- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Immunofluorescence of GFAP and TNF-α in the Mouse Hypothalamus
Published: Vol 11, Iss 13, Jul 5, 2021 DOI: 10.21769/BioProtoc.4078 Views: 6120
Reviewed by: Xiaoyi ZhengGisela HelferHe Liu
Original research article
The authors used this protocol in:

Jan 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Immunofluorescence is a reliable method for identifying specific proteins in neuronal and glial cell populations of the hypothalamus. Several immunofluorescence protocols are available to detect protein markers and neuropeptides in the hypothalamus; however, published methods may vary in subtle details that can potentially impact the final outcome of the procedure. Here, we provide a detailed protocol suitable for thin cryostat sections, which has been successful for specific antibodies directed against key markers of hypothalamic neurons and glial cells. We include every detail concerning brain tissue collection, processing, sectioning, and labeling with optimal dilutions of antibodies with the aim of reducing non-specific background. Our background-optimized immunostaining protocol has been routinely used in the lab and allows efficient detection of specific neuropeptides, glial cells, and markers of inflammation and endoplasmic reticulum stress in the hypothalamus.
Keywords: HypothalamusBackground
Elucidating the protein organization in neurons and glia is essential for understanding hypothalamic function. Immunofluorescence is a powerful tool to investigate changes in the expression of neuronal and glial proteins and to map activated brain areas at the cellular level. This imaging technique enables the visualization of highly specific protein (antigen) targets using a combination of primary and secondary antibodies. Antigenic determinants of interest in brain tissue are first treated with a monoclonal or polyclonal primary antibody, targeted toward peptides from the species/protein of interest generated in a host (mouse-, rabbit-, chicken-, rat-, donkey-derived, etc., are available), followed by a secondary antibody directed toward the host of the primary antibody and conjugated to a synthetic or natural fluorophore. The antibody targets often labeled in brain tissue sections are glial fibrillary acidic protein (GFAP), neuronal and axonal neurofilaments, neuropeptides, microtubule-associated proteins, blood-brain barrier proteins, pre- and post-synaptic proteins, myelin, markers of inflammation, and numerous antigens associated with specific diseases. Among the useful fluorescent markers for the visualization of antigens in brain tissue are rhodamine, fluorescein, the Alexa Fluor series, and the cyanine dyes. Counterstaining for nuclei using a variety of popular DNA-binding dyes (such as TO-PRO-3 iodide) follows treatment with the secondary antibody.
This method has been previously used by our laboratory to identify neuronal subpopulations in various hypothalamic nuclei involved in energy homeostasis (Dalvi et al., 2017). In particular, using double immunofluorescence staining, we identified GFAP in the hypothalamus and tumor necrosis factor (TNF)-α, a marker of inflammation, in the hypothalamic arcuate nucleus following acute lipid infusion and chronic high-fat diet feeding in mice, which may contribute to a significant alteration in content or expression of appetite-regulating neuropeptides. To the best of our knowledge, this was the first description of TNF-α induction demonstrated by immunofluorescence in the mouse hypothalamic arcuate nucleus after exposure to a high-fat diet. This method can be applied to the identification of other markers of inflammation and endoplasmic reticulum (ER) stress in the hypothalamic regions provided that reliable antibodies are available for detection. This protocol details a generalized procedure for staining frozen brain cryosections ranging from 20 to 30 µm in thickness. Figures 1 and 2 presented in this protocol are confocal images that reveal the expression of GFAP and TNF-α in the hypothalamic arcuate nucleus in 25 µM brain sections of mice treated acutely with lipid or chronically with a high-fat diet (Dalvi et al., 2017).
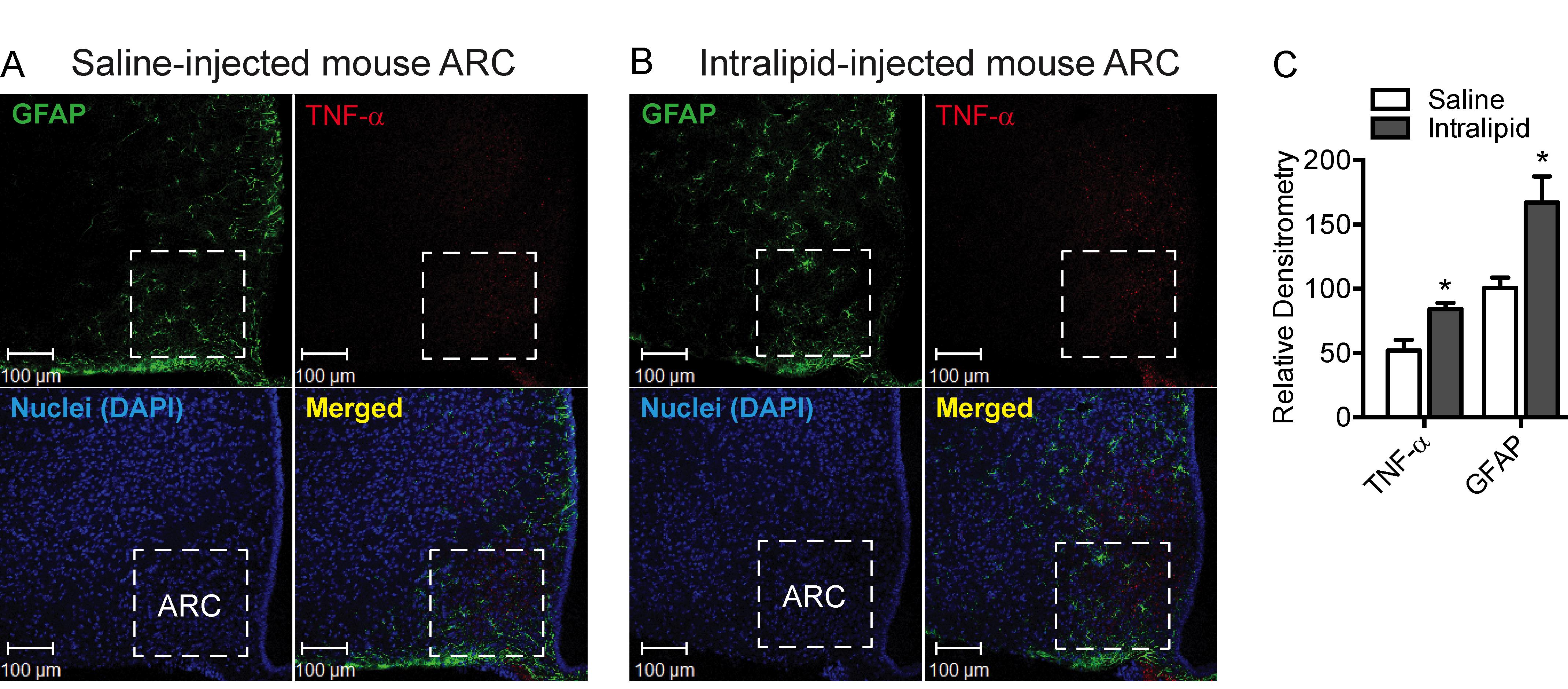
Figure 1. Effect of acute intralipid treatment at 24 h post-infusion in C57BL/6 mice on hypothalamic arcuate nucleus (ARC) reactive astrocytosis and expression of TNF-α, a marker of inflammation. A-B.Representative images showing immunofluorescence for the expression of GFAP (green) and TNF-α (red) in the hypothalamic ARC of C57BL6 mice at 24 h following acute infusion with saline (A) or Intralipid (B). C. Quantification of the immunofluorescence intensity for GFAP and TNF-α expression in the ARC of C57BL/6 mice at 24 h following acute infusion with saline or Intralipid. Data in the bar graph are expressed as the mean ± SEM (n = 5-6 animals/group); *P < 0.05 versus control. Images in A and B and the graph in C are adapted and modified from Dalvi et al. (2017).
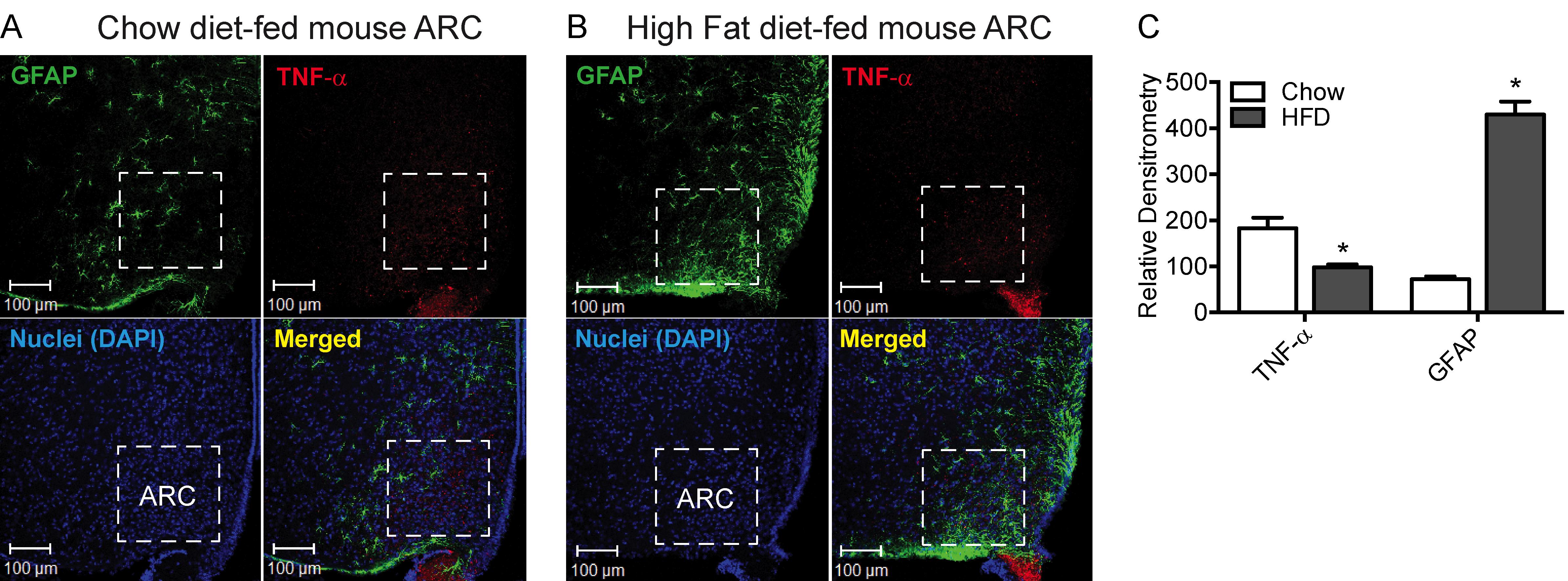
Figure 2. Effect of high-fat diet (HFD, 60% kcal) feeding for 8 weeks in CD-1 mice on reactive astrocytosis and expression of TNF-α in the hypothalamic arcuate nucleus (ARC). A-B. Representative images showing immunofluorescence for the expression of GFAP (green) and TNF-α (red) in the hypothalamic ARC of CD-1 mice fed chow (A) or an HFD (B) for 8 weeks. C. Quantification of immunofluorescence intensity of GFAP and TNF-α expression in the ARC of CD-1 mice fed either chow or an HFD for 8 weeks. Data in the bar graph are expressed as the mean ± SEM (n = 5-6 animals/group); *P < 0.05 versus control. Images in A and B and the graph in C are adapted and modified from Dalvi et al. (2017).
Materials and Reagents
50 ml Falcon tubes
Aluminum foil
Fine paint brushes
Cryostat blades (Leica Biosystems, catalog number: 3802106-TRI-FACET/LOPRO/CTD Blade-10PK)
12-well plates to store frozen sections (BD Falcon, Corning, catalog number: 353043)
24-well plates to store frozen sections (BD Falcon, Corning, catalog number: 353047)
Coverslips (25 × 50 mm) (VWR International, catalog number: 48393-059)
SuperFrost Plus slides (positively charged) (Thermo Fisher Scientific, catalog number: 12-550-15)
Wax pen (PAP Pen) (Abcam, catalog number: ab2601)
Fresh 4% paraformaldehyde in PBS (75 ml per mouse) (Millipore Sigma, catalog number: 158127-100G)
0.9% saline (Millipore Sigma, catalog number: S8776-100ML)
PBS pH 7.4 (Thermo Fisher Scientific, Gibco, catalog number: 10010023)
15% and 30% sucrose in PBS (300 g sucrose in 1 L PBS can be stored in the fridge for up to a month) (Millipore Sigma, catalog number: S7903-1KG)
Cryoprotectant solution (Millipore Sigma, catalog number: G7893-500M)
Tissue embedding solution (Tissue-Tek® O.C.T. compound) (Sakura® Finetek, VWR, catalog number: 25608-930)
Tween 20 for preparation of PBS-T (0.06% Tween-20 in PBS) (Millipore Sigma, catalog number: P1379-500ML)
Triton X-100 for preparation of PBS-Triton-X100 (0.4% Triton-X100 in PBS) (Millipore Sigma, catalog number: X100-500ML)
Normal donkey serum for preparation of blocking buffer (5% serum in PBS-Triton-X100) (Abcam, catalog number: ab7475)
Normal donkey serum for preparation of antibody buffer (2% serum in PBS-Triton-X100) (Abcam, catalog number: ab7475)
Nuclear stain: TOPRO (TO-PRO-3 iodide 642/661) (Life Technologies, catalog number: T3605)
Pro-Long Gold anti-fade mountant (mounting medium) (Thermo Fisher Scientific, catalog number: P10144)
Clear nail polish (or equivalent for sealing coverslips)
Primary antibodies:
Anti-GFAP: Chicken-derived polyclonal mouse antibody against GFAP (1:500 dilution) (Abcam, catalog number: ab4674)
Anti-TNF-α: Goat-derived polyclonal mouse antibody against TNF-α (1:50 dilution) (Santa Cruz, catalog number: SC-1350)
Secondary antibodies (fluorescently tagged secondary antibody against species from which primary antibodies are obtained):
Donkey anti-Chicken IgY (H+L) Secondary Antibody, FITC (will interact with the GFAP chicken-derived anti-mouse primary antibody) (Thermo Fisher Scientific, Invitrogen, catalog number: SA1-72000)
Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 (will interact with the TNF-α goat-derived anti-mouse primary antibody) (Thermo Fisher Scientific, Invitrogen, catalog number: A-11057)
Equipment
Scalpel
Tweezers
Fine scissors
Spatula
-20°C freezer
Adult Mouse Brain Slicer Matrix (Zivic Instruments, catalog number: BSMAS001-1)
Mouse perfusion apparatus (VWR, Leica Biosystems, catalog number: 10030-382)
Cryostat (Leica, model: CM3050S or similar)
Confocal laser scanning microscope ((Zeiss, model: LSM 510 or similar)
Software
AxioVision 3.0 imaging software (Carl Zeiss GmbH, Jena, Germany)
ImageJ software, National Institutes of Health, Bethesda, MD, USA (Schneider et al., 2012)
GraphPad Prism software, Inc., La Jolla, CA, USA
Procedure
Isolation and preparation of brain tissue for immunofluorescence
Perfusion of mice and brain extractionOn the morning of the perfusion (or the day before), make fresh 4% paraformaldehyde (PFA) (Gage et al., 2012).
Note: Prepare this solution either fresh or no more than 72 h in advance and store at 4°C.
Using a transcardiac approach, perfuse with 20 ml cold 0.9% saline (or PBS) followed by 20 ml cold 4% PFA (up to 25 ml maximum over 5-10 min by slow perfusion).
After the mouse is perfused, extract the brain using a scalpel, tweezers, fine scissors, and spatula (Papouin et al., 2018), gently place the brain into a 50-ml Falcon tube filled with 4% PFA (30-40 ml), and shake for at least 6-8 h (but not more) post-perfusion at 4°C.
Prepare 15% and 30% sucrose in PBS (300 g sucrose in 1 L PBS can be stored in the fridge for up to a month).
Place brains in cold 15% sucrose solution in a 50-ml Falcon tube at 4°C on a shaker overnight or until they sink.
Place brains in a cold 30% sucrose solution in a 50-ml Falcon tube at 4°C on a shaker for 2-3 days or until they sink. The brains should then be cut on a cryostat within a week after freezing (as described below).
Once brains have settled to the bottom of the tube, snap-freeze them in an isopentane bath (as described below).
For investigation of the hypothalamus, before freezing the entire brain, cut it coronally anterior and posterior to the hypothalamus, such that the rostral (anterior) cut is just at the optic chiasm and the caudal (posterior) cut is just at the end of the hypothalamus (between the hypothalamus and pons). Use the mouse brain atlas (Paxinos et al., 2001) and a mouse brain slicer matrix to orient the brain anatomy and to accurately cut the brain. The middle cut parts of the brain will contain the hypothalamus.
Get a small container and fill it halfway with dry ice. Add isopentane to cover the ice. Place a small beaker containing some isopentane in the middle of the bath. Then one by one, place the three cut parts from each brain into the beaker to freeze them. They should turn white within 10-15 s.
Once frozen, wrap the brains in aluminum foil, label them, and leave at -80°C until ready to section.
Sectioning the brains
Note: Brain sections can be either collected in a 12- or 24-well plate or directly on a glass slide, depending on the region of interest, availability of time, and the cost of antibody. The sections in a 12- or 24-well plates can be stored at -20°C until further use, but sections on a slide need to be processed right after collection. If processing many sections, ideally use a 24-well plate for processing (but this will require more antibody). However, if processing only a few sections, use a slide to use less antibody.
Turn on the cryostat (Leica CM-3050-S) well in advance.
Note: The machine takes about two hours to cool to -24°C. Make sure this temperature is reached before continuing. Throughout sectioning, ensure that the cabinet temperature remains between -18°C and -24°C. A temperature range may be required, as the optimal temperature for cryostat sectioning depends on the nature of the tissue, whether the tissues have been freshly frozen or pre-fixed with subsequent cryoprotection, the type of cryostat, and how well perfusion worked, etc. If there are problems with cutting, the temperature can be increased/decreased. In particular, if there is a problem with sectioning of pre-fixed brains, try cutting at a lower temperature, as the pre-fixed brains need a very low temperature.
Take the brains to the cryostat on dry ice. If only sections of the hypothalamus are needed, take out only the middle cut part of the brain corresponding to the hypothalamus.
Note: Work quickly or the brains will deform. Place a pair of forceps on the dry ice with the brains.
Clean the cover and platform of the cryostat with ethanol.
Note: The lever on the side of the platform needs to be lifted toward you to unlock the platform.
Take out the tissue embedding platforms (O.C.T. molds) from the cryostat with the embedding solution (O.C.T. compound). Cover the top of the platform with the solution, moving in a spiral motion, and place the platform on the dry ice.
Using the cold forceps, quickly transfer the brain to the platform with its caudal end sitting on it and the free rostral end facing upward. Then add more embedding solution around the brain and on top to cover it. Leave these on the dry ice until the solution begins to freeze (will turn white). Place the platform in the holes on the left side of the interior of the cryostat (Figure 3A, 3B).
Note: Another common method is to pre-freeze tissue in embedding solution on the embedding platform (i.e., O.C.T. compound within O.C.T. mold) before sectioning and to store at -80°C. However, this method has not been tested in our lab, so we are unable to predict the effect of pre-embedding and storage at -80°C on the protein staining.
Once all the brains have been moved into the machine, close the lid and leave it for about an hour so the brains and the cryotome blade reach the same temperature.
Once brains have frozen, position the blade using the black handle at the side of the stage.
Note: Use extreme caution around the cryostat blades as they are dangerously sharp.
Label the cover and sides of a 24-well plate (include thickness of samples, date, etc.). Fill each well at least halfway with cryoprotectant solution using a larger Pasteur pipette. Use one 24-well plate per each mouse brain.
Lock the embedding platform with sample into the front holder and adjust into position.
Note: You can move the entire apparatus closer to the blade by pushing the button at the front of the cryostat, rather than just turning the dial.
Ensure the machine is set to the desired thickness using the dial on the cryostat.
Note: 20-30 µm thickness is often used for immunofluorescence.
Proceed with sectioning the brain (Figure 3C). Use the fine paint brush to transfer sections to the 24-well plate (Figure 3D, 3E).
Note: You can discard the first few slices as they will likely consist of just mounting medium and not the brain tissue.
Once the brain becomes visible, place the sections in sequential wells within the 24-well plate containing cryoprotectant (Figure 3E, 3F).
Notes:
Use the mouse brain atlas (Paxinos et al., 2001) to orient the anatomy of the brain (hypothalamic) slices; this needs practice. First, the two lateral ventricles appear at the beginning of the sectioning, and subsequently the third ventricle appears that has hypothalamus on each side.
In adult mouse, the hypothalamus is about 5 mm in length, so there will be about 250 sections with 20 µm thickness and about 166 sections with 30 µm thickness. Thus, if brain sections are collected sequentially in each well of a 24-well plate, each well will contain 6-10 sections from throughout the hypothalamus.
Alternatively, using the atlas, a certain number of brain sections (i.e., about 6-10 sections) can be collected sequentially in each well of a 24-well plate. It must be noted how the sections are collected to avoid confusion at the time of processing for immunofluorescence.
Try to minimize the contact area between the brush and the section when transferring them from the cryostat to the plate to avoid damage. Also, try to lower the section into the center of the well so as to avoid making contact with the sides.
Once complete, cover the plate with plastic wrap and store in a -20°C freezer.
Note: Be sure to clean the cryostat and embedding platforms after use (most importantly, remove the blade to preserve its lifespan).

Figure 3. Steps of sectioning a mouse brain and transferring cut sections to cryoprotectant solution in a 24-well plate. A. Tissue embedding platforms (O.C.T. molds) are used, the top of the platforms are covered with the solution moving in a spiral motion, and the platforms are placed on the dry ice. Using cold forceps, the brain is quickly transferred onto the platform with its caudal end sitting on it and the rostral free end facing upward. More embedding solution is added around the brain and on top to cover it sufficiently. B. The platforms with brains frozen on top of them are left on the dry ice until the solution begins to freeze (will turn white). Once all the brains have been moved into the cryostat machine, the lid of the cryostat is closed and the brains are left inside for an hour so that the brains and the cryotome blade reach the same temperature. C. Using the desired thickness on the cryostat (20-30 µM thickness is often used for immunofluorescence), sectioning of the brain tissue is performed. D and E. Using a fine paint brush, the cut frozen brain sections are transferred to a 24-well plate filled with cryoprotectant solution. F. A magnified image of a single well of a 24-well plate filled with cryoprotectant solution and floating brain sections inside.
Labeling of brain sections: Blocking and incubation with primary and secondary antibodies
Take out 24-well plates containing brain sections from the -20°C freezer (each 24-well plate contains brain slices from an individual mouse in cryoprotectant). Take a new 24-well plate and fill all wells with PBS-T (Figure 4A). Transfer brain sections from one or two wells from an individual mouse to one separate well of the new 24-well plate filled with PBS-T (Figure 4B). Use fine paint brushes to transfer the brain sections from the cryoprotectant into PBS-T. The transfer from cryoprotectant to PBS-T will cause the cryoprotectant and embedding medium to diffuse, and the brain sections will spread.
Note: Prepare at least 2 wells for the negative controls of the primary antibody. This provides more control sections for imaging (the sections can easily get damaged during sequential frequent washing) (Figure 4B).
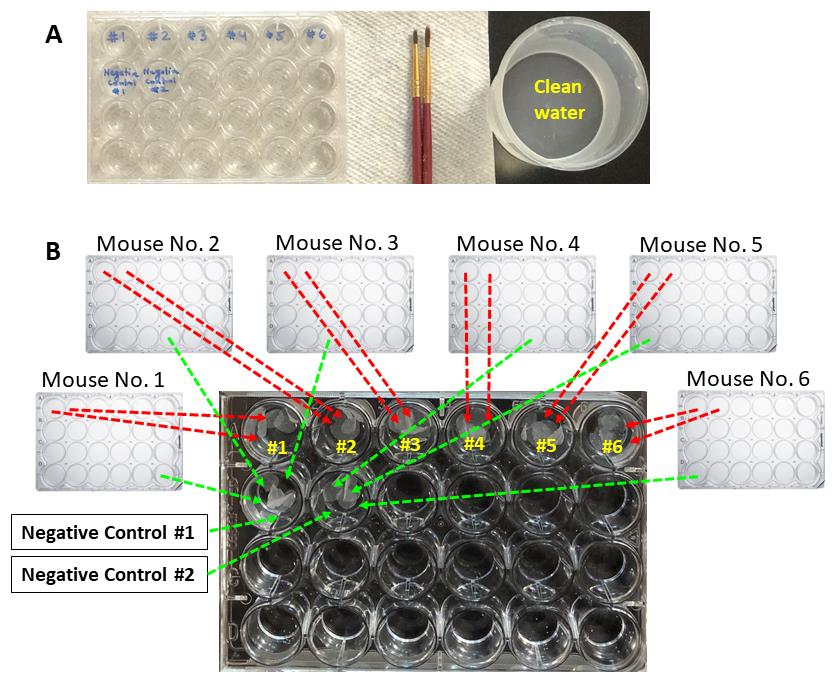
Figure 4. Transferring cut sections of brains stored in cryoprotectant solution in a 24-well plate to a new 24-well plate containing 0.06% Tween-20 in PBS (PBS-T). A. A new 24-well plate filled with PBS-T is prepared, and fine paint brushes and containers with clean water for washing paint brushes are kept ready. B. All 24-well plates containing brain sections immersed in cryoprotectant from an individual mouse are taken out, and brain sections from one or two wells from an individual mouse are transferred to a separate well of the new 24-well plate filled with PBS-T. Fine paint brushes are used to transfer the brain sections from the cryoprotectant into PBS-T. At least 2 wells for the negative controls of the primary antibody are prepared using brain sections from 1 to 3 mice.Wash 4× with PBS-T, 15 minutes per wash on a gyrating platform. For the “wash” steps, each time transfer the brain sections to a new well filled with PBS-T using fine paint brushes (see Video 1). Use separate brushes for the negative control brain sections and start washing with those sections first to avoid any contamination.
 Video 1. Transferring brain sections to a new well using fine paint brush during the “wash” steps.
Video 1. Transferring brain sections to a new well using fine paint brush during the “wash” steps.Note: Secure the plates properly and monitor the speed – if the speed increases rapidly, the plates may be dislodged if not secured properly.
In between the washes:
Thaw aliquots of serum. The serum should be the same as the host species of your secondary antibody.
Note: If the host species of both secondary antibodies was donkey, use donkey serum
Prepare blocking buffer using PBS-T.
Prepare extra blocking buffer because it can be diluted further to prepare the primary antibodies.
For blocking the potential background in the tissue, use 1 ml blocking solution per well, and the remaining can be diluted and used to prepare the primary antibodies.
Transfer brain sections into the blocking buffer and incubate for a minimum of 2 h at room temperature or optimally overnight at 4°C.
Note: 10% serum in PBS-T can also be used to block the tissue but for a minimum of 1 h.
After blocking, transfer the brain sections into the antibody buffer with the appropriate dilution of the primary antibody(s).
Note: Diluted primary antibodies can be mixed together and incubated simultaneously. Add 0.5 to 1.0 ml antibody buffer to each well in a 24-well plate. Using less antibody will decrease costs.
Incubate for 12-24 h at 4°C.
Wash 4× with PBS-T buffer for 15 min per wash on a gyrating platform at room temperature.
Note: Start with the primary antibody negative control sections first to avoid contamination with the primary antibodies.
Block the sections with 5% blocking buffer for 1 h to decrease background.
Note: It is acceptable to skip this step, if necessary, as the tissue should already be sufficiently blocked.
Transfer sections to the antibody buffer with an appropriate dilution of the secondary antibodies and nuclear stain.
Notes:
Typically, 1:500 dilution is used for FITC secondary antibodies, and 1:1,000 dilution is used for Alexa Fluor secondary antibodies and the nuclear stain (TOPRO).
Use some sections from each group for negative secondary antibody controls. Make a separate 24-well plate with these sections and use a separate fine paint brush to avoid any contamination with the secondary antibodies.
Incubate for 1 h at room temperature or overnight at 4°C.
Note: Cover with aluminium foil to avoid bleaching of the fluorescent tag.
Wash 4× with PBS-T following secondary antibody incubation on a gyrating platform for 15 min per wash.
Note: Cover with aluminium foil when possible to avoid bleaching of the fluorophore.
After the final wash, transfer the sections to PBS, and slowly mount the slices onto SuperFrost Plus slides using fine paint brushes.
Note: Try to minimize exposure of slices to ambient lighting, maintaining the foil cover over the sections when possible. Place the slides inside a slide holder box with a lid to avoid exposing the brain sections to light. The entire procedure of primary and secondary antibody conjugation can be done on a slide. After placing a section on a slide, circle it with wax pen and stain the slides following the above procedure. This will use much less reagents and cut costs substantially with only a few slices.
Wait until all sections on the slide are dry.
Spin the anti-fade mounting media at 1200 rpm for 10 min at 4°C in a centrifuge so that any solid particles will settle at the bottom of the tube and will not create any background signal.
Dispense the minimum amount of anti-fade mounting media and mount a coverslip.
Dry for 24 h at room temperature or 4°C in the dark, and seal edges with clear nail polish.
Capture images from the immunostained sections on a Zeiss LSM 510 confocal upright microscope fitted with a color digital camera and AxioVision 3.0 imaging software. Capture fluorescent images using an identical exposure time without saturating the pixel intensities (Figure 1A, 1B, 2A, 2B).
Data analysis
Use the ImageJ software (Schneider et al., 2012) to measure the intensity of immunofluorescence (densitometry) in a manner such that the samples are not identified to the assessor (blinded).
To measure fluorescence intensity, background is first determined based on the average intensity in several areas without any detectable immunoreactivity, and intensity is then determined as the signal-to-background intensity ratio using the ImageJ software.
In each group of control or treated animals, use the mean value of intensity measurements of all sections from an individual animal for statistical analysis.
Express the results as the relative intensity of markers of glial or neuronal cells in the hypothalamus for each group of mice or, if two antibodies are used, the coexpression of the markers of inflammation and endoplasmic reticulum stress in the hypothalamic arcuate nucleus for each group of mice.
Analyze the data using the GraphPad Prism software. Compare the two groups using the two-tailed Student’s t-test. For more than two groups, perform statistical analysis using either one-way or two-way analysis of variance (ANOVA), as appropriate, and determine statistical significance by a post-hoc analysis using the Bonferroni method, Holm-Sidak method, or Student’s t-test with P < 0.05 (Figures 1C, 2C).
Notes
Alternatives
If perfusion with 4% PFA is not possible, or if the perfusion was not successful, the brains should be post-fixed overnight in 4% PFA at 4°C.
Brain sections can be incubated at room temperature for 2-24 h depending on the prominence of expression of the protein of interest.
For double-labeled immunofluorescence, far-red fluorescent nuclear stain is recommended, but for single-labeled immunofluorescence, TOPRO should be used. Alternatively, other nuclear stains, such as DAPI or Hoechst, can be used depending on the wavelengths of the other antibodies being used.
Troubleshooting
If the brains are not properly cryoprotected, there is a higher chance of getting freezing artifacts.
If the antibody dilution is not correct, there is a possibility that the antibody signal will be lost and there will be too much background for accurate analysis. You may need to adjust the dilutions of primary, as well as secondary antibodies, to determine the optimal concentrations required.
It is recommended to be familiar with the thorough topographic anatomy of the organ under investigation, especially with the brain. There are reference books available for this task. Use the mouse brain atlas (Paxinos et al., 2001).
For better resolution, do not use a spinning disk imaging microscope.
Acknowledgments
We thank the Canadian Institutes for Health Research (CIHR), Canadian Diabetes Association (CDA), and Canada Research Chairs (CRC) Program for funding this study (DDB). Scholarship support through the Banting and Best Diabetes Centre (BBDC), National Sciences and Engineering Research Council (NSERC), and the Ontario Graduate Scholarship (OGS) Program (to PSD) was much appreciated. This protocol was adapted from Dalvi et al. (2017).
Competing interests
The authors declare no competing interests.
Ethics
All animal experiments were performed with approval from the Animal Care Committee of the University of Toronto, Canada. Standard approved housing conditions consisted of a 12 h light-dark cycle and housing with food and water ad libidum, with the recommended environmental enrichment.
References
- Dalvi, P. S., Chalmers, J. A., Luo, V., Han, D. Y., Wellhauser, L., Liu, Y., Tran, D. Q., Castel, J., Luquet, S., Wheeler, M. B. and Belsham, D. D. (2017). High fat induces acute and chronic inflammation in the hypothalamus: effect of high-fat diet, palmitate and TNF-α on appetite-regulating NPY neurons. Int J Obes (Lond) 41(1): 149-158.
- Gage, G. J., Kipke, D. R. and Shain, W. (2012). Whole animal perfusion fixation for rodents. J Vis Exp (65): 3564.
- Papouin, T. and Haydon P. G. (2018). Obtaining Acute Brain Slices. Bio-protocol 8(2): e2699.
- Paxinos, G. and Franklin, K. (2001). The mouse brain in stereotaxic coordinates. ISBN: 0125476361, 9780125476362. Academic Press.
- Schneider, C. A., Rasband, W. S. and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9(7): 671-675.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Dalvi, P. S. and Belsham, D. D. (2021). Immunofluorescence of GFAP and TNF-α in the Mouse Hypothalamus. Bio-protocol 11(13): e4078. DOI: 10.21769/BioProtoc.4078.
Category
Neuroscience > Cellular mechanisms > Microglia
Biochemistry > Protein > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.