- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Novel Method to Map Small RNAs with High Resolution
Published: Vol 11, Iss 16, Aug 20, 2021 DOI: 10.21769/BioProtoc.4128 Views: 4882
Reviewed by: Alessandro DidonnaIgnacio LescanoAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2020
Advertisement





Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Analyzing cellular structures and the relative location of molecules is essential for addressing biological questions. Super-resolution microscopy techniques that bypass the light diffraction limit have become increasingly popular to study cellular molecule dynamics in situ. However, the application of super-resolution imaging techniques to detect small RNAs (sRNAs) is limited by the choice of proper fluorophores, autofluorescence of samples, and failure to multiplex. Here, we describe an sRNA-PAINT protocol for the detection of sRNAs at nanometer resolution. The method combines the specificity of locked nucleic acid probes and the low background, precise quantitation, and multiplexable characteristics of DNA Point Accumulation for Imaging in Nanoscale Topography (DNA-PAINT). Using this method, we successfully located sRNA targets that are important for development in maize anthers at sub-20 nm resolution and quantitated their exact copy numbers.
Graphic abstract:
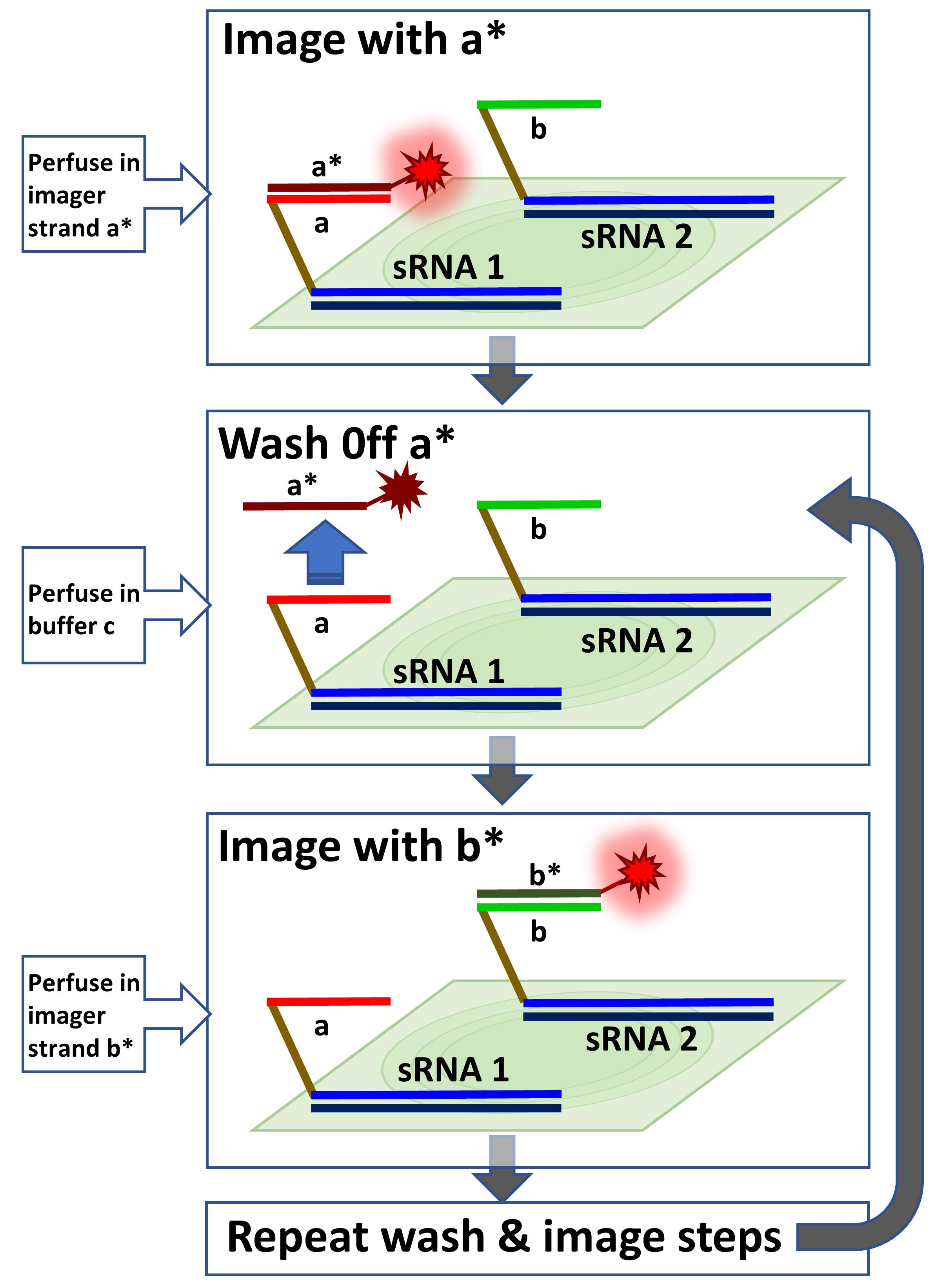
Multiplexed sRNA-PAINT. Multiple Vetting and Analysis of RNA for In Situ Hybridization (VARNISH) probes with different docking strands (i.e., a, b, …) will be hybridized to samples. The first probe will be imaged with the a* imager. The a* imager will be washed off with buffer C, and then the sample will be imaged with b* imager. The wash and image steps can be repeated sequentially for multiplexing.
Background
Light diffraction limits restrict imaging fluorophores closer than ~200 nm (Hell, 2009). Several imaging technologies have been created to overcome this limitation by exploiting transient fluorophore ON and OFF states (blinking), including stimulated emission depletion (STED) (Blom and Widengren, 2017), photoactivated localization microscopy (PALM) (Shroff et al., 2008), structured illumination microscopy (SIM) (Heintzmann and Huser, 2017), and stochastic optical reconstruction microscopy (STORM) (Bates et al., 2013). STORM increases the imaging resolution to around 30 nm (Xu and Liu, 2019); however, it relies on the stochastic blinking of fluorophores. As a result, multiplexing is often limited by the choice of fluorophores with ideal photophysical properties, including high photon numbers, low duty cycles, and high survival fractions, such as Alexa Fluor 647 and Dyomics 654 (Dempsey et al., 2011). Moreover, image quantitation is often challenging due to the unpredictability of the blinking properties of the chosen fluorophores and photobleaching (Schnitzbauer et al., 2017; Huang et al., 2020).
DNA Point Accumulation for Imaging in Nanoscale Topography (DNA-PAINT) was invented for ultra-high, sub-5 nm spatial detection of target protein molecules (Jungmann et al., 2014; Schnitzbauer et al., 2017; Strauss and Jungmann, 2020). It utilizes the base-pairing nature of single-stranded DNA molecules to their reverse complement strands. In the DNA-PAINT design, an approximately 10 nucleotide (nt)-long docking strand is linked to a target molecule via a biotin-streptavidin bridge or DBCO-sulfo-NHS ester chemical reaction (Schnitzbauer et al., 2017). An imager strand, conjugated to a fluorophore, is perfused in during the imaging process. The melting temperature (Tm) of the imager and docking strands is designed to be ~15°C. As a result, the imager strand-docking strand pairs will constantly bind and unbind at room temperature, which contributes to the blinking events, similar to the other super-resolution microscopy imaging techniques. The predictability of the binding duration and unlimited docking-imager strand combinations make DNA-PAINT ideal for quantitation and multiplexing (Jungmann et al., 2016).
sRNA-PAINT was created based on DNA-PAINT to detect small RNAs (sRNAs) that are usually ~10 nm in size (Huang et al., 2020). Our method preserves the nanometer resolution, quantitation, and multiplexing advantages of DNA-PAINT. In addition, we have introduced efficient and specific detection of sRNA targets with locked nucleic acid (LNA) (McTigue et al., 2004) and probe immobilization with 1-ethyl-1-(3-dimethylaminopropyl) carbodiimide (EDC) (Pena et al., 2009). sRNA-PAINT can be used for the multiplexed detection of sRNAs, their precursor mRNAs, and proteins in the sRNA biogenesis pathway when employed in combination with DNA-PAINT for understanding the relative localization patterns of small RNAs and their biogenesis machinery.
Materials and Reagents
20-ml glass scintillation vials (Electron Microscopy Sciences, catalog number: 72632)
50-ml Falcon tubes (Thermo Fisher Scientific, Corning, catalog number: 14-432-22)
0.5 M ethylenediaminetetraacetic acid (EDTA) solution (pH 8.0) (Thermo Fisher Scientific, Fisher BioReagentsTM, catalog number: BP2482-500)
1 M Tris-HCl solution (pH 8.0) (Thermo Fisher Scientific, catalog number: AAJ22638AP)
16% paraformaldehyde (Electron Microscopy Sciences, catalog number: RT15710) (store at 4°C)
1-ethyl-3- (3-dimethylaminopropyl) carbodiimide (EDC) (store at -20°C with a nitrogen seal)
200 proof ethanol (Thermo Fisher Scientific, Fisher BioReagentsTM, catalog number: 64-17-5) (store in a flammable cabinet)
Deionized formamide (Millipore Sigma, catalog number: S4117) (store at 4°C)
Denhardt’s solution (50×) (Millipore Sigma, catalog number: D2532) (store at -20°C)
Dextran sulfate sodium salt (Millipore Sigma, catalog number: 67578) (store at 4°C)
Histoclear II (VWR, Electron Microscopy Sciences, catalog number: 101412-878)
NaCl (Millipore Sigma, catalog number: NIST975A)
Paraffin wax pellets (Electron Microscopy Sciences, Paraplast X-tra, catalog number: 19214)
Saline-sodium citrate (SSC) buffer (20×) (Thermo Fisher Scientific, Fisher BioReagentsTM, catalog number: BP1325)
Sodium phosphate (Millipore Sigma, catalog number: 324283)
tRNA (Millipore Sigma, catalog number: R1753) (store at -20°C)
Ethanol gradient solutions (see Recipes)
PHEM buffer (2×) (see Recipes)
Fixation buffer (see Recipes)
Enzyme solution (see Recipes)
Glycine solution (see Recipes)
TE buffer (see Recipes)
TE-protease solution (see Recipes)
Dextran sulfate solution (50%) (see Recipes)
Highly abundant sRNA (see Recipes)
Hybridization salt (see Recipes)
Hybridization buffer (see Recipes)
TBS buffer (pH 7.5) (see Recipes)
tRNA solution (100 mg/ml) (see Recipes)
Buffer C (see Recipes)
EDC buffer (see Recipes)
EDC solution (see Recipes)
Equipment
DH40iL culture dish incubation system (Warner Instruments, model: 640388)
Dissecting microscope (Zeiss M2BIO Dissecting Microscope)
Flat-bottomed containers (Pyrex storage, 6 cup-1.5L)
Fluigent Aria perfusion (Fluigent, Parts no: CB_SY_AR_1)
Forceps (Electron Microscopy Sciences, catalog number: 72997-20)
Glass staining dishes (Electron Microscopy Sciences, catalog number: 71426-DL)
Grace Bio-Labs HybriSlip hybridization covers (Millipore Sigma, catalog number: 726024)
Heat incubator (Fisher Scientific Isotemp incubator)
Heat block for 1.5-ml tubes (BioExpress, Mini Dry Bath, model number: AS-BSH200-471)
Hybridization oven (UVP HB-1000 hybridizer, model number: 95-0030-01)
Masterflex C/L peristaltic pump (60 RPM) (Cole-Parmer Instrument, model: 77120-62)
Paraffin microtome (Microm, model: MRK0403643)
Parafilm (Thermo Fisher Scientific, BemisTM, catalog number: PM998)
Plastic wrap (Thermo Fisher Scientific, FisherbrandTM Clear Plastic Wrap, catalog number: 12-640)
Quick-release magnetic chamber for 25-mm low-profile round coverslips (Warner Instruments, model: 641943)
Razor blades (Thermo Fisher Scientific, FisherbrandTM Razor Blades, catalog number: 22-305654)
Slide warmer (Thermo Fisher Scientific, FisherbrandTM Slide Drying Bench, catalog number: 11-474-470)
Stainless-steel glass rack (Electron Microscopy Sciences, catalog number:71426-R)
Superfrost glass slides (Thermo Fisher Scientific, FisherbrandTM Tissue Path SuperfrostTM Plus Gold Slides, catalog number: 15-188-48)
Tissue embedding and processing cassettes (Electron Microscopy Sciences, catalog number: 70070)
Vacuum bell jar (Jelo Tech, model number: F42400-2221 with a Nisshin vacuum gauge, model number: B53595)
Vacuum pump (Welch, model number: K48ZZMEM31)
ValveLink8.2 Perfusion System (AutoMate Scientific, Berkeley, CA)
Watercolor paintbrushes (#0)
Wide Spectral Band 600 ± 100 nm Gold Fiducials coverglass (600-100AuF; Hestzig LLC, Leesburg, VA)
Single-molecule localization microscope: Zeiss Elyra PS.1 super-resolution microscope (Carl Zeiss, Lberkochen, Germany) or Andor Dragonfly (Oxford Instruments, Abingdon, United Kingdom).
Software
ImageJ ThunderSTORM (https://zitmen.github.io/thunderstorm/) (Ovesny et al., 2014)
Picasso (https://github.com/jungmannlab/picasso) (Schnitzbauer et al., 2017)
Procedure
Sample preparation
Dissect the samples using a dissecting microscope with forceps and razor blades. This step can be omitted for cell cultures.
Prepare the fixation buffer immediately prior to dissection. Place the dissected samples in an adequate amount of fixation buffer in 20-ml glass scintillation vials at room temperature. The volume of the fixative needs to be at least 15 times the volume of the sample.
For plant samples, vacuum-infiltrate the samples (without the vial cap) for 15 min at 0.1 mPa in a vacuum bell jar. Repeat this step at least 3 times until the samples completely sink to the bottom of the fixation buffer.
Store the fixed samples overnight at 4°C in fixation buffer. Samples can be stored at 4°C in fixation buffer for up to one week before embedding.
Wash off the fixation buffer by incubating the samples in PBS (1×) buffer 3 times for 20 min each.
Dehydrate the samples by incubation in an ethanol series of 25%, 50%, 75%, and 100% for 30 min each.
Incubate the samples in 75% ethanol/25% histoclear, 50% ethanol/50% histoclear, and 25% ethanol/75% histoclear solution for 1 h each.
Wash the samples 3 times in 100% histoclear for 1 h each.
Add 5 ml fresh histoclear to the sample and then add paraplast to fill the jar. Incubate at 58°C overnight.
The next day, add fresh paraplast to fill the jar. Repeat this step every hour until the melted wax fills up the jar.
Wash the sample with freshly melted wax every 3 h. Repeat this step 3 times.
Transfer the paraffin-embedded sample to the processing cassettes in the right orientation for sectioning.
Cool the samples at room temperature. Store embedded samples at 4°C. Samples can be stored for up to 6 months.
Section the samples using a paraffin microtome. The suggested section thickness is 6-10 µm.
Transfer 1-2 sections carefully to the center of a gold fiducial-labeled coverglass using a paintbrush.
Completely dry the samples by leaving the coverglass at 37°C for 2 days.
Probe design
VARNISH (Vetting and Analysis of RNA for in situ Hybridization) probes are designed automatically on our sRNA-PAINT probe design website hosted at the Donald Danforth Plant Science Center server: https://wasabi.ddpsc.org/~apps/varnish/.
Enter the sRNA target sequence in the “Paste your small RNA sequence” field. Input the desired salt concentration and hybridization temperature (or use the default settings).
Enter or select the docking strand from the docking strand dropdown menu.
Enter or select the linker sequence.
The results of the probe design will be sent to you via email with a link for ordering.
In situ hybridization and washes
Deparaffinize the samples twice by incubation in 100% histoclear for 10 min each.
Rehydrate the samples by immersing the slides in an ethanol series of 100%, 95%, 80%, 70%, 50%, 30%, 10% (vol/vol), and water for 1 min each.
Incubate the slides in 250 ml protease solution at 37°C for 20 min.
Neutralize the samples in 250 ml 0.2% glycine solution for 20 min at room temperature.
Wash the slides twice in PBS (1×) buffer for 2 min each.
Dehydrate the samples by immersing the slides in water and ethanol series of 10%, 30%, 50%, 70%, 80%, 95%, and 100% for 1 min each.
Immerse the slides in 100% ethanol for an additional 1 min.
Store the samples in fresh 100% ethanol at 4°C for at least 4 h. Slides can be stored at 4°C for 1 week.
Add 1 µl 250 µM probe to 9 µl water and 10 µl formamide. Heat denature at 90°C for 3 min and immediately chill on ice.
Add 80 µl hybridization buffer to the solution mix in Step C9. Mix gently by pipetting without causing bubbles.
Apply 100 µl probe mix to the sample slide and cover with hybridization membrane. Alternatively, simply place the slide (sample side down) on a wet paper towel that has been covered with a layer of parafilm such that the sample side touches the parafilm (Figure 1). Avoid bubbles during this step.
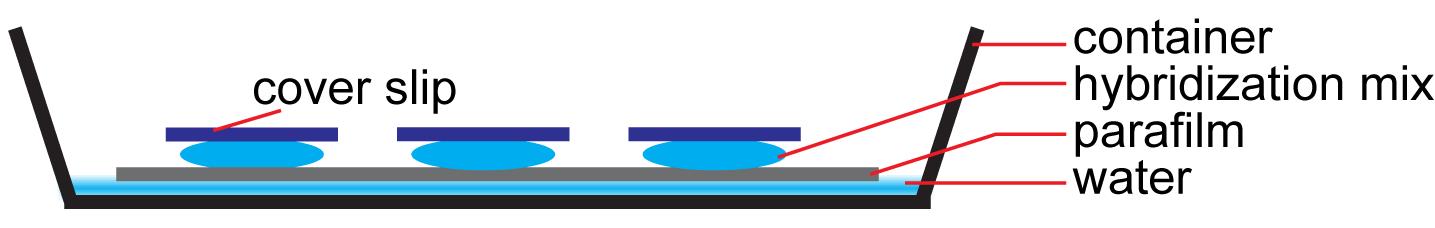
Figure 1. Assembly of the sRNA-PAINT hybridization chamberPlace the slide in a moist chamber and seal with plastic wrap. A simple moist chamber can be made by placing a sheet of parafilm on top of a wet paper towel in a flat-bottomed container.
Hybridize at 53°C overnight (or at least 16 h).
Pre-heat 0.2× SSC buffer to the same hybridization temperature as in Step C13.
After hybridization, wash the samples twice with 0.2× SSC for 1 h each.
Wash the samples in TBS (1×) buffer for 5 min at room temperature.
To immobilize the hybridized probes, incubate the slides twice in freshly prepared EDC buffer at room temperature for 10 min each.
Immerse the slides in EDC solution for 1 h and 15 min at room temperature.
Neutralize the slides in 0.2% glycine solution for 30 min.
Wash the slides twice in TBS (1×) buffer for 10 min each.
Store the slides at 4°C in TBS (1×) buffer until imaging.
sRNA-PAINT imaging
Secure one slide on the imaging perfusion chamber with the sample side facing up (Figure 2).
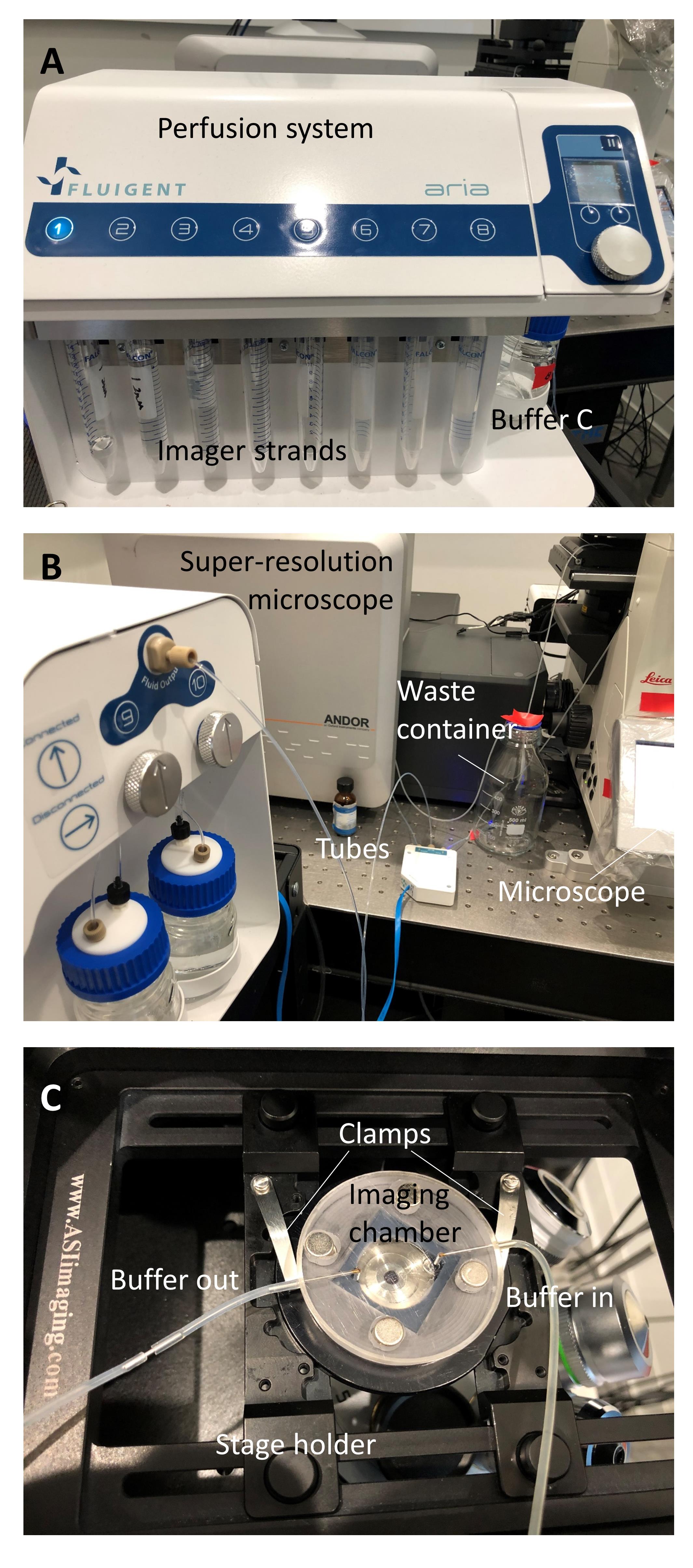
Figure 2. Assembly of the sRNA-PAINT imaging system. A. System for buffer storage and perfusion. B. Multiple tubes connect the perfusion unit to the imaging chamber. C. Imaging chamber setup for buffer exchange during image acquisition.Start the perfusion system and adjust the perfusion rate so that at least 2 ml imager solution is perfused through the sample during a 20-min imaging acquisition time.
Image the sample on a microscope capable of total internal fluorescence microscopy (TIRFM) fitted with a 63× or 100× objective that is compatible with TIRFM. Use a 100-200 ms camera exposure time and collect about 10,000-20,000 frames for each imager strand. Save the images in a format that can be opened with Bio-Formats.
Switch the perfusion input to buffer C and wash off the imager strand.
Image the sample under the same conditions as in Step D3 to collect a buffer control image.
Switch the perfusion input to a non-specific imager strand.
Image the sample under the same conditions as Step D3 to collect a non-specific imager strand control image.
Data analysis
Super-resolution imaging construction (Figure 3) using the ImageJ plugin ThunderSTORM: open ImageJ → Plugins → ThunderSTORM → Run analysis. Adjust the conditions and click on “Preview” to make sure that all specific spots are detected without any background being selected (usually, the default setting works well for most images). Click “OK.” This will generate an image file and a .txt file with the locations of all spots. Save the image file and .txt file.
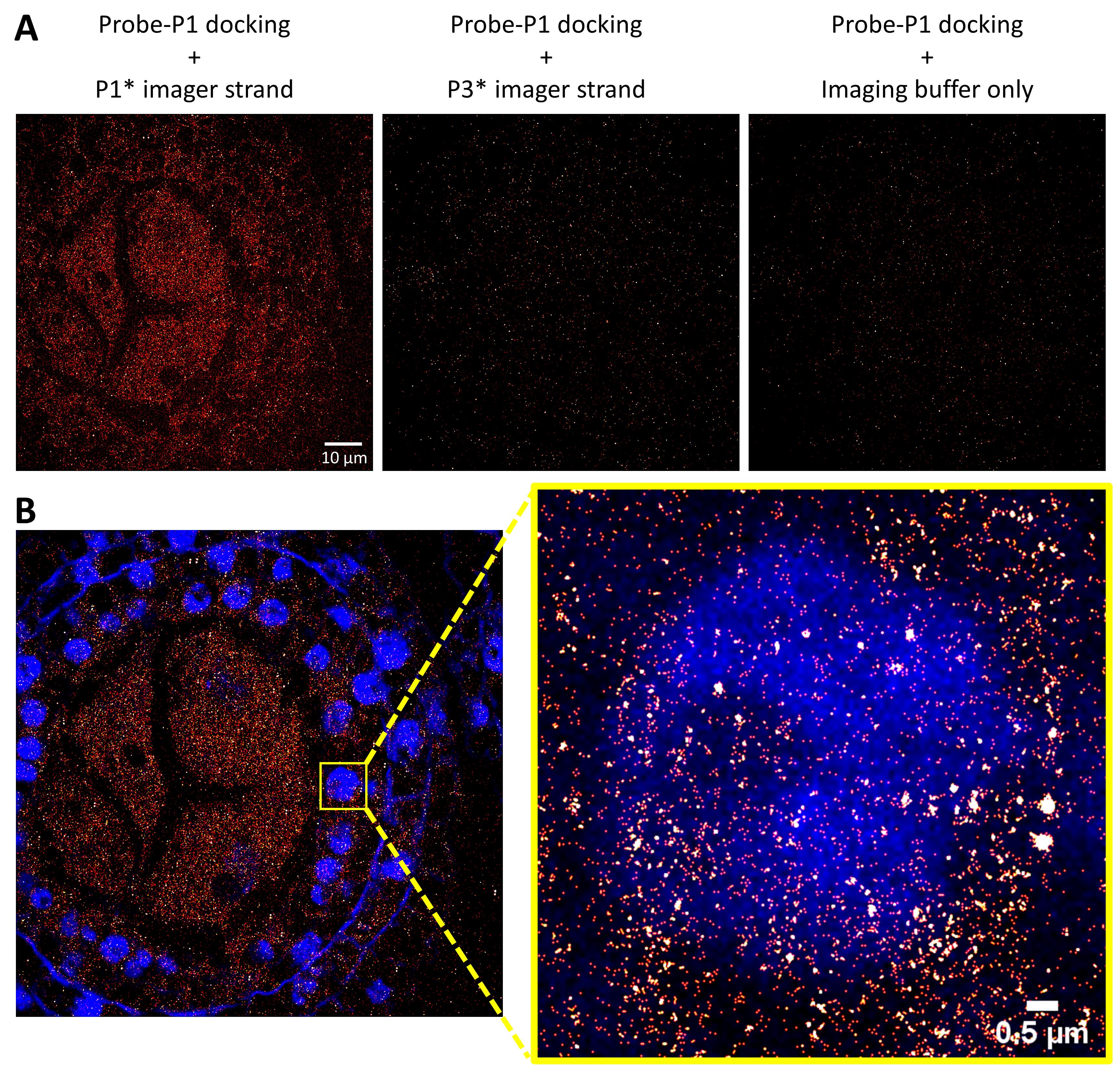
Figure 3. sRNA-PAINT imaging of a 24-nt phasiRNA in a 1.5-mm maize anther. A. sRNA-PAINT specifically detects the target signal. Alexa Fluor 647 was conjugated to imager strands. sRNA-PAINT images were acquired at a 1024×1024 pixel size and processed using the ThunderStorm ImageJ plugin. The final images are displayed at a 1024×1024 pixel size using equal brightness and contrast. Scale bar = 10 µm for all images. B. sRNA-PAINT achieved nanometer resolution of small RNAs. Nuclei are stained with DAPI (blue). Small RNA localization is shown as red/white dots.Quantitation using the Picasso software: a detailed user manual for the Picasso software is described by Schnitzbauer et al. (2017). Briefly, open Picasso: Localize and choose a suitable “Min. Net Gradient” so that all the spots are detected. Run “Localize (Identify & fit).” This will generate a .hdf5 file, which will be used in Picasso: Render for unit number calculation. The “Min. Net Gradient” sets the threshold to distinguish signal vs. background and needs to be consistent for all images. For sRNA copy number calculation, the non-specific background binding sites (buffer control, non-specific imaging strand, and ideally, a non-specific VARNISH probe) need to be subtracted from the copy number for each sRNA (Figure 4). In the example used in this manuscript, we selected an 80-nm pixel size as the area of interest to match the size of cells in the meiocyte. The average background detected was 17.10 ± 7.14 copies for the buffer control (Figure 3A, last panel) and 15.59±5.52 for the non-specific imager strand (Figure 3A, mid panel). In the final calculation, 17.10 was subtracted from the copy number for each cell layer.
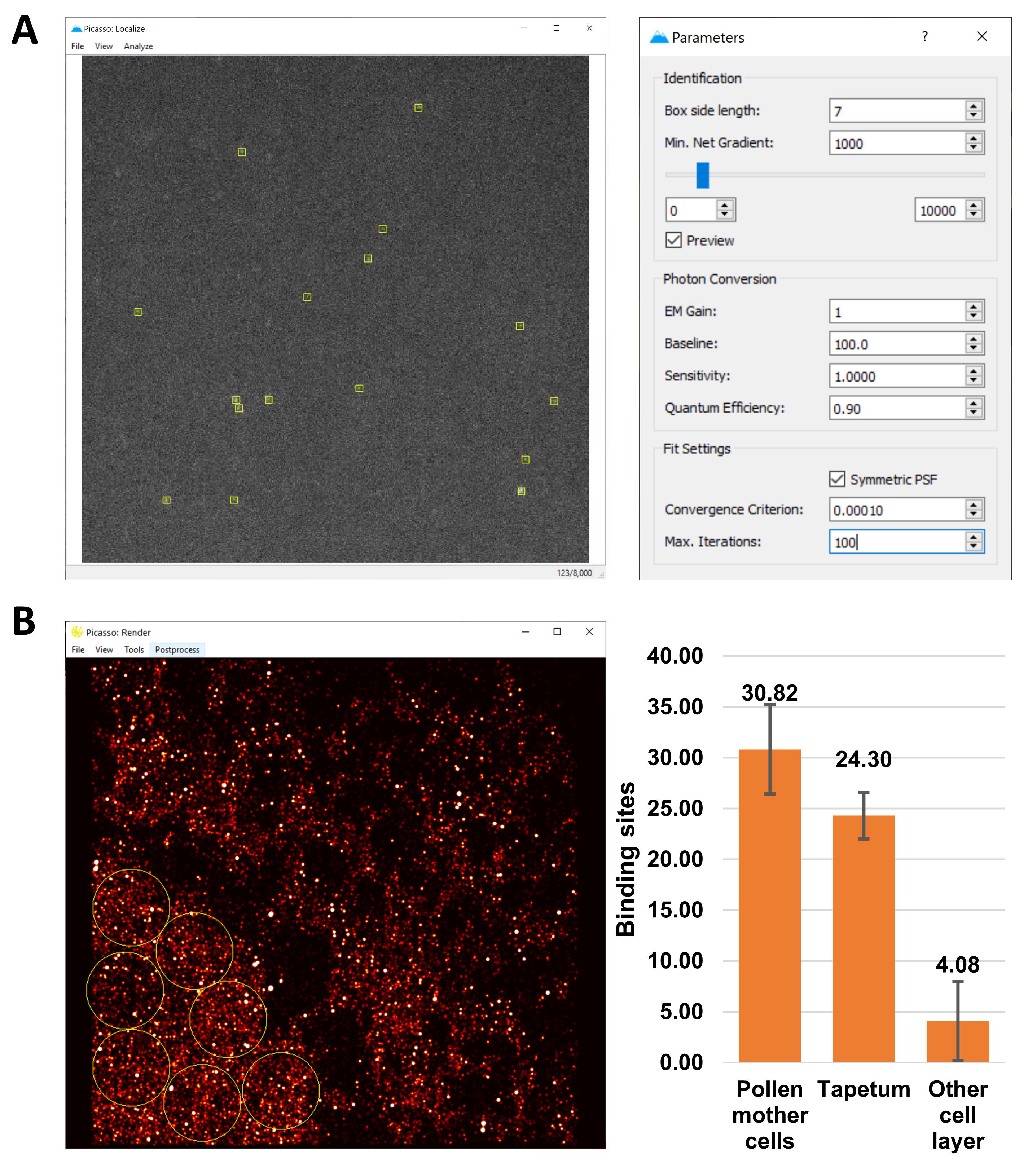
Figure 4. Quantitation of sRNA-PAINT using the Picasso software. A. Spot localization using Picasso: Localize function. B. Rendering and quantitation of the number of unit calculation using Picasso: Render. Yellow circles show the selected quantitation areas as an example of how the quantitation step was carried out in pollen mother cells.
Notes
Protease treatment: The protease concentration can be adjusted based on the signal intensity. Normally, a 20-min incubation is sufficient at the provided concentration (130 µg/ml). A longer incubation time and an increased protease concentration can be combined if low signal intensity is observed.
Probe concentration: Normally, 10 µM is sufficient for most sRNA detections. A probe concentration series can also be performed with 1 µM, 10 µM, and 100 µM to determine the most suitable concentration for sRNA targets.
Hybridization temperature: Generally, 50-60°C is suitable for most VARNISH probes that are designed with a TM above 80°C. The ideal hybridization temperature can be determined by the melting temperature (Tm) of each VARNISH probe.
EDC treatment: EDC treatment is used to attach the 5’ end of the VARNISH probes to the sample. EDC treatment has been shown to decrease VARNISH probe wash-off during multiplexed exchange-PAINT.
Imager strand concentration: Imager strand concentration should be determined experimentally. Start with a lower concentration around 0.5 nM and gradually increase the imager strand until a good blinking rate (15-30 molecules per frame) is reached for each imaging frame.
Choices of imager strands: Imager strands chosen for each sample should not interfere with the sample nonspecifically. To test imager strand (non-) specificity, add 2 nM each imager strand to the sample before hybridization and image under the same conditions that will be used in later image acquisition steps. Only imager strands that do not interfere with samples should be used in subsequent experiments.
Perfusion system: The perfusion systems that we have tested are the Fluigent Aria perfusion unit using air pressure, the ValveLink8.2 perfusion system using gravity flow, and a syringe pump system using syringe pressure force. The Fluigent system works best for multiplexed sRNA-PAINT, but it is costly. The syringe pump system is convenient and cost-efficient, and is an ideal starting unit for 1-2 imager strands.
Recipes
Ethanol gradient solutions
To generate 10%, 30%, 50%, 70%, 80%, 95%, and 100% ethanol series, mix 50 ml, 150 ml, 250 ml, 350 ml, 400 ml, 475 ml, and 500 ml ethanol and bring to 500 ml with nanopure water.
PHEM buffer (2×)
Add 18.14 g PIPES, 6.5 g HEPES, 3.8 g EGTA, and 0.99 g MgSO4 to 400 ml water and bring to a final volume of 500 ml. The final concentration of each ingredient is: 60 mM PIPES, 5 mM HEPES, 10 mM EGTA, 2 mM MgSO4, pH 8.
Autoclave and store the solution at 4°C.
Fixation buffer
Add 10 ml 16% paraformaldehyde and 20 ml 2× PHEM buffer to a 50-ml tube.
Add water to bring up to 50 ml. This step should be carried out in a fume hood. Prepare immediately before use.
Enzyme solution
Dissolve 0.5 g protease in 10 ml water.
Pre-digest the solution at 37°C for 4 h. The final stock concentration is 50 mg/ml.
Store 650-µl aliquots at -20°C.
Glycine solution
Add 5 ml 10% (w/v) glycine solution to 245 ml PBS buffer (1×). The final concentration of glycine solution is 0.2% (w/v).
Filter sterilize and store the solution at 4°C.
TE buffer
Add 100 ml 100 mM Tris-HCl and 100 ml 10 mM ethylenediaminetetraacetic acid (EDTA) to 800 ml water. Store at room temperature.
TE-protease solution
Add 650 µl enzyme solution to 250 ml TE buffer. Warm to 37°C. Prepare immediately before use.
Dextran sulfate solution (50%)
Add 5 g dextran sulfate to 7 ml water.
Dissolve dextran sulfate using 80°C heat.
Bring up to 10 ml with water.
Aliquot to 2.5 ml and store at -20°C.
Highly abundant sRNA
A highly abundant sRNA with a specific localization pattern can be used as a positive control. For example, miR2275 has been used as a positive control for sRNA-PAINT in maize anthers in our previous experiments due to its high abundance during meiosis and specific accumulation in the tapetal cell layer in meiotic anthers.
Hybridization salt
Add 1.75 g NaCl and 164 mg sodium monophosphate to 1 ml Tris-HCl (pH 8.0) and 1 ml 0.5 M EDTA.
Bring up to 10 ml with water.
Store 1.25-ml aliquots at -20°C.
Hybridization buffer
Add 1.25 ml hybridization salts, 5 ml deionized formamide, 2.5 ml 50% dextran sulfate, 250 μl 50× Denhardt’s solution, and 125 μl tRNA solution to 875 μl water.
Filter-sterilize and store 1-ml aliquots at -20°C.
TBS buffer (pH 7.5)
Add 100 ml 500 mM Tris-HCl and 30 ml 5 M NaCl to water.
Bring up to 1 L.
TBS is stable at 4°C for 3 months.
tRNA solution (100 mg/ml)
Add 1 g tRNA to 1 ml water. Mix and store at -20°C.
Buffer C
Add 100 ml 5 M NaCl to 900 ml PBS buffer (1×).
EDC buffer
Add 400 µl 1-methylimidazole, 112.5 µl 12M HCl, and 2.4 ml 5 M NaCl to 30 ml water.
Bring up to 40 ml with water.
Prepare immediately before use.
EDC solution
Add 0.31 g EDC to 40 ml EDC buffer.
Acknowledgments
This project was supported by the US NSF Plant Genome Research Program (awards 1649424, 1611853, and 1754097). We thank the members of the Meyers and Caplan labs for their support, and Joanna Friesner for editorial assistance. We thank Virginia Walbot (Stanford University) and the members of her lab for supplying maize materials and for useful discussions on anther small RNAs. We thank Prof. Ralf Jungmann for his support in DNA-PAINT, qPAINT, and analysis using Picasso. Microscopy equipment was acquired with a shared instrumentation grant (S10 OD016361), and access was supported by the NIH-NIGMS (P20 GM103446), the NSF (IIA-1301765), and the State of Delaware. The original research paper that developed sRNA-PAINT is published in Nucleic Acids Research: doi.org/10.1093/nar/gkaa623.
Competing interests
No competing interests.
References
- Bates, M., Jones, S. A. and Zhuang, X. (2013). Stochastic optical reconstruction microscopy (STORM): a method for superresolution fluorescence imaging. Cold Spring Harb Protoc 2013(6): 498-520.
- Blom, H. and Widengren, J. (2017). Stimulated Emission Depletion Microscopy. Chem Rev 117(11): 7377-7427.
- Dempsey, G. T., Vaughan, J. C., Chen, K. H., Bates, M. and Zhuang, X. (2011). Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods 8(12): 1027-1036.
- Heintzmann, R. and Huser, T. (2017). Super-Resolution Structured Illumination Microscopy. Chem Rev 117(23): 13890-13908.
- Hell, S. W. (2009). Microscopy and its focal switch. Nat Methods 6(1): 24-32.
- Huang, K., Demirci, F., Batish, M., Treible, W., Meyers, B. C. and Caplan, J. L. (2020). Quantitative, super-resolution localization of small RNAs with sRNA-PAINT. Nucleic Acids Res 48(16): e96.
- Jungmann, R., Avendano, M. S., Dai, M., Woehrstein, J. B., Agasti, S. S., Feiger, Z., Rodal, A. and Yin, P. (2016). Quantitative super-resolution imaging with qPAINT. Nat Methods 13(5): 439-442.
- Jungmann, R., Avendano, M. S., Woehrstein, J. B., Dai, M., Shih, W. M. and Yin, P. (2014). Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat Methods 11(3): 313-318.
- McTigue, P. M., Peterson, R. J. and Kahn, J. D. (2004). Sequence-dependent thermodynamic parameters for locked nucleic acid (LNA)-DNA duplex formation. Biochemistry 43(18): 5388-5405.
- Ovesny, M., Krizek, P., Borkovec, J., Svindrych, Z. and Hagen, G. M. (2014). ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 30(16): 2389-2390.
- Pena, J. T., Sohn-Lee, C., Rouhanifard, S. H., Ludwig, J., Hafner, M., Mihailovic, A., Lim, C., Holoch, D., Berninger, P., Zavolan, M. and Tuschl, T. (2009). miRNA in situ hybridization in formaldehyde and EDC-fixed tissues. Nat Methods 6(2): 139-141.
- Schnitzbauer, J., Strauss, M. T., Schlichthaerle, T., Schueder, F. and Jungmann, R. (2017). Super-resolution microscopy with DNA-PAINT. Nat Protoc 12(6): 1198-1228.
- Shroff, H., Galbraith, C. G., Galbraith, J. A. and Betzig, E. (2008). Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods 5(5): 417-423.
- Strauss, S. and Jungmann, R. (2020). Up to 100-fold speed-up and multiplexing in optimized DNA-PAINT. Nat Methods 17(8): 789-791.
- Xu, J. and Liu, Y. (2019). Imaging Higher-order Chromatin Structures in Single Cells Using Stochastic Optical Reconstruction Microscopy. Bio-protocol 9(3): e3160.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Huang, K., Demirci, F., Meyers, B. C. and Caplan, J. L. (2021). A Novel Method to Map Small RNAs with High Resolution. Bio-protocol 11(16): e4128. DOI: 10.21769/BioProtoc.4128.
Category
Plant Science > Plant molecular biology > RNA > RNA detection
Plant Science > Plant cell biology > Cell imaging
Cell Biology > Cell imaging > Super resolution imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.