl of Penicillin-Streptomycin (100×)
50 µM β-mercaptoethanol
25 mM HEPES
1 mM Na pyruvate
100 µM nonessential amino acids
20 U/ml recombinant human IL-2
Bring volume up to 500 ml with RPMI media
0.2% Triton X-100
Add 2 ml of 100% Triton X-100 to 100 ml of 10× PBS and bring volume up to 1 L with ddH2O.
Blocking buffer for permeabilization
Add 0.5 ml of goat serum (or serum originating from the same species as the secondary fluorescent conjugated antibody) and 10 µl of Triton X-100 in 1 ml of 10× PBS and bring the volume to 10 ml with ddH2O.
4% Paraformaldehyde (PFA)
Dilute the 16% paraformaldehyde in 10× PBS by adding 12.5 ml of 16% of PFA to 5 ml of 10× PBS and bring the volume to 50 ml by adding ddH2O.
70% Ethanol
Dilute 100% ethanol in distilled water by adding 700 ml of 100% ethanol in 300 ml distilled water.
Mowiol
Add 2.4 g of MOWIOL® 4-88 to 4.75 ml of glycerol and stir to mix.
Add 6 ml of water and stir for several hours at room temperature.
Add 12 ml of 0.2 M Tris (pH 8.5) and heat to 50°C for 10 min with occasional mixing.
After the MOWIOL 4-88 dissolves, clarify the solution by centrifugation at 5,000 × g for 15 min.
Acknowledgments
The authors would like to thank Binita Dam and Johan Ajnabi for critical review of the manuscript. Work in the Jamora Laboratory is supported by inStem core funds and with past funding from the Department of Biotechnology of the Government of India (BT/PR8738/AGR/36/770/2013); the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), NIH (5R01AR053185-03); and the American Cancer Society (15457-RSG-08-164-01-DDC). The animal work was partially supported by the National Mouse Research Resource (NaMoR) (BT/PR5981/MED/31/181/2012; 2013-2016) from the Department of Biotechnology. We thank the staff of the NCBS/inStem Animal Care and Resource Centre for assistance with animal husbandry, and the NCBS/inStem Central Imaging and Flow Cytometry Facility (CIFF) for help with image acquisition. We dedicate this protocol to the memory of our colleague and friend Professor Wendy Havran who was a pioneer and leader in the field of γδT-cell biology.
This protocol was utilized in the following manuscript: Lee, P., Gund, R., Dutta, A., Pincha, N., Rana, I., Ghosh, S., Witherden, D., Kandyba, E., MacLeod, A., Kobielak, K., Havran, W. L. and Jamora, C. (2017). Stimulation of hair follicle stem cell proliferation through an IL-1 dependent activation of γδT-cells. Elife 6: e28875.
Competing interests
No conflicts of interest to be declared.
Ethics
All work with animals was carried out at the NCBS/inStem Animal Care and Resource Centre and protocols were approved by the Institutional Animal Ethics Committee.
References
- Badarinath, K., Dutta, A., Hegde, A., Pincha, N., Gund, R. and Jamora, C. (2019). Interactions Between Epidermal Keratinocytes, Dendritic Epidermal T-Cells, and Hair Follicle Stem Cells. Methods Mol Biol 1879: 285-297.
- Boismenu, R. and Havran, W. L. (1994). Modulation of epithelial cell growth by intraepithelial γδ T cells. Science 266(5188): 1253-1255.
- Cruz, M. S., Diamond, A., Russell, A. and Jameson, J. M. (2018). Human αβ and γδ T Cells in Skin Immunity and Disease. Front Immunol 9: 1304.
- De Craene, B., Denecker, G., Vermassen, P., Taminau, J., Mauch, C., Derore, A., Jonkers, J., Fuchs, E. and Berx, G. (2014). Epidermal Snail expression drives skin cancer initiation and progression through enhanced cytoprotection, epidermal stem/progenitor cell expansion and enhanced metastatic potential. Cell Death Differ 21(2): 310-320.
- Du, F., Nakamura, Y., Tan, T. L., Lee, P., Lee, R., Yu, B. and Jamora, C. (2010). Expression of snail in epidermal keratinocytes promotes cutaneous inflammation and hyperplasia conducive to tumor formation. Cancer Res 70(24): 10080-10089.
- Edelbaum, D., Mohamadzadeh, M., Bergstresser, P. R., Sugamura, K. and Takashima, A. (1995). Interleukin (IL)-15 promotes the growth of murine epidermal γδ T cells by a mechanism involving the β- and γc-chains of the IL-2 receptor. J Invest Dermatol 105(6): 837-843.
- Fischer, A. H., Jacobson, K. A., Rose, J. and Zeller, R. (2008). Cryosectioning tissues. CSH Protoc 2008: pdb prot4991.
- Gay, D., Kwon, O., Zhang, Z., Spata, M., Plikus, M. V., Holler, P. D., Ito, M., Yang, Z., Treffeisen, E., Kim, C. D., Nace, A., Zhang, X., Baratono, S., Wang, F., Ornitz, D. M., Millar, S. E. and Cotsarelis, G. (2013). Fgf9 from dermal γδ T cells induces hair follicle neogenesis after wounding. Nat Med 19(7): 916-923.
- Gund, R., Zirmire, R., J, H., Kansagara, G. and Jamora, C. (2021). Histological and Immunohistochemical Examination of Stem Cell Proliferation and Reepithelialization in the Wounded Skin. Bio-protocol 11(2): e3894.
- Gustafsson, K., Herrmann, T. and Dieli, F. (2020). Editorial: Understanding γδ T Cell Multifunctionality - Towards Immunotherapeutic Applications. Front Immunol 11: 921.
- Havran, W. L., Grell, S., Duwe, G., Kimura, J., Wilson, A., Kruisbeek, A. M., O'Brien, R. L., Born, W., Tigelaar, R. E. and Allison, J. P. (1989). Limited diversity of T-cell receptor gamma-chain expression of murine Thy-1+ dendritic epidermal cells revealed by V gamma 3-specific monoclonal antibody. Proc Natl Acad Sci U S A 86(11): 4185-4189.
- Shapiro, H. M. (2003). Practical Flow cytometry. John Wiley & Sons, Inc.
- Jameson, J. and Havran, W. L. (2007). Skin γδ T-cell functions in homeostasis and wound healing. Immunol Rev 215: 114-122.
- Jameson, J., Ugarte, K., Chen, N., Yachi, P., Fuchs, E., Boismenu, R. and Havran, W. L. (2002). A role for skin γδ T cells in wound repair. Science 296(5568): 747-749.
- Kashem, S. W. and Kaplan, D. H. (2018). Isolation of Murine Skin Resident and Migratory Dendritic Cells via Enzymatic Digestion. Curr Protoc Immunol 121(1): e45.
- Lee, P., Lee, D. J., Chan, C., Chen, S. W., Ch'en, I. and Jamora, C. (2009) Dynamic expression of epidermal caspase 8 simulates a wound healing response. Nature 458(7237):519-23.
- Lee, P., Gund, R., Dutta, A., Pincha, N., Rana, I., Ghosh, S., Witherden, D., Kandyba, E., MacLeod, A., Kobielak, K., Havran, W. L. and Jamora, C. (2017). Stimulation of hair follicle stem cell proliferation through an IL-1 dependent activation of γδT-cells. Elife 6: e28875.
- Li, F., Adase, C. A. and Zhang, L. J. (2017). Isolation and Culture of Primary Mouse Keratinocytes from Neonatal and Adult Mouse Skin. J Vis Exp (125): 56027.
- Nielsen, M. M., Lovato, P., MacLeod, A. S., Witherden, D. A., Skov, L., Dyring-Andersen, B., Dabelsteen, S., Woetmann, A., Odum, N., Havran, W. L., Geisler, C. and Bonefeld, C. M. (2014). IL-1beta-dependent activation of dendritic epidermal T cells in contact hypersensitivity. J Immunol 192(7): 2975-2983.
- Nowak, J. A. and Fuchs, E. (2009). Isolation and culture of epithelial stem cells. Methods Mol Biol 482: 215-232.
- Sharp, L. L., Jameson, J. M., Cauvi, G. and Havran, W. L. (2005). Dendritic epidermal T cells regulate skin homeostasis through local production of insulin-like growth factor 1. Nat Immunol 6(1): 73-79.
- Wohn, C. T., Pantelyushin, S., Ober-Blobaum, J. L. and Clausen, B. E. (2014). Aldara-induced psoriasis-like skin inflammation: isolation and characterization of cutaneous dendritic cells and innate lymphocytes. Methods Mol Biol 1193: 171-185.
Article Information
Copyright
![]() Rana et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Rana et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Rana, I., Badarinath, K., Zirmire, R. K. and Jamora, C. (2021). Isolation and Quantification of Mouse γδT-cells in vitro and in vivo. Bio-protocol 11(17): e4148. DOI: 10.21769/BioProtoc.4148.
- Lee, P., Gund, R., Dutta, A., Pincha, N., Rana, I., Ghosh, S., Witherden, D., Kandyba, E., MacLeod, A., Kobielak, K., Havran, W. L. and Jamora, C. (2017). Stimulation of hair follicle stem cell proliferation through an IL-1 dependent activation of γδT-cells. Elife 6: e28875.
Category
Immunology > Immune cell isolation > Maintenance and differentiation
Immunology > Immune cell staining > Flow cytometry
Cell Biology > Cell imaging > Fixed-tissue imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.