- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Cell-free Translation: Preparation and Validation of Translation-competent Extracts from Saccharomyces cerevisiae
Published: Vol 11, Iss 18, Sep 20, 2021 DOI: 10.21769/BioProtoc.4093 Views: 5475
Reviewed by: Xin XuKomuraiah MyakalaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2021
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Cell-free translation is a powerful technique for in vitro protein synthesis. While cell-free translation platforms prepared from bacterial, plant, and mammalian cells are commercially available, yeast-based translation systems remain proprietary knowledge of individual labs. Here, we provide a detailed protocol for simple, fast, and cost-effective preparation of the translation-competent cell-free extract (CFE) from budding yeast. Our protocol streamlines steps combined from different procedures published over the last three decades and incorporates cryogenic lysis of yeast cells to produce a high yield of the translationally active material. We also describe techniques for the validation and troubleshooting of the quality and translational activity of the obtained yeast CFE.
Graphic abstract:
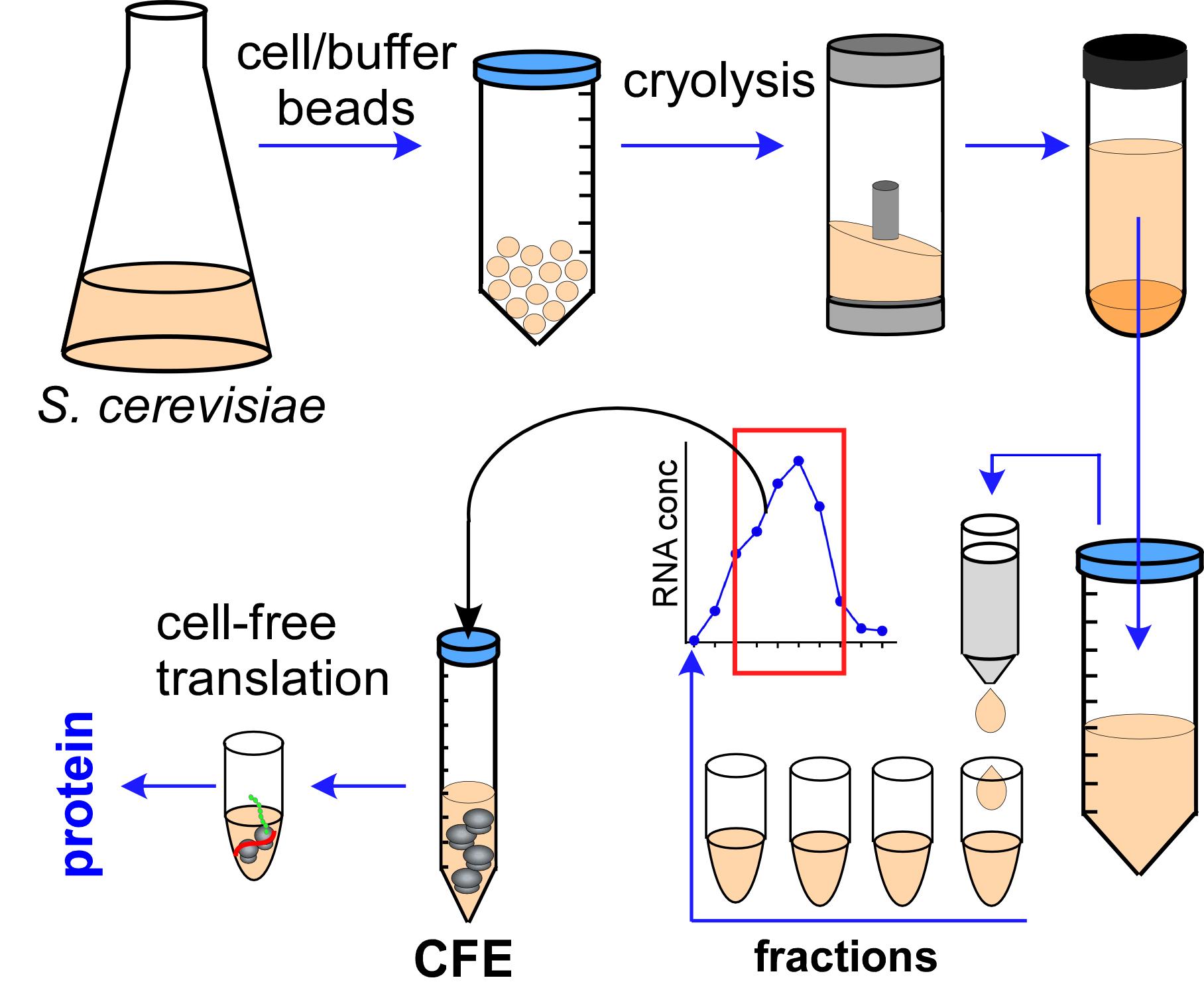
The flow of Cell-Free Extract (CFE) preparation procedure.
Background
Cell-free systems for protein synthesis make use of specially prepared cell extracts that retain macromolecular components required for translation, including ribosomes, tRNAs, aminoacyl tRNA synthetases, protein translation factors, and, in some cases, endogenous cellular mRNAs. The cell-free reactions can be charged with an mRNA of choice supplemented with radiolabeled or otherwise modified amino acids that become incorporated into nascent polypeptide chains during translation.
Cell-free translational platforms have been developed and optimized over the years for a plethora of organisms, such as E. coli (Shrestha et al., 2012; Kim et al., 2019), Bacillus subtilis (Kelwick et al., 2016), Pseudomonas putida (Wang et al., 2018), Streptomyces strains (Li et al., 2017), Leishmania trypanosomes (Kovtun et al., 2011), fungi Neurospora crassa (Wu et al., 2018), tobacco Bright Yellow 2 (BY-2) cells (Buntru et al., 2014), insect cells (Ezure et al., 2014), Chinese hamster ovary cells (CHO) (Brödel et al., 2014; Thoring and Kubick, 2018), and HeLa cells (Witherell, 2001; Mikami et al., 2010). For eukaryotic organisms, the most well-known and widely used cell-free translation systems have been those based on the rabbit reticulocyte (Pelham and Jackson, 1992; Olliver and Boyd, 1996) and wheat germ lysates (Harbers, 2014); both types are commercially available nowadays in several formulations. Besides generating both labeled and unlabeled proteins in a cell-free context, these reagents aided significantly in studying molecular mechanisms of protein synthesis, providing insights into individual steps of translation, translational efficiency, fidelity, and ribosome-associated protein quality control. In addition, cell-free translation systems have been instrumental in studies of protein evolution, enzyme engineering, high-throughput screens, and production of large quantities of proteins of interest for functional and structural analysis (reviewed in Carlson et al., 2012; Chong, 2014; Gregorio et al., 2019).
The budding yeast Saccharomyces cerevisiae represents an excellent source for translation-competent lysates. This unicellular eukaryotic organism combines the benefits of cost-effective microbial culture with an assortment of well-developed genetic tools: strains of interest can be easily generated with genes deleted (non-essential genes), depleted (essential genes), or overproduced. Moreover, a multitude of genetically modified yeast strains is already available from research laboratories and commercial sources. Despite many benefits, yeast-based translation extracts are not commercially available at present. Although several laboratory protocols have been published, many are too lengthy or incorporate cumbersome experimental manipulations, the rationale for which is not always clear. Guided by the published protocols, we developed a streamlined procedure, described below, that allows efficiently generating translation-competent S. cerevisiae extracts. Our main goal was to improve and simplify this technology to make it more user-friendly, reliable, reproducible, and affordable for any laboratory.
Unlike the previously documented lysis procedures that utilize enzymatic cell wall digestion and spheroplasting of yeast cells (Gasior et al., 1979; Tuite and Plesset, 1986) or the mechanical yeast cell lysis (Hodgman and Jewett, 2013; Hussain and Leibowitz, 1986; Schoborg et al., 2014; Tarun and Sachs, 1995; Tuite and Plesset, 1986; Tuite et al., 1980), we adapted a cryogenic lysis technique. This approach has several advantages over conventional lysis procedures: the translationally important components are protected from excessive degradation; precise amounts of cellular material can be taken for cryogenic grinding/milling and lysis, improving reproducibility between lysate preparations or among different strains. In addition, frozen yeast can be stored at -80°C either prior to or after cryogrinding for an extended time, creating breakpoints in the procedure. In our protocol, we adapted two consecutive centrifugation steps (at 30,000 × g and at 100,000 × g) as in procedures described by Hofbauer et al. (1982) and Tuite and Plesset (1986). These steps allow separation of the translationally active cellular fraction from heavy particles, lipids and polysaccharides. After a gel filtration step on Sephadex G-25 columns, we use a glycerol-containing buffer, which in our hands results in longer storage of an active lysate. Unlike many published protocols (Hodgman and Jewett, 2013; Hussain and Leibowitz, 1986; Schoborg et al., 2014; Tarun and Sachs, 1995; Tuite and Plesset, 1986), we omitted treatment of the cell-free extract (CFE) with micrococcal nuclease, designed to remove the endogenous mRNA. Although the cellular mRNA might interfere with translation of the reporter, we find that for many types of experiments, this step is unnecessary, and it also tends to lead to high variability between different CFE batches.
Here, we provide a detailed protocol for preparing translationally competent lysates from budding yeast, which has been applied in our work to study unconventional translation initiation mechanisms driven by 5’-UTRs (Trainor et al., 2021).
Materials and Reagents
PD-10 columns Sephadex G-25 (20 × 80 mm) (GE Healthcare, catalog number: 17085101); store at room temperature
15 ml conical centrifuge tubes (RNase-free) (VWR, catalog number: 10026-076) or equivalent
50 ml conical centrifuge tubes (RNase-free) (VWR, catalog number: 10026-078) or equivalent
1.7 ml microcentrifuge tubes (RNase-free) (Denville, catalog number: C2170) or equivalent
PCR tubes (USA Scientific, catalog number: 1402-2900) or equivalent
Ultracentrifuge tubes (Beckman Coulter, model: 10.4 ml polycarbonate bottles with cap assembly (16 × 76 mm) for the Ti80 rotor; catalog number: 355603)
Volumetric Serological pipettes, RNase-free, for 25 ml (Denville Scientific Inc, catalog number: P7129) and for 5 ml (Denville Scientific Inc, catalog number: P7127) or equivalent
25 mm Syringe filter (Corning, catalog number: 431224)
Rapid flow bottle top PES filter, 500 ml (Thermo Scientific, catalog number: 73520-994)
Sterile Pasteur pipettes (Thomas Scientific, SteriPettes, catalog number: 1215D97) or equivalent
BY4741 strain (Fisher Scientific; Dharmacon Inc, YEAST PARENT STRAIN BY4741, YSC1048); store in YPD/20% glycerol stock at -80°C
Peptone (Research Products International, catalog number: P20250); store at room temperature
Yeast extract (Thermo Scientific, catalog number: J23547-A1); store at room temperature
Dextrose (VWR, BDH Chemicals, catalog number: BDH9230); store at room temperature
Bacto-agar (VWR, Life Sciences, catalog number: J637); store at room temperature
RNase away solution (Thomas Scientific, catalog number: 1236B62); store at room temperature
HEPES free acid (VWR Life Sciences, catalog number: 0511); store at room temperature
Potassium acetate, C2H3O2K (Bio Basic, catalog number: PB0438); store at room temperature
Magnesium acetate tetrahydrate, C4H6MgO4·4H2O (Sigma, catalog number: M5661); store at room temperature
Mannitol powder (VWR BDH Chemicals, catalog number: BDH9248); store at room temperature
Dithiothreitol (DTT) (Sigma, catalog number: D0632); store at 4°C
Renilla luciferase plasmid, pRL-null (Promega, catalog number: E2271); store at -20°C
Firefly plasmid, pGEM-luc (Promega, catalog number: E154A); store at -20°C
Nano luciferase plasmid, pF4Ag NanoLuc (Addgene, catalog number: 137777); store at -20°C
DNA clean & concentrator kit (ZYMO RESEARCH, catalog number: D4004); store at room temperature
mMESSAGE mMACHINETM T7 Transcription kit (Thermo Fisher Scientific, Invitrogen, catalog number: AM1344); store at -20°C
Creatine kinase, rabbit muscle (BioVision, catalog number: P1301); store at -20°C
Creatine phosphate (VWR, catalog number: 97061-328); store at -20°C
GTP solution, 100 mM (Thermo Fisher Scientific, catalog number: R0461); store at -20°C
RiboLock RNase inhibitor, 40 U/μl (ThermoFisher Scientific, catalog number: EO0381); store at -20°C
Renilla Luciferase Assay System (Promega, catalog number: E2810); store at -20°C
Firefly Luciferase Assay Systems (Promega, catalog numbers: E1500, E4030, E4550) store at -20°C
Nano-Glo Luciferase Assay Systems (Promega, catalog numbers: N1110, N1120) store at -20°C
Renilla luciferase monoclonal antibody (Thermo Fisher Scientific, catalog number: SR07-830), store at 4°C
TAP polyclonal antibody (Thermo Fisher Scientific, catalog number: CAB1001), store at 4°C
Rpl3 monoclonal antibody (ScRPL3, Developmental Studies Hybridoma Bank, University of Iowa), store at 4°C
EasyTag EXPRESS35S Protein Labeling mix, [35S]-, 2 mCi, 11 mCi/ml (PerkinElmer, catalog number: NEG772002MC), store at 4°C
Trichloroacetic acid, TCA (Sigma, catalog number: T6399); store at 4°C
UltraPure Agarose (Invitrogen, catalog number: 16500); store at room temperature
Formamide (Sigma, catalog number: 47670-25ML-F); store in 1 ml aliquots at -80°C
SYBR gold nucleic acid gel stain (Thermo Fisher Scientific, Invitrogen, catalog number: S11494); store at -20°C
Optional: Hybond-N+ Nylon membrane (GE Healthcare, catalog number: NS0921); store at room temperature
Optional: TRI-REAGENT-LS (Molecular Research Center Inc., catalog number: TS 120); store at 4°C
HCl (GFS Chemicals, catalog number: 43491); store at room temperature
Glycerol (VWR Life Sciences, catalog number: BDH1172); store at room temperature
Liquid nitrogen
DreamTaq PCR master mix (2×) (ThermoFisher Scientific, catalog number: K1071); store at -20°C
GeneRuler DNA ladder mix (Thermo Fisher Scientific, catalog number: SM0333); short term storage 4°C, long term storage -20°C
ATP solution, 100 mM (Thermo Fisher Scientific, catalog number: R0441); store at -20°C
1 mM solution of 20 Essential amino acids, complete (Promega, catalog number: L4461); store at -20°C
1 mM solution of Essential amino acids minus Methionine and Cysteine (Promega, catalog number: L5511); store at -20°C
Formaldehyde solution, 37% (GFS Chemicals, catalog number: 40301); store at room temperature
EDTA, 0.5 M sterile solution, RNase-free (VWR Life Sciences, catalog number: E522-100ML); store at room temperature
Yeast medium (see Recipes)
YPD medium
YPD-agar plates
Stock solutions (see Recipes)
1 M HEPES-KOH, pH 7.6
2 M KOAc
300 mM MgOAc
100 mM MgOAc
1 M DTT
50% glycerol
Working solutions (see Recipes)
Buffer A
Buffer A/glycerol
Buffer A/mannitol
Buffer A/mannitol/DTT
Buffer A/glycerol/DTT
Creatine kinase dilution buffer
Creatine kinase solution
10× Energy mix
FAE solution
Equipment
-80°C freezer
-20°C freezer
Lab water purification system (PureLab, model: Elga water polisher system) or equivalent
pH meter (Sartotius, model: PB-11-P11.1) or equivalent
Optional: Laminar Flow Hood (NuAir, NU-201-430)
Microbiological incubator (Binder World, Series ED) or equivalent
Incubator shaker (Eppendorf, model: New Brunswick Innova® 43) or equivalent temperature-controlled shaker that can fit a 4 L flask
Preparative centrifuge (Beckman Coulter, model: J2-MI) or equivalent
Beckman JLA-10.500 rotor (Beckman Coulter) or equivalent
Preparative ultracentrifuge (Beckman Coulter, model: Le-80)
Beckman ultracentrifuge rotor (Beckman Coulter, model: Ti80)
Eppendorf centrifuge with the A-4-62 rotor, refrigerated (Eppendorf, model: 5810R)
Benchtop centrifuge (Eppendorf, model: 5420)
Freezer/Mill® (SPEX SamplePrep, model: 6700) or equivalent Cryogenic grinder
Freezer/Mill® accessories: small grinding vial set (SPEX SamplePrep, catalog number: 6751)
Toplanding balance (Sartorius, catalog number: CP3202P) or equivalent
Pipetman (Gilson, P20, P200, and P1000) or equivalent
Vortexer (any model)
Spectrophotometer (Eppendorf, catalog number: 6131-26951) or equivalent
500 ml Erlenmeyer flask
4 L Erlenmeyer flask
PPCO centrifuge bottles with sealing closure, 500 ml (Thomas Scientific, catalog number: 2625H84) or equivalent
Thermocycler (Eppendorf, model: Mastercycler®) or equivalent
Dry bath incubator (Eppendorf, model: Thermomixer R)
Luminometer (Promega; model: Glomax 20/20) or equivalent
Liquid scintillation counter (Beckman Ls System, model: Ls6000Ta) or equivalent
Power supply (Bio-Rad, model: PowerPac Universal Power Supply, catalog number: 1645070) or equivalent
Agarose gel electrophoresis equipment (Mansour and Pestov, 2013)
Gel rocker platform (Thomas Scientific, BioRockerTM 3D Mini Rockers, catalog number: 1155J96) or equivalent
Typhoon imager (GE, model: Typhoon 5 Biomolecular Imager), or any gel-imaging system (for example, Cole-Parmer, model: DelLogic 100 imaging system, or equivalent)
Protein gel electrophoresis equipment (Bio-Rad, model: Mini-PROTEAN tetra Vertical Electrophoresis Cell, catalog number: 1658004) or equivalent
Optional: polyacrylamide gel electrophoresis equipment (Hoefer Inc, model: SE260 Mighty Small II Delux Mini Vertical electrophoresis Unit)
Optional: Transfer Cell (Bio-Rad, Trans-Blot® SD Semi-Dry Transfer Cell, catalog number: 1703940) or equivalent
Optional: Hybridization oven (UVP, model: HB-1000 Hybridizer) or equivalent
Procedure
The preparation of a translationally active yeast CFE can be divided into three stages: (1) Growth, harvesting, and cryogenic disruption of yeast cells; (2) Isolation of a translationally active fraction from the total cell lysate; and (3) Validation of the CFE activity in translation. The procedure described below is for a 1 L yeast culture of BY4741 cells. This is the minimal volume we recommend for a CFE preparation. The protocol can also be scaled up if desired.
Growth, harvesting, and cryogenic disruption of yeast cells
The objective of this part of the procedure is to prepare cryomilled yeast cell powder from which the CFE will be derived.
Growing yeast on a plate (2-4 days in advance)
Streak the yeast strain of interest on a YPD-agar plate to achieve single-colony growth. Incubate the plate in an incubator at the temperature optimal for the strain until colonies are ~1-1.5 mm in diameter. We usually use the BY4741 strain and its derivatives for this procedure. Depending on the strain's genotype, colonies may take between 2 to 4 days to grow at 30°C.
General preparation and starting a yeast culture (Day 1, ~2-3 h)
Make sure that yeast media and all solutions required for the procedure have been prepared and stored at the appropriate temperature.
Autoclave 2-3 L of deionized H2O and store it at 4°C (referred hereafter to as "cold diH2O").
Autoclave large centrifuge bottles (400 ml or analogous). Place the autoclaved centrifuge bottles, a bag of 50 ml, a bag of 15 ml conical centrifuge tubes, and a bag of 1.7 ml microcentrifuge tubes at -20°C.
Inoculate one large yeast colony into 100 ml of YPD medium in a sterile 500 ml flask. Place the flask into an orbital shaker and grow the culture for 14-20 h at 30°C with moderate shaking (~160 rpm).
Growing and harvesting yeast cells (Day 2, ~6-7 h)
Inoculate the overnight culture started at Step A2d into 1 L of fresh YPD in a 4 L flask to obtain an ODλ=600nm ~0.8. Incubate this culture at 30°C for 5-6 h with constant shaking until ODλ=600nm ~2.5.
In our experience, any deviation from these parameters may significantly lower either the CFE yield or its activity in translation reactions. For slow-growing or non-BY4741 strains, optimization of the dilution factor may be required. Avoid prolonged incubation of the day culture, which can deplete dextrose from the medium and alter cell metabolism.
While the culture is growing, cool down to 4°C all rotors and centrifuges used at Steps A3c-A3e below (e.g., a JLA-10.500 preparative rotor for the large centrifuge bottles with a Beckman J2-MI centrifuge, and an A-4-62 rotor for 50 ml tubes used with an Eppendorf 5810R centrifuge).
When the cell culture density reaches ODλ=600nm ~2.5, pour the cell culture into the prechilled large centrifuge bottles. Harvest cells at 3,700 × g for 6 min at 4°C (5,500 rpm in a Beckman JLA-10.500 rotor).
Discard the supernatant, resuspend the cell pellets in a total volume of 250 ml of cold diH2O. Pellet diH2O-washed cells at 3,700 × g for 6 min at 4°C (5,500 rpm in a Beckman JLA-10.500 rotor).
To resuspend cell pellets, we use a volumetric 25 ml plastic pipet and gently swirl centrifuge bottles periodically until a homogenous cell suspension is formed. Do not vortex the cell pellets. We typically divide cell suspensions obtained at this step between two centrifuge bottles, which makes it easier to resuspend cell pellets at the next step.
Discard the supernatant, resuspend the cell pellets in a total volume of 100 ml of cold diH2O. Working on ice, transfer the cell suspension into two cold, sterile 50 ml centrifuge tubes. Pellet cells at 3,220 × g for 5 min at 4°C (4,000 rpm in an Eppendorf 5810R centrifuge with the A-4-62 rotor).
Preparation of the frozen cell/buffer beads (Day 2, ~2 h)
Keep centrifuge tubes containing yeast cells on ice during all steps. Use appropriate safety precautions and wear eye protection when working with liquid nitrogen.
Wash cell pellets once with cold buffer A/mannitol and once with cold buffer A/mannitol/DTT (25 ml per tube; centrifuge cells as in Step A3e above). Combine cell suspensions in one tube during the last wash.
Use a Pasteur pipet attached to a vacuum flask to aspirate off the excess buffer from the tube. Determine the weight of the yeast cell pellet. Use an empty 50 ml centrifuge tube to tare the balance. Normally, we obtain ~5 g of the wet cell material from a 1 L yeast culture grown to a final ODλ=600nm ~2.5.
Resuspend the yeast pellet in buffer A/mannitol/DTT at a 2:3 volume (ml):pellet weight (g) ratio.
For example, for 5 g of yeast cells, use 3.3 ml of the buffer. The cell suspension will be very thick. Make sure you keep the cell suspension on ice.
Pour ~25 ml of liquid nitrogen into clean 50 ml "collector" tubes secured in a plastic rack. Place the rack inside an ice bucket or a clean styrofoam container partially filled with liquid nitrogen (as shown in Figure 1A). Wait for ~10 min to ensure a thorough cooldown of the entire setup.
Collect the cell suspension from Step A4c above into a 5 ml volumetric plastic pipet. Keeping the pipet tip ~5-6 cm above the liquid nitrogen's level to avoid tip freezing (Figure 1A), eject the cell suspension dropwise into a liquid nitrogen-filled collector tube. To form individual cell/buffer beads, allow each drop to freeze before adding the next one; adding drops too quickly will cause clump formation, which complicates loading the beads into a grinding vial at a later step of the procedure. Add more liquid nitrogen into the collector tube if needed.
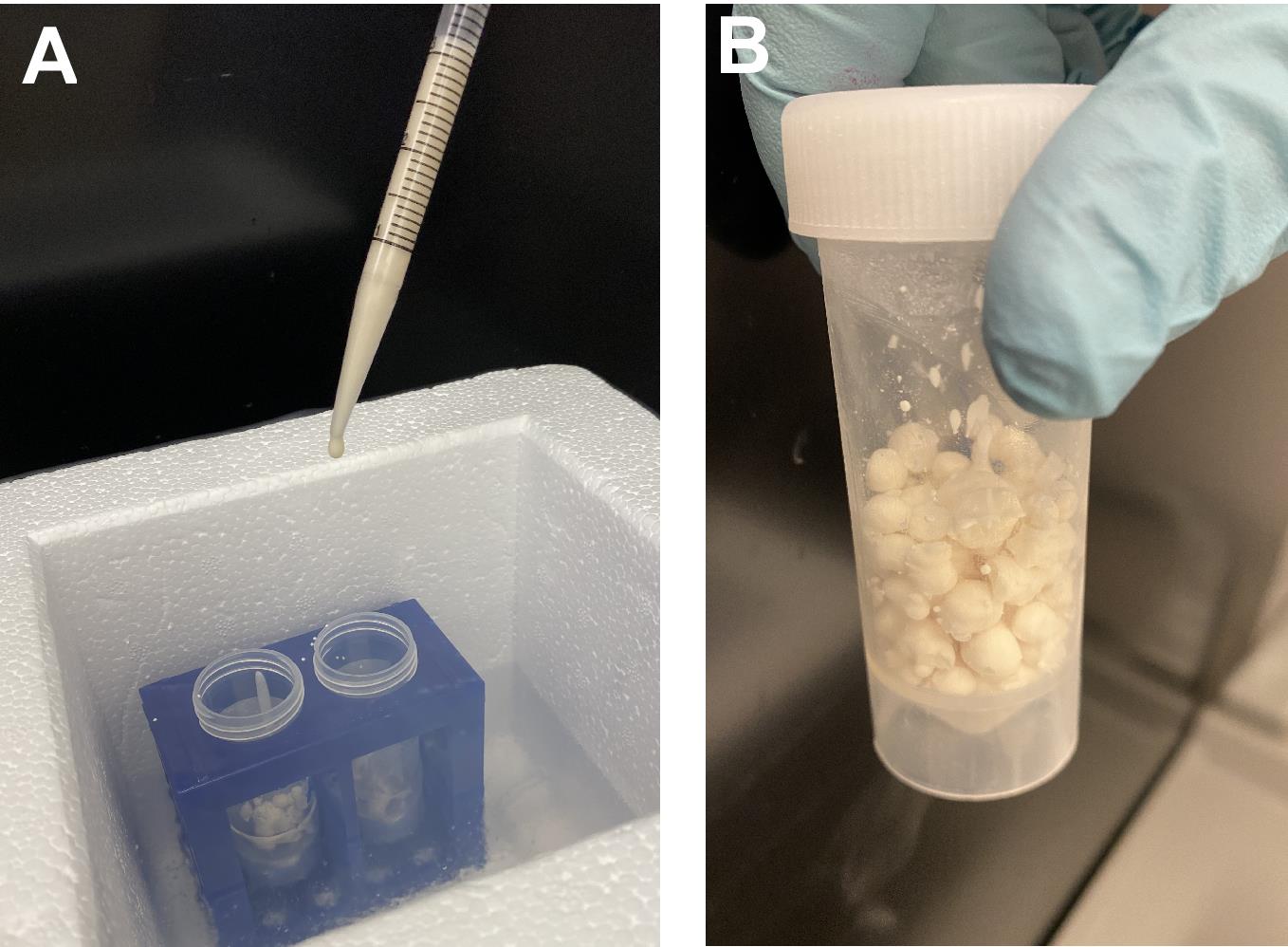
Figure 1. Illustration of the frozen cell/buffer beads preparation for cryolysis. To ensure well-separated beads, slowly eject cell suspension with an interval of 3-5 s between drops. Using two collector tubes and switching between them for each drop (as shown on panel A) can speed up the process while allowing the drops to freeze properly. Cell/buffer beads should then be combined in a single tube for storage, as shown in panel B.When the last drop is frozen, carefully discard excess liquid nitrogen from the collector tubes. Cap the tubes loosely to allow outgassing and place the frozen cell/buffer beads (Figure 1B) at -80°C for storage. The frozen cell/buffer beads can be used immediately for the next steps. For a break in the procedure, beads can be kept at -80°C from overnight to several weeks. Typically, we obtain ~7-8 g of the cell/buffer beads from 5 g of the wet cell pellet Step A4b.
Cryogenic grinding of yeast cells (Day 2 or Day 3, ~2 h)
In this section, we provide the instructions and illustration for the SPEX SamplePrep Freezer/Mill® Model 6700. Other, more advanced Freezer/Mill models are available commercially. Use appropriate safety precautions and wear eye protection when working with liquid nitrogen.
Fill the cryogenic mill's chamber with liquid nitrogen following the manufacturer's instructions.
Place grinding vials with the small end plugs attached and impactor rods placed inside (see Figure 2A, parts a, b, and c) vertically into a styrofoam container partially filled with liquid nitrogen (Figure 2B). Be sure to only submerge the vials halfway in liquid nitrogen, as excessive cooling of their tops will shrink them too much and prevent locking of the large end plugs (Figure 2A, part d) at a later step. Allow the chamber and vials to cool down for 15-30 min. Normally, for 7-8 g of the cell/buffer beads obtained in Step A4f, we use two small polycarbonate grinding vials fitted with individual stainless-steel impactor rods.
Blank the weight of a grinding vial (with a rod inside) on a balance. Shake cell/buffer beads inside the vial to get 4-4.5 g of the beads per vial (Figure 2D). Place the vial filled with beads back into the liquid nitrogen in a vertical position for 2-3 min (Figure 2C). Work fast to avoid thawing the cell/buffer beads.
Close the vial with a large end plug (Figure 2E) and position the locked vial horizontally inside the liquid nitrogen container so that the vial is completely submerged (Figure 2F).
Repeat Steps A5c-A5d with the remaining vial(s).
Place a grinding vial loaded with beads inside the cryomill chamber (Figure 3A), lock the chamber, and wait 10 min for the system’s complete cooling. Grind the cells using the maximum speed of the SPEX Freezer/Mill® 6700 and the following settings: 1 min grinding, 1 min rest, for a total of 8-10 cycles. Add liquid nitrogen to the chamber if needed, as multiple cycles could lead to partial nitrogen evaporation. After the last cycle, wait 10 min before transferring the vial to a -80°C freezer. We recommend storing powdered cells inside unopened grinding vials (Figure 3B) at -80°C for at least 30 min (overnight is also an option) to allow for outgassing of any residual nitrogen. For long-term storage, the powder can be stored at -80°C for 2-3 months.

Figure 2. Preparation of grinding vials for cryomilling. A. Parts of a grinding vial: polycarbonate vial (a); small (bottom) end plug (b), impactor rod (c), large (top) end plug (d). Parts b and d may look slightly different depending on cryomill model. B. Prechilling grinding vials in liquid nitrogen. C, D. Grinding vials with cell/buffer beads inside. E. A vial closed with the top end plug. F. Locked grinding vials loaded with beads, ready for cryomilling.
Figure 3. Cryogenic cell grinding. A. Cryogenic mill filled with liquid nitrogen and a grinding vial loaded into the device. B. Powdered yeast cells inside a grinding vial. C. Correct placement of grinding vials containing yeast powder in an ice bucket, making the top ends exposed to the room-temperature air.
Isolation of a translationally active fraction from the cell lysate
The main objective of this part of the procedure is to separate the translationally active fraction of the cell lysate from other lysate components that are unnecessary, and potentially inhibitory, for a translation reaction. Desalting on Sephadex G-25 columns removes low molecular weight compounds from the CFE, including endogenous amino acids and nucleotides. All steps described in (B) must be performed on the same day.
Cell lysis (Day 3, ~6 h)
Prechill a fixed-angle rotor (e.g., a Beckman Ti80 rotor) and an ultracentrifuge to 4°C.
Transfer grinding vials containing cryomilled yeast powder (Figure 3B) from -80°C to an ice bucket. Position the vials vertically so that the top end plugs are exposed to the room-temperature air (Figure 3C). Place two 50 ml conical tubes and four capped small polycarbonate ultracentrifuge tubes into the ice bucket.
After keeping the grinding vials on ice for ~5 min, remove the top end plugs and shake the yeast powder from the vials into a prechilled 50 ml tube. Keeping the vials on ice for too long may cause thawing of the yeast powder, making it resemble a paste. In that case, use a clean RNase-free spatula to transfer the material into the 50 ml tube.
Add 2-3 ml of buffer A/mannitol/DTT to the 50 ml tube.
Use 3-4 ml of buffer A/mannitol/DTT to collect the residual amounts of the cell material left on the walls of the grinding vial and the spatula. Add the recovered cell material to the 50 ml tube containing the bulk of the cryomilled yeast powder (B1c).
Pipet the mixture up and down to obtain a homogenous slurry of the broken yeast cells and the buffer. The lysate will be very thick at this point. Add additional buffer A/mannitol/DTT to the final volume of 9 ml and pipet to mix. Transfer the lysate into the prechilled small ultracentrifuge tube. Rinse the 50 ml tube with 1 ml of the remaining buffer to collect the leftover material and transfer it into the ultracentrifuge tube. The total volume of the lysate must be 10 ml. Close the ultracentrifuge tube with a cap.
If the lysate is not completely thawed and slushy, incubate the ultracentrifuge tubes containing the yeast suspension on ice for 5-15 min.
Place the ultracentrifuge tubes into a rotor and centrifuge them at 30,000 × g (17,000 rpm in Beckman Ti80 rotor) for 15 min at 4°C.
Carefully remove the tubes from the centrifuge rotor and place them on ice vertically. You will see three fractions, as shown in Figure 4. Transfer 6 ml of the middle fraction ("S30") into a new prechilled small polycarbonate ultracentrifuge tube while avoiding any lipid layer at the top. We successfully used a Pipetman P1000 set at 1 ml to collect the middle fraction. However, to avoid repeated crossing through the top layer, one can also use a 10 ml syringe with an attached needle. Be careful not to aspirate the top or bottom fractions. Keep tubes on ice and work fast to minimize the lysate exposure to room temperature.

Figure 4. Formation of three distinct fractions after the 30,000 × g centrifugation. Top: milky-white layer of lipids. Middle (marked as S30): the yellowish soluble fraction that contains the translationally active material. Bottom: large pellet containing unbroken cells, cellular debris, and DNA.Place the ultracentrifuge tubes into the rotor and centrifuge them at 100,000 × g (30,000 rpm in a Beckman Ti80 rotor) for 35 min at 4°C.
While the tubes are spinning, it is a good time to start preparing gel-filtration columns used for lysate desalting (see Step B2a below).
When centrifugation is complete (B1j), carefully remove the ultracentrifuge tubes from the rotor and place them on ice vertically for 5-10 min (see Step B1m). Avoid disturbing the tube contents. Carefully collect 2.5 ml of the S100 fraction as depicted in Figure 5 (also, see Step B1m) and transfer it into a prechilled 15 ml conical tube.

Figure 5. Formation of four distinct layers in yeast cell lysate following the 100,000 × g centrifugation. Top: Large yellowish layer containing lipids. Middle (marked as S100): translucent layer that is the desired polysome-free cell lysate fraction. Bottom: A largely immobile pellet composed of heavy cellular components, such as the ER and polysomes (traced in pink), and a mobile yellowish fraction (traced in blue).Keeping the tubes on ice for 5-10 min helps to better visualize the four layers. To collect the intermediate S100 fraction, a Pasteur pipet or a syringe with an attached needle can be used. The collection of S100 while avoiding the upper or bottom layers is a challenging step as these layers are mobile and easily intermix with the S100 fraction. Be especially careful not to contaminate S100 with the mobile pellet fraction (traced in blue in Figure 5).
Lysate desalting and concentration (Day 3, 45-60 min for column equilibration + ~1 h for desalting)
Remove caps from the PD-10 Sephadex G-25 columns, then cut off the bottom tips at a 45° angle. Allow the storage buffer to run out by gravity flow. Equilibrate each column with 5 × 5 ml of cold buffer A/glycerol/DTT.
During column equilibration, label ten 1.7 ml microcentrifuge tubes for collecting fractions (1-10), plus two extra tubes for the flowthrough. Place the tubes on ice.
Transfer the equilibrated columns, microcentrifuge tubes, and lysates from Step B1l into a cold (4°C) room. Make sure that the columns have no buffer left at the top. Place the microcentrifuge tubes in a rack, arrange them so that the first flowthrough tube is directly underneath the outlet of a PD-10 Sephadex G-25 column clamped to a laboratory stand.
Carefully apply 2.5 ml of lysate from Step B1l to the top of the column and allow the sample to completely enter the column bed. Collect the flowthrough using two microcentrifuge tubes. For elution, pipet 5 ml of buffer A/glycerol/DTT onto the column bed and begin collecting fractions into 1.7 ml microcentrifuge tubes labeled 1-10. Collect ~0.5 ml per tube. Cap each tube. Gently vortex or pipet up and down the tube content. Place all the tubes on ice.
Measure the RNA concentration in each tube: dilute a portion of each fraction with H2O 1:100 and measure the absorbance at λ = 260 nm using a spectrophotometer. To calculate RNA concentration (in μg/ml), multiply the absorbance by the RNA extinction coefficient (ϵRNA = 40) and by the dilution factor (100). Plot the RNA concentration in each fraction on a graph, as shown in Figure 6.
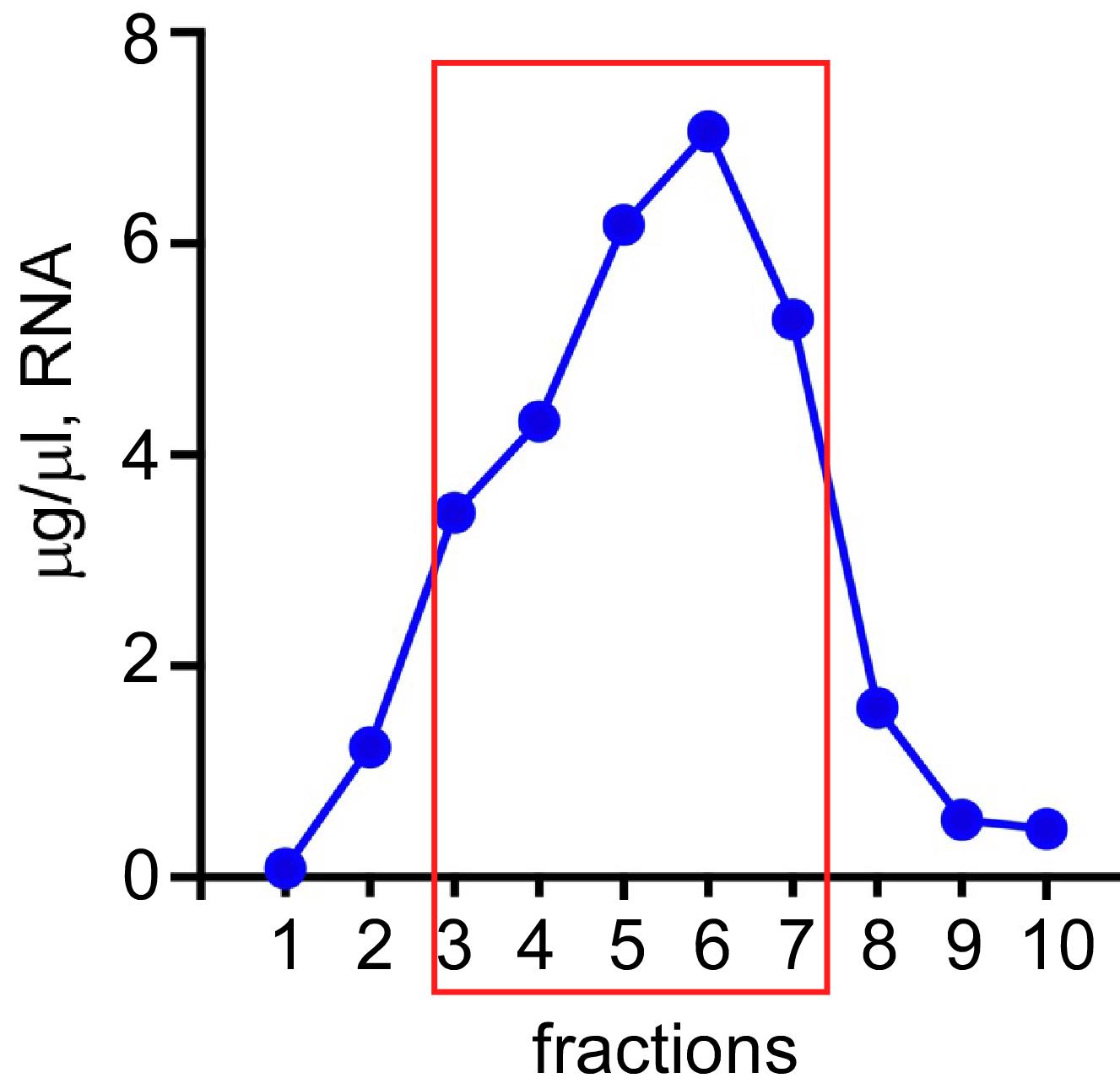
Figure 6. Graphical representation of the total RNA distribution in the G-25 column elution fractions. RNA concentration in each fraction was determined spectrophotometrically: a portion of each fraction was diluted with H2O (1:100), and the absorbance was measured at λ = 260 nm. RNA concentration was calculated (ϵRNA = 40), converted to μg/μl, and plotted on a graph. We collect fractions with at least 60-75% RNA compared to the highest containing fraction. Typically, these are fractions 3-7 (boxed in red).Combine fractions with the highest RNA concentration in one 15 ml tube and thoroughly mix the content by gentle vortexing or pipetting it up and down. Working on ice, aliquot the resulting CFE into prechilled 1.7 ml microcentrifuge tubes, 100 μl per tube. Snap-freeze the CFE aliquots in liquid nitrogen and place the tubes at -80°C for storage. An example of the yeast CFE preparation procedure is in Table 1.
Table 1. An example of the yeast Cell-Free Extract (CFE) preparation procedure

Using the described procedure, from 1 L of yeast culture grown to ODλ=600nm ~2.5 (column 4), we generate 7-8 g of cell/buffer beads (column 6). The cryogenic lysis of the cell/buffer bead material is performed in two separate vials, resulting in two 2.5 ml aliquots of S100, followed by the two desalting/concentrating steps using two individual PD-10 Sephadex G-25 columns. The volume of the generated eluate from one column with the highest RNA concentration is 2.5 ml. By combining eluates from two columns, we obtain 5 ml of the CFE, which is enough for ~660 standard-size reactions.
Validation of the CFE activity in translation
To determine the translational activity of a newly prepared CFE, set up a translation reaction with an mRNA reporter (section 1 below) or, alternatively, use [35S]-methionine/cysteine labeling of translation products derived from the endogenous mRNA present in the CFE (section 2). An optional analysis of RNA integrity (section 3) may be useful for troubleshooting the extract preparation procedure.
Validation of the translational activity of CFE using luciferase reporters
Generation of the reporter mRNA
The protocol below is designed for the Renilla luciferase construct, as used in Trainor et al. (2021). Refer to Table 2 for details on using other luciferases, such as Firefly and NanoLuc®, as the protein reporters in CFE-based translation reactions.
Table 2. Synthesis and detection of Firefly and NanoLuc® luciferase-based protein reporters
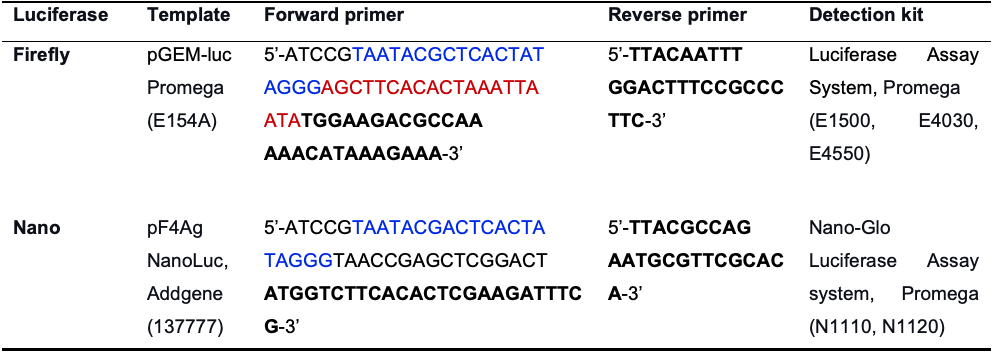
The T7 promoter sequence is marked in blue; the CYC1 5’-UTR translational enhancer used to increase translation of the Firefly luciferase is marked in red.Amplify the Renilla luciferase gene by PCR using the following primers: the forward primer (5’-ATCCGTAATACGACTCACTATAGGGTAACCGAGCTCGGACTATGACTTCGAAAGTTTATGATCCAGAA), the reverse primer (5’-TCACGACATTTG TTCATTTTTGAGAACTCG-3’). The forward primer contains a T7 polymerase promoter sequence (bolded); the start ATG codon is highlighted in red.
Set up a PCR reaction in a total volume of 50 μl by mixing 25 μl of the DreamTaq PCR master mix (2×), 1 μl of 20 μM forward primer, 1 μl of 20 μM reverse primer, 1 μl of the template plasmid (10 ng/μl), and 22 μl of H2O. A Renilla luciferase gene-containing plasmid can be purchased from Promega (see Materials). Amplify DNA using the following PCR program: 27-30 cycles at 94°C for 30 s (denaturation), 54°C for 30 s (annealing), and 72°C for 1 min (polymerization).
Upon completion of the PCR, check 5 μl of the reaction on a 1% agarose gel. The Renilla luciferase gene is 940 bp. Use a molecular weight marker to determine the size of the PCR product. If you detect additional bands, repeat the PCR reaction with an increased primer annealing temperature to achieve a single 940 bp band.
Purify the PCR product using the DNA clean & concentrator kit spin columns following the manufacturer's protocol. Elute the purified PCR product from a column with 16 μl of H2O. Measure DNA concentration using a spectrophotometer.
Generate m7G-capped mRNA using 1 μg of the purified PCR DNA (from Step C1a.iii) in a transcription/capping reaction with an mMESSAGE mMACHINETM T7 transcription kit. Incubate the reaction at 37°C for 2 h. Follow the kit protocol for purifying the transcription product, including DNase treatment for 15 min at 37°C and RNA precipitation with LiCl. Finish with washing the precipitated RNA with 80% EtOH and air-drying the RNA pellet.
Resuspend the RNA pellet in 30-40 μl of RNase-free H2O by repeated pipetting. Keep the RNA on ice. Measure RNA concentration using a spectrophotometer as described in Step B2e. Aliquot 4-5 μl of the generated RNA into PCR tubes and freeze these aliquots at -80°C. For consistency, we do not recommend freeze-thaw cycles of the reporter mRNA.
Perform the in vitro translation reactions
This protocol is based on Wu and Sachs (2014) with some minor modifications. To ensure reproducibility and for statistical analysis, assemble each reaction in triplicate.
Prepare the 2.5× master mix (required volume: 80 μl for 12 reactions) by assembling its components in the order indicated in Table 3. Keep the master mix on ice.
Table 3. Preparation of the 2.5× master mix for translation reactions
Thaw an mRNA aliquot on ice, adjust the concentration to 200-300 ng/μl using RNase-free H2O. Keep the diluted mRNA on ice.
Thaw an aliquot of the CFE by warming the tube in your hand and periodically tapping it gently to mix the content. As soon as the CFE is thawed, place the tube on ice.
Set up the desired number of translation reactions by first adding 7.5 μl of CFE to 1.7 ml microcentrifuge tubes. Add 6 μl of the 2.5× master mix to each tube. Add 1.5 μl (300-450 ng) of the mRNA into each tube. Reactions can be assembled on a bench at room temperature. The total volume of each reaction is 15 μl. Mix the reaction components by gently tapping the tubes and centrifuge the tubes in a tabletop centrifuge for 10 s. Place the tubes into a dry bath incubator set at 21°C. Incubate the reactions for 1-2 h.
Reagent Volume 1 H2O 45.8 μl 2 2M KOAc 5 μl 3 0.1M MgOAc 4 μl 4 10× Energy mix (see Recipes) 20 μl 5 1 mM amino acids 2 μl 6 RiboLock (40 units/μl) 2 μl 7 Creatine kinase (10 units/μl) 1.2 μl Analyze the translation reaction products by a luciferase assay and/or western blotting
For a Renilla luciferase assay, use reagents from a Renilla Luciferase Assay System kit (Promega). Follow the protocol provided with the kit.
To visualize proteins generated in the translation reaction on a gel, perform quantitative western blotting with fluorescent secondary antibodies, for example, as detailed in Ghosh and Shcherbik (2020), or by using HRP-fused secondary antibodies and an ECL detection system (Trainor et al., 2021). The Renilla luciferase-specific antibodies are commercially available (see Materials).
Labeling of the nascent polypeptides with [35S]-Met/Cys as a readout of the CFE translational activity
Use appropriate safety precautions when working with radioactive materials.
Prepare the 2× labeling master mix (100 μl for 12 reactions) by assembling its components in the order indicated in Table 4. Keep the master mix on ice.
Table 4. Preparation of the 2× labeling master mix for the labeling of nascent proteins with [35S]-Met/Cys
Reagent Volume 1 H2O 56.8 μl 2 2M KOAc 5 μl 3 0.1M MgOAc 4 μl 4 10× Energy mix (see Recipes) 20 μl 5 1 mM amino acids (-Met, Cys) 2 μl 6 RiboLock (40 units/μl) 2 μl 7 Creatine kinase (10 units/μl) 1.2 μl 8 EasyTag EXPRESS35S Protein Labeling mix, [35S], 11 mCi/ml 9 μl Thaw an aliquot of the CFE by warming the tube in your hand and gently tapping the tube to mix the content. As soon as the CFE is thawed, place the tube on ice.
Set up the translation reactions by first adding the CFE to 1.7 ml microcentrifuge tubes, 7.5 μl per tube. Next, add 7.5 μl of the 2× labeling master mix to each tube. The total volume of each reaction is 15 μl. Mix the reactions by gently tapping the tubes and centrifuge the tubes in a tabletop centrifuge for 10 s. Place the tubes into a dry bath incubator set at 21°C. Incubate the reactions for 1-2 h.
Precipitate [35S]-labeled polypeptides with TCA. For example, follow the protocol described in https://www.promega.com/-/media/files/resources/protocols/technical-manuals/0/rabbit-reticulocyte-lysate-system-protocol.pdf.
Measure the activity of the [35S-Met/Cys]-labeled polypeptides by scintillation counting.
Analysis of RNA integrity
If a newly prepared CFE batch has a low translational activity, we recommend examining both the CFE and the generated mRNA reporter by gel analysis and (optionally) Northern hybridizations. This will address the potential RNase contamination of the reagents and equipment used in the procedure, which may lead to degradation of the translation reaction components.
Analysis of rRNA and tRNA integrity
Thaw one tube of the frozen CFE and measure its RNA concentration. For gel analysis, take an aliquot of the CFE and dilute it with RNase-free water to 2.5 μg/μl RNA. Mix 1 μl of this diluted CFE with 4 μl of the FAE solution (see Recipes) and heat the sample at 70°C for 5 min.
Be advised that the CFE obtained through the protocol described above contains a high amount of rRNA and tRNA. Dilute the CFE with H2O 100-fold before measuring RNA concentration, as described in (B2.e). After heating the sample with FAE (Shedlovskiy et al., 2017), it can be directly analyzed by the denaturing gel electrophoresis.
Analyze large molecular weight RNA species (25S and 18S rRNAs) by electrophoresis on formaldehyde-agarose gels as described in Mansour and Pestov (2013). To separate small molecular weight RNA species (tRNAs, 5S and 5.8S rRNAs), use polyacrylamide denaturing gels as described in Shcherbik et al. (2016).
Dilute the SYBR Gold dye stock 1:10,000 (10 μl of SYBR Gold in 100 ml of H2O). Pour this solution into a clean plastic tray and stain the gel by incubating it for 20-30 min with slow rocking at room temperature. Visualize SYBR Gold-stained RNA bands using a suitable gel-imaging system or a laser fluorescence scanner.
(Optional) Transfer RNA from the gel to a nylon membrane and hybridize the blot with probes specific for different yeast rRNAs and tRNAs (probe sequences are provided in Table 5). Follow the hybridization protocol described in Pestov et al. (2008). We recommend first to use probes for 25S and 18S rRNAs, the large rRNA species whose degradation in lysates can be easily detected.
Table 5. Sequences of probes used to detect yeast rRNAs and tRNAs
Probe Sequence 25S rRNA 5’-TCCTACCTGATTTGAGGTCAAAC-3’ 18S rRNA 5’-AGAATTTCACCTCTGACAATTG-3’ 5.8S rRNA 5’-AAATGACGCTCAAACAGGCATG-3’ 5S rRNA 5’-TAACTACAGTTGATCGGACGG-3’ tRNAGlu 5’-TGGCTCCGATACGGGGAGTCG-3’ tRNAVal 5’-TGGTGATTTCGCCCAGGA-3’ tRNAAla 5’- GGTGGACGAGTCCGGAATCG-3’
Examine the quality of m7G-capped mRNA prior to and after the translation reaction
If a low translational activity is detected in a CFE using a protein reporter, there is a possibility that the reporter mRNA might have a secondary structure resulting in abortive synthesis by T7 polymerase. Another possibility is that mRNA generated in vitro might be degraded either during the synthesis or during translation reaction. To test these possibilities, proceed with the following steps:
After synthesizing the mRNA reporter (C1.a), analyze 2 μg of the generated RNA by agarose gel electrophoresis followed by SYBR Gold staining, as described in Steps C3a.iii. The detection of a single band would indicate that the mRNA is intact. If you see multiple bands, repeat mRNA synthesis using the mMESSAGE mMACHINETM T7 Transcription kit; however, heat the PCR DNA product before adding into the reaction at 65°C for 5 min followed by an immediate cooldown on the ice for 2 min. If this does not improve the quality of the generated mRNA, change all the solutions to rule out the possibility of RNase contamination. Follow other troubleshooting steps described in the mMESSAGE mMACHINETM T7 Transcription kit manual.
After the completion of the translation reaction (C1.b), extract RNA from the reaction using TRI-REAGENT-LS according to the manufacturer's protocol. Resuspend the RNA pellet in 50 μl of FAE (Shedlovskiy et al., 2017) and analyze 2.5 μg of RNA by northern hybridization using a probe specific for the mRNA reporter. Detection of a single band indicates that the mRNA is stable in the CFE-based translation reaction. The appearance of extensive degradation products may indicate the RNase contamination of reagents, in which case we recommend changing the solutions and reviewing the laboratory setup to ensure an RNase-free environment.
Data analysis
If the researcher chooses to use a luciferase reporter to assess the activity of the CFE, the amount of the generated products can be quickly estimated using a commercially available luciferase assay. This is a simple, quantitative, and reliable method to verify the translational competency of a newly prepared CFE batch. An example of a luciferase-based assay is shown in Figure 7.
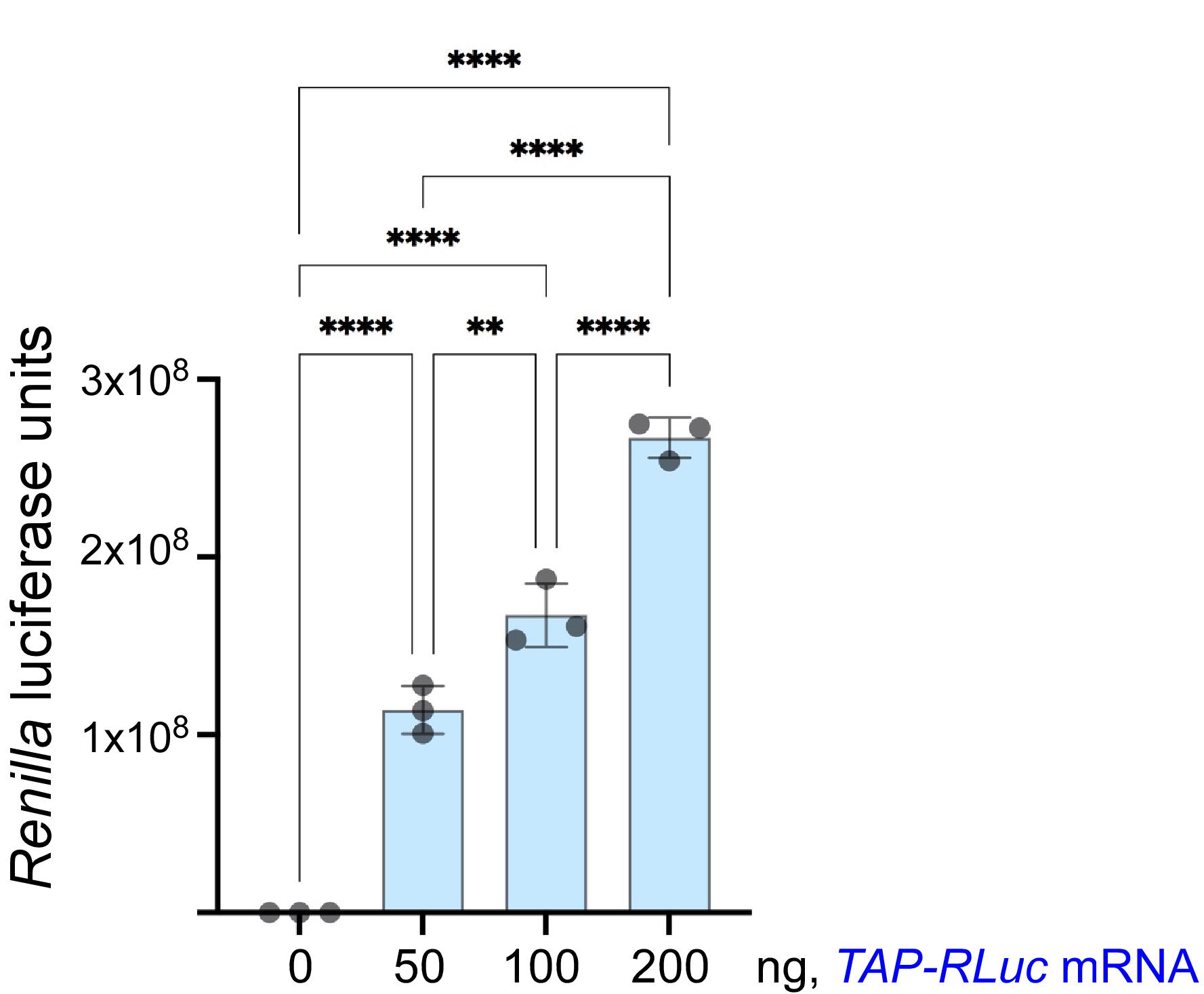
Figure 7. Detection of the TAP-RLuc reporter synthesis in a yeast CFE-based reaction. The indicated amounts of the TAP-RLuc mRNA reporter (Trainor et al., 2021) were added to CFE-based translation reactions set in triplicate. The reactions were incubated at 21°C for 2 h, and the products were analyzed using a commercial Renilla Luciferase Assay System (Promega). Luminescence was measured for 10 s after the addition of the detection reagent. Bars show mean values; error bars represent SEM. One-way ANOVA tests were used to compare the samples. **, P < 0.01; ****, P < 0.0001.An alternative way to estimate protein amounts synthesized from a reporter in a CFE-based reaction is to separate the translation products on a gel followed by immunoblotting with the appropriately selected antibodies. In our hands, a strong signal for a TAP-fused protein reporter is evident after a 30-min translation reaction performed with an active CFE (Figure 8). This method can be used with any protein reporter for which antibodies are available for immunodetection.
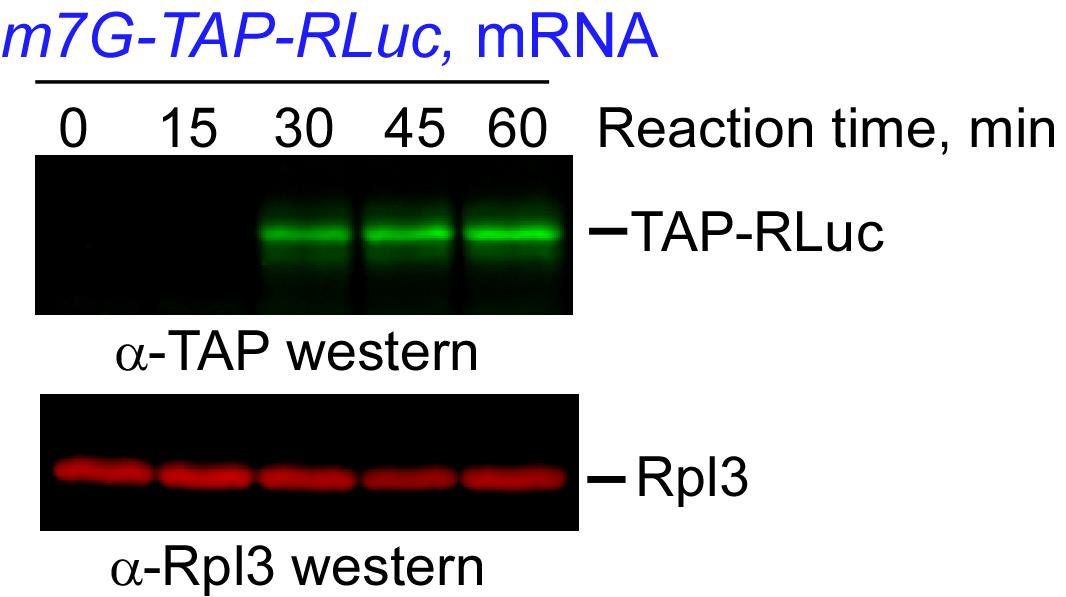
Figure 8. Immunoblot detection of a TAP-fused reporter translated in a yeast CFE. Aliquots of 2 μl were taken from a translation reaction at the indicated times, mixed with 2 μl of 2× SDS-PAGE loading dye, and resolved on a 10% SDS-polyacrylamide gel. Proteins were transferred onto a nitrocellulose membrane, blocked in 5% milk prepared in PBS, and incubated with a mixture of primary antibodies against TAP (rabbit polyclonal) and yeast Rpl3 (mouse monoclonal), the latter used as an internal control. The molecular weight of TAP-RLuc is ~56 kDa, Rpl3 is ~44 kDa. The blots were washed with 0.1% Tween-20 in PBS and incubated with IRDye-800CW anti-rabbit and IRDye-680RD anti-mouse antibodies. Fluorescent signals were detected using a Typhoon 5 imager at 800 nm and 680 nm.A CFE can also be tested by monitoring the amount of [35S-Met/Cys]-labeled polypeptides synthesized over the course of a translation reaction with endogenous mRNA. Cold methionine and cysteine should be added to a separate translation reaction to serve as a control (Figure 9).
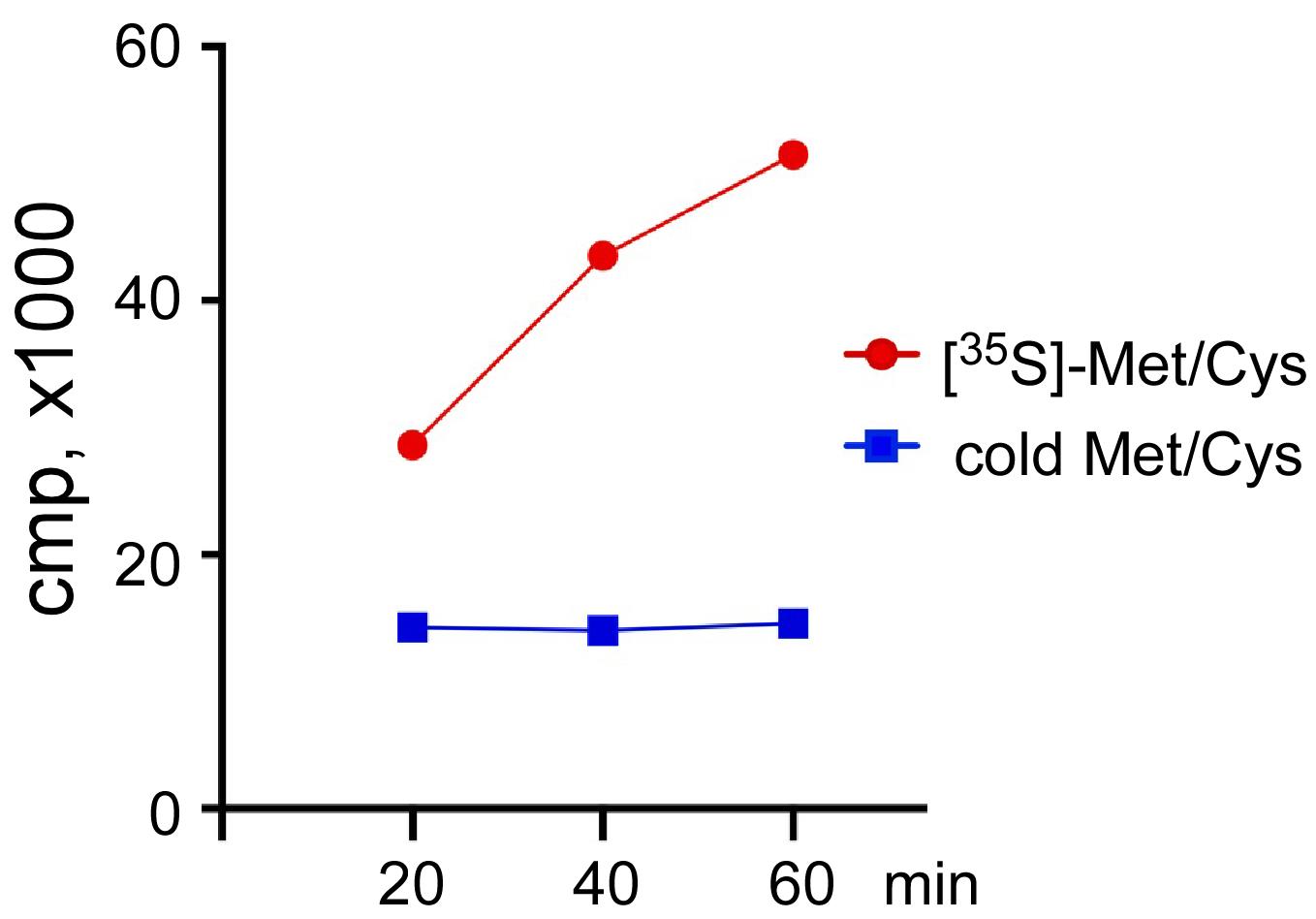
Figure 9. Incorporation of [35S]-Met/Cys into polypeptides synthetized in a CFE. [35S]-Met/Cys (red) or cold Met/Cys (blue) were added to two CFE-based translation reactions. Three 4 μl aliquots were collected every 20 min, and proteins were precipitated with TCA. The amount of [35S]-Met/Cys incorporated into polypeptide was estimated by scintillation counting of the counts per minute (CPM) in the TCA precipitates.The integrity of RNAs in CFE can be quickly assessed by SYBR Gold staining and/or Northern blotting (Figure 10). With our CFE preparation protocol, we expect to see only trace amounts of degradation of rRNAs, as indicated by additional faint bands appearing on gels below the main 25S and 18S bands, as compared with RNAs isolated directly from cells using the fast formamide-based extraction method (Shedlovskiy et al., 2017). No degradation of tRNAs or 5.8S rRNA was detected (Figure 10, compare “Cells” lanes and “CFE” lanes).
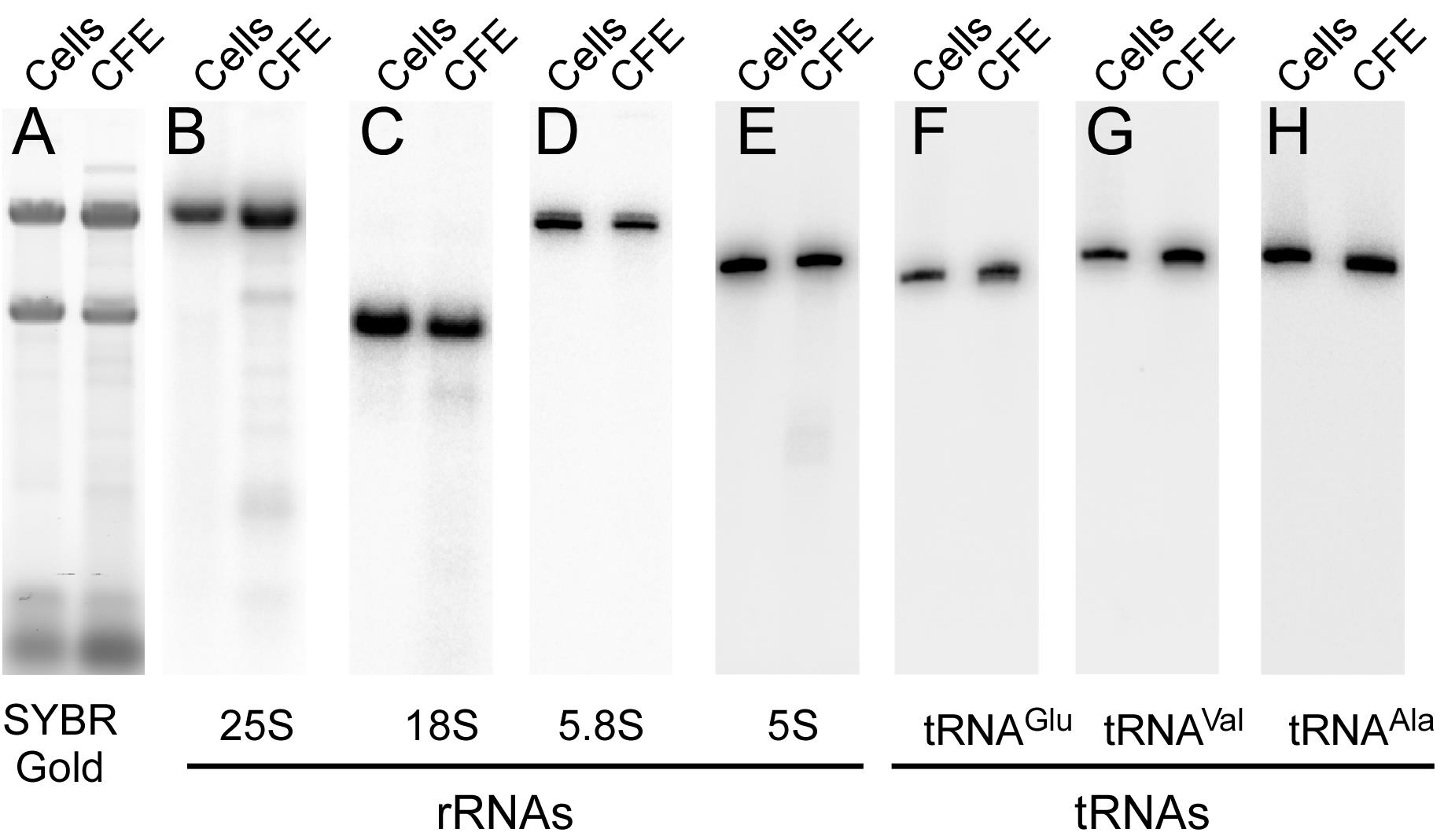
Figure 10. Determination of RNA integrity in a CFE. RNA derived from a CFE was resolved electrophoretically side-by-side with total RNA extracted from the same yeast strain by a one-step hot formamide method (Shedlovskiy et al., 2017) (lanes labeled “Cells”). Samples were resolved on 1.2% agarose gels containing 1.3% formaldehyde (panels A-C) or on 8M urea-10% polyacrylamide gels (panels D-H). A. RNA resolved on an agarose gel stained with SYBR Gold. B-C. Northern hybridizations of RNA resolved on agarose gels with [32P]-labeled probes against 25S and 18S rRNAs. D-H. Northern hybridizations of RNA resolved on polyacrylamide gels with probes against small rRNAs and tRNAs indicated at the bottom.
Notes
A critical parameter in the preparation of a yeast CFE is the culture growth conditions. Previously published protocols vary greatly with respect to how yeast culture should be grown. By testing different protocols, we identified culture conditions optimal for the preparation of translation extracts from BY4741 cells. The starting (overnight) yeast culture should reach ODλ=600nm ~10-12; when diluted to ODλ=600nm = 0.8, the culture should be next grown for no longer than 5-6 h at 30°C to reach ODλ=600nm ~2.4-2.5. In our hands, these conditions result in the highest yield and strong translational activity of the yeast lysate.
We recommend sterilizing yeast growth media used in this protocol by filtration through a 0.2 µm PES filter instead of autoclaving. Sterilization by filtration better controls the concentration of components in the medium and minimizes variability between CFE preparations.
To further improve translational activity, one may wish to modify the genetic background of the yeast strain used to generate the CFE. For example, deletion of the ribosome inactivating factor STM1 was reported to increase the activity of CFEs prepared from S. cerevisiae (Brodiazhenko et al., 2018). We also found that deletion of the secretory endoribonuclease gene RNY1 increases the stability of RNAs, such as rRNAs and tRNAs, in yeast lysates (Shcherbik, 2013).
The described protocol is optimized for use with translation reporters. To measure absolute amounts of the synthesized protein (in mg/ml), additional steps (such as affinity purification from the CFE translation mixture) are necessary.
To design primers for a gene of interest other than Luciferases, we recommend incorporating a T7 promoter sequence (shown in blue, below) in the forward primer sequence upstream of the 5’-end of a coding sequence of a gene. To increase the expression level of a protein in the cell-free translation system described here, the CYC1 5’-UTR enhancer sequence (marked in red below) can be incorporated in the forward primer sequence downstream of the T7 promoter and upstream of the coding region of a gene; however, this step is optional. The Tm for the forward primer should be calculated only for a sequence that anneals to a gene of interest (shown in black in bold lettering). We were successful using the Tm-calculator tool from New England Biolabs: https://tmcalculator.neb.com/#!/main. In this tool, a researcher can choose a polymerase that is available in a lab. We were successful using Phusion and Taq polymerases.
5’-ATCCGTAATACGCTCACTATAGGGAGCTTCACACTAAATTAATAATGXXXXXXXXXX-3’
For the reverse primer, a sequence complementary to the 3’-end coding sequence of a gene of interest is sufficient. Tm of the reverse primer should match the Tm of the forward primer. In our experience, Tm of 54-56°C works well for Phusion and Taq polymerases.
Recipes
Yeast medium
YPD medium
To make 1 L of the liquid YPD medium:
Mix 20 g of peptone, 10 g of yeast extract, 20 g of dextrose, add deionized H2O to 1 L.
Adjust pH to 6.0 with 1 M HCl.
Sterilize medium by filtration through a 0.2 µm PES filter.
Filtered YPD can be stored at RT in the dark for up to 2-3 months.
YPD-agar plates
Add 20 g of bacto-agar per 1 L of YPD medium; sterilize by autoclaving.
Stock solutions
1 M HEPES-KOH, pH 7.6
Take 238.3 g of HEPES acid, add deionized H2O to 0.9 L, adjust pH to 7.6 with 50% (w/v) KOH, and adjust volume to 1 L; sterilize by autoclaving. Store at room temperature or at 4°C.
2 M KOAc
Take 98.14 g of KOAc, add deionized H2O to 500 ml, and sterilize by autoclaving. Store at room temperature or at 4°C.
300 mM MgOAc
Take 6.4 g of magnesium acetate tetrahydrate (MgOAc·4H2O), add deionized H2O to 100 ml, and sterilize by autoclaving. Store at room temperature or at 4°C.
100 mM MgOAc
Take 2.14 g of magnesium acetate tetrahydrate (MgOAc·4H2O), add deionized H2O to 100 ml, and sterilize by autoclaving. Store at room temperature or at 4°C.
1 M DTT
Take 1.5 g of the DTT powder and add deionized H2O to 10 ml; vortex until you see no crystals. Sterilize the DTT solution by filtration through a 0.2 µm nylon syringe filter. Store at -20°C in 1-ml aliquots.
50% glycerol
Mix 500 ml of glycerol and 500 ml of deionized H2O, and sterilize the 50% glycerol solution by autoclaving. Store at room temperature or at 4°C.
Working solutions
Buffer A, 1 L
Mix 30 ml of 1 M HEPES pH 7.6, 50 ml of 2 M KOAc, and 10 ml of 300 mM MgOAc. Use sterile H2O to adjust the volume to 1L. Store at 4°C.
Buffer A/glycerol, 1 L
Mix 30 ml of 1M HEPES pH 7.6, 50 ml of 2 M KOAc, 10 ml of 300 mM MgOAc, and 400 ml of 50% glycerol solution. Use sterile H2O to adjust the volume to 1 L. Store at 4°C.
Buffer A/mannitol, 1 L
Mix 30 ml of 1M HEPES pH 7.6, 50 ml of 2 M KOAc, 10 ml of 300 mM MgOAc, and 750 ml of deionized H2O. Weight 85 g of mannitol, add to the solution, and adjust the volume to 1 L. Sterilize by autoclaving. Store at 4°C.
Buffer A/mannitol/DTT
Add DTT from the 1 M stock solution to buffer A/mannitol to a final concentration of 2 mM. This solution should be freshly made on the day of the procedure and kept on ice.
Note: For the yeast powder derived from 7-8 g of cell/buffer beads, 20 ml of buffer A/mannitol/DTT is required.
Buffer A/glycerol/DTT
Add DTT from the 1M stock solution to buffer A/glycerol to a final concentration of 2 mM. This solution should be freshly made on the day of the procedure and kept on ice.
Creatine kinase dilution buffer
Mix 0.1 ml of 1M HEPES pH 7.6 and 0.25 ml of 2 M KOAc. Add 50% sterile glycerol up to 10 ml (9.65 ml). Mix by vortexing and store at -20°C.
Creatine kinase solution
Add 0.1 ml of the creatine kinase dilution buffer to 1, 000 units of lyophilized creatine kinase to achieve the concentration of 10 units/μl. Store at -20°C with other enzymes.
10× Energy mix
Mix 0.2 ml of 1 M HEPES pH 7.6, 0.1 ml of 100 mM ATP, 0.01 ml of 100 mM GTP, 0.4 ml of 500 mM creatine phosphate, 0.02 ml of 1M DTT, and 0.27 ml of sterile H2O. The total volume is 1 ml. Store at -80°C in 0.03-ml aliquots.
FAE solution
Add 0.02 ml of 0.5M EDTA, pH 8.0 to 1 ml of formamide. Store frozen at -20°C.
Acknowledgments
This work was supported by the National Institutes of Health Grant R01GM114308 (to N. S.). We are acknowledging the original research paper (Trainor et al., 2021) published by our lab, wherefrom this protocol has been derived. We are thankful to Dan Smethurst for technical assistance. We would like to express our gratitude to Misha Anikin and Randy Strich for the stimulating discussion and useful suggestions.
Competing interests
The authors declare no competing interests.
References
- Brödel, A. K., Sonnabend, A. and Kubick, S. (2014). Cell-free protein expression based on extracts from CHO cells. Biotechnol Bioeng 111(1): 25-36.
- Brodiazhenko, T., Johansson, M. J. O., Takada, H., Nissan, T., Hauryliuk, V. and Murina, V. (2018). Elimination of Ribosome Inactivating Factors Improves the Efficiency of Bacillus subtilis and Saccharomyces cerevisiae Cell-Free Translation Systems. Front Microbiol 9: 3041.
- Buntru, M., Vogel, S., Spiegel, H. and Schillberg, S. (2014). Tobacco BY-2 cell-free lysate: an alternative and highly-productive plant-based in vitro translation system. BMC Biotechnol 14: 37.
- Carlson, E. D., Gan, R., Hodgman, C. E. and Jewett, M. C. (2012). Cell-free protein synthesis: applications come of age. Biotechnol Adv 30(5): 1185-1194.
- Chong, S. (2014). Overview of cell-free protein synthesis: historic landmarks, commercial systems, and expanding applications. Curr Protoc Mol Biol 108: 16.30.11-11.
- Ezure, T., Suzuki, T. and Ando, E. (2014). A cell-free protein synthesis system from insect cells. Methods Mol Biol 1118: 285-296.
- Gasior, E., Herrera, F., Sadnik, I., McLaughlin, C. S. and Moldave, K. (1979). The preparation and characterization of a cell-free system from Saccharomyces cerevisiae that translates natural messenger ribonucleic acid. J Biol Chem 254(10): 3965-3969.
- Ghosh, A. and Shcherbik, N. (2020). Cooperativity between the Ribosome-Associated Chaperone Ssb/RAC and the Ubiquitin Ligase Ltn1 in Ubiquitination of Nascent Polypeptides. Int J Mol Sci 21(18).
- Gregorio, N. E. and Levine, M. Z. (2019). A User's Guide to Cell-Free Protein Synthesis. Methods Protoc 2(1): 24.
- Harbers, M. (2014). Wheat germ systems for cell-free protein expression. FEBS Lett 588(17): 2762-2773.
- Hodgman, C. E. and Jewett, M. C. (2013). Optimized extract preparation methods and reaction conditions for improved yeast cell-free protein synthesis. Biotechnol Bioeng 110(10): 2643-2654.
- Hofbauer, R., Fessl, F., Hamilton, B. and Ruis, H. (1982). Preparation of a mRNA-dependent cell-free translation system from whole cells of Saccharomyces cerevisiae. Eur J Biochem 122(1): 199-203.
- Hussain, I. and Leibowitz, M. J. (1986). Translation of homologous and heterologous messenger RNAs in a yeast cell-free system. Gene 46(1): 13-23.
- Kelwick, R., Webb, A. J., MacDonald, J. T. and Freemont, P. S. (2016). Development of a Bacillus subtilis cell-free transcription-translation system for prototyping regulatory elements. Metab Eng 38: 370-381.
- Kim, J., Copeland, C. E., Padumane, S. R. and Kwon, Y. C. (2019). A Crude Extract Preparation and Optimization from a Genomically Engineered Escherichia coli for the Cell-Free Protein Synthesis System: Practical Laboratory Guideline. Methods Protoc 2(3): 68.
- Kovtun, O., Mureev, S., Jung, W., Kubala, M. H., Johnston, W. and Alexandrov, K. (2011). Leishmania cell-free protein expression system. Methods 55(1): 58-64.
- Li, J., Wang, H., Kwon, Y. C. and Jewett, M. C. (2017). Establishing a high yielding streptomyces-based cell-free protein synthesis system. Biotechnol Bioeng 114(6): 1343-1353.
- Mansour, F. H. and Pestov, D. G. (2013). Separation of long RNA by agarose-formaldehyde gel electrophoresis. Anal Biochem 441(1): 18-20.
- Mikami, S., Kobayashi, T. and Imataka, H. (2010). Cell-free protein synthesis systems with extracts from cultured human cells. Methods Mol Biol 607: 43-52.
- Olliver, L. and Boyd, C. D. (1996). In vitro translation of messenger RNA in a rabbit reticulocyte lysate cell-free system. Methods Mol Biol 58: 477-484.
- Pestov, D. G., Lapik, Y. R. and Lau, L. F. (2008). Assays for ribosomal RNA processing and ribosome assembly. Curr Protoc Cell Biol Chapter 22: Unit 22.11.
- Pelham, H. R., Jackson, R. J. (1976). An efficient mRNA-dependent translation system from reticulocyte lysates.Eur J Biochem 67(1): 247-256.
- Schoborg, J. A., Hodgman, C. E., Anderson, M. J. and Jewett, M. C. (2014). Substrate replenishment and byproduct removal improve yeast cell-free protein synthesis. Biotechnol J 9(5): 630-640.
- Shcherbik, N. (2013). Golgi-mediated glycosylation determines residency of the T2 RNase Rny1p in Saccharomyces cerevisiae. Traffic 14(12): 1209-1227.
- Shcherbik, N., Chernova, T. A., Chernoff, Y. O. and Pestov, D. G. (2016). Distinct types of translation termination generate substrates for ribosome-associated quality control. Nucleic Acids Res 44(14): 6840-6852.
- Shedlovskiy, D., Shcherbik, N. and Pestov, D. G. (2017). One-step hot formamide extraction of RNA from Saccharomyces cerevisiae. RNA Biol 14(12): 1722-1726.
- Shrestha, P., Holland, T. M. and Bundy, B. C. (2012). Streamlined extract preparation for Escherichia coli-based cell-free protein synthesis by sonication or bead vortex mixing. Biotechniques 53(3): 163-174.
- Tarun, S. Z., Jr. and Sachs, A. B. (1995). A common function for mRNA 5' and 3' ends in translation initiation in yeast. Genes Dev 9(23): 2997-3007.
- Thoring, L. and Kubick, S. (2018). Versatile Cell-Free Protein Synthesis Systems Based on Chinese Hamster Ovary Cells. Methods Mol Biol 1850: 289-308.
- Trainor, B. M., Ghosh, A., Pestov, D. G., Hellen, C. U. T. and Shcherbik, N. (2021). A translation enhancer element from black beetle virus engages yeast eIF4G1 to drive cap-independent translation initiation. Sci Rep 11(1): 2461.
- Tuite, M. F. and Plesset, J. (1986). mRNA-dependent yeast cell-free translation systems: theory and practice. Yeast 2(1): 35-52.
- Tuite, M. F., Plesset, J., Moldave, K. and McLaughlin, C. S. (1980). Faithful and efficient translation of homologous and heterologous mRNAs in an mRNA-dependent cell-free system from Saccharomyces cerevisiae. J Biol Chem 255(18): 8761-8766.
- Wang, H., Li, J. and Jewett, M. C. (2018). Development of a Pseudomonas putida cell-free protein synthesis platform for rapid screening of gene regulatory elements. Synth Biol (Oxf) 3(1): ysy003.
- Witherell, G. (2001). In vitro translation using HeLa extract. Curr Protoc Cell Biol Chapter 11: Unit 11.18.
- Wu, C., and Sachs, M.S. (2014). Preparation of a Saccharomyces cerevisiae cell-free extract for in vitro translation. Methods Enzymol 539: 17-28.
- Wu, C., Dasgupta, A., Shen, L., Bell-Pedersen, D. and Sachs, M. S. (2018). The cell free protein synthesis system from the model filamentous fungus Neurospora crassa. Methods 137: 11-19.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Trainor, B. M., Komar, A. A., Pestov, D. G. and Shcherbik, N. (2021). Cell-free Translation: Preparation and Validation of Translation-competent Extracts from Saccharomyces cerevisiae. Bio-protocol 11(18): e4093. DOI: 10.21769/BioProtoc.4093.
Category
Microbiology > Microbial genetics > RNA
Biochemistry > RNA > mRNA translation
Biochemistry > Protein > Expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.