- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Single-molecule Fluorescence Technique to Monitor the Co-transcriptional Formation of G-quadruplex and R-loop Structures
Published: Vol 11, Iss 13, Jul 5, 2021 DOI: 10.21769/BioProtoc.4069 Views: 4395
Reviewed by: ilgen MenderChristina Yan Ru TanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
G-quadruplexes (GQ) and R-loops are non-canonical nucleic acid structures related to gene regulation and genome instability that can be formed during transcription; however, their formation mechanisms remain elusive. To address this question, we developed a single-molecule fluorescence technique to monitor the formation of G-quadruplex and R-loop structures during transcription. Using this technique, we found that R-loop formation precedes GQ formation and that there exists a positive feedback loop between G-quadruplex and R-loop formation.
Keywords: Single-molecule fluorescenceBackground
G-quadruplexes (GQ) consist of stacked G-tetrads that are formed from four Hoogsteen base-paired guanines (Gellert et al., 1962). GQ formed at certain hotspots in the genome (Biffi et al., 2013; Marsico et al., 2019) play regulatory roles (Siddiqui-Jain et al., 2002; Falabella et al., 2019) and are related to certain diseases (Biffi et al., 2014; De Magis et al., 2019). R-loops are three-stranded nucleic acid structures composed of a double-stranded DNA-RNA hybrid and a single-stranded DNA. R-loops are also related to gene regulation and genome instability (Aguilera and García-Muse 2012; Santos-Pereira and Aguilera 2015). Interestingly, GQ and R-loops are formed during transcription and often coexist (Duquette et al., 2004).
The biological roles of GQ and R-loops and their formation mechanisms have been extensively studied over the last few decades. GQ and R-loop formation has previously been detected using bisulfite sequencing (Roy and Lieber 2009) or sequencing after immunoprecipitation (Chan et al., 2014; Hansel-Hertsch et al., 2018). Fluorescent ligands that bind specifically to GQ have also been used to observe GQ formation (Kreig et al., 2015). These techniques have provided understanding of the hotspots where GQ and R-loops are often formed (Sanz et al., 2016), in addition to the enzymes that regulate the formation of GQ and R-loops (Masse and Drolet 1999); however, their exact formation mechanisms remain to be elucidated.
Here, we report single-molecule fluorescence techniques that monitor the cotranscriptional formation of GQ and R-loops. To detect GQ formation, we used FRET (Fluorescence Resonance Energy Transfer), and to detect R-loop formation, we used the fluorescently labeled S9.6 antibody (Figure 1). We monitored GQ and R-loop formation during transcription and found that R-loop formation precedes GQ formation and there exists a positive feedback loop between GQ and R-loop formation.
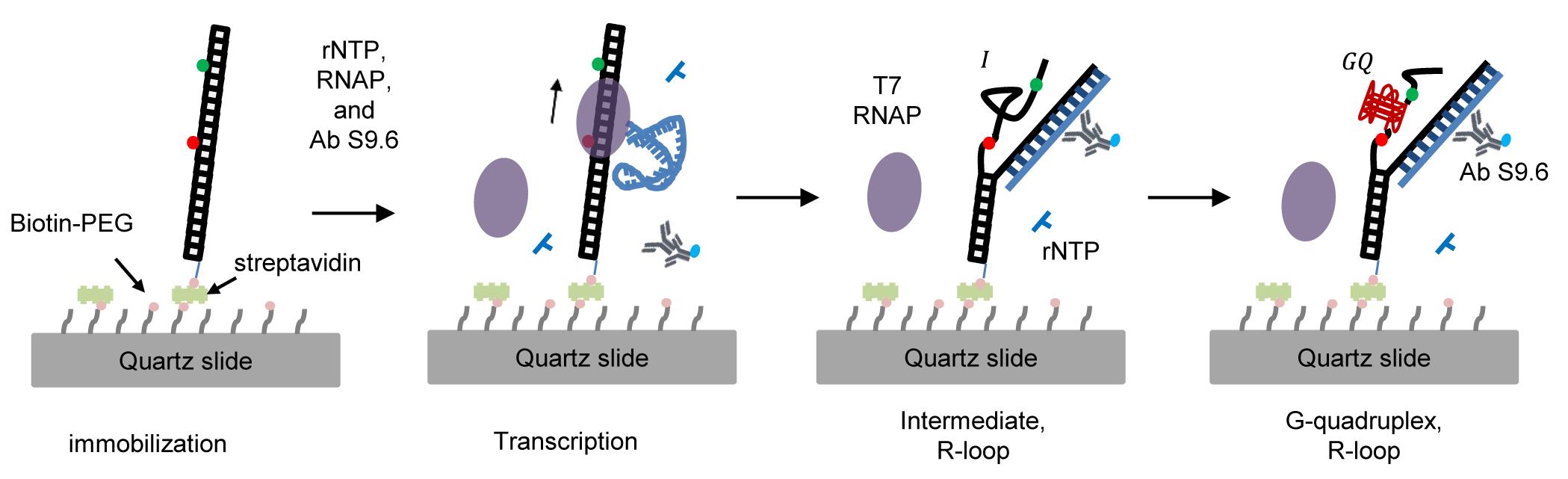
Figure 1. Single-molecule fluorescence technique to monitor the cotranscriptional formation of GQ coupled with R-loops. GQ and R-loop formation is monitored by high FRET efficiency and labeled S9.6 antibody, respectively.
Materials and Reagents
Double-sided tape (3M, catalog number: 137-ROK)
Polyethylene tubing (BD Intramedic, Fisher Scientific, catalog number: 427411)
Quartz microscope slide (H. FINKENBEINER Inc., USA, catalog number: custom-order, size : 1’’ × 3’’ × 1mm)
Micro cover glass (VWR, USA, catalog number: 48393230)
DNA oligos (Figure 2)
Non-template strand (1/2; purchased from IDT):
ATCAGGTCTAATACGACTCACTATAGGAAGAGAAAGT/iCy5/TTCTGGGAGGG
Non-template strand (2/2; purchased from IDT):
/5Phos/AGGGAGGGTGTA/iCy3/CTGATGCGTTCCACTCGC
Template strand (1/1; purchased from IDT):
GCGAGTGGAACGCATCAGTACACCCTCCCTCCCTCCCAGAAACTTTCTCTTCCTATAGTGAGTCGTATTAGACCTGAT/biotin/
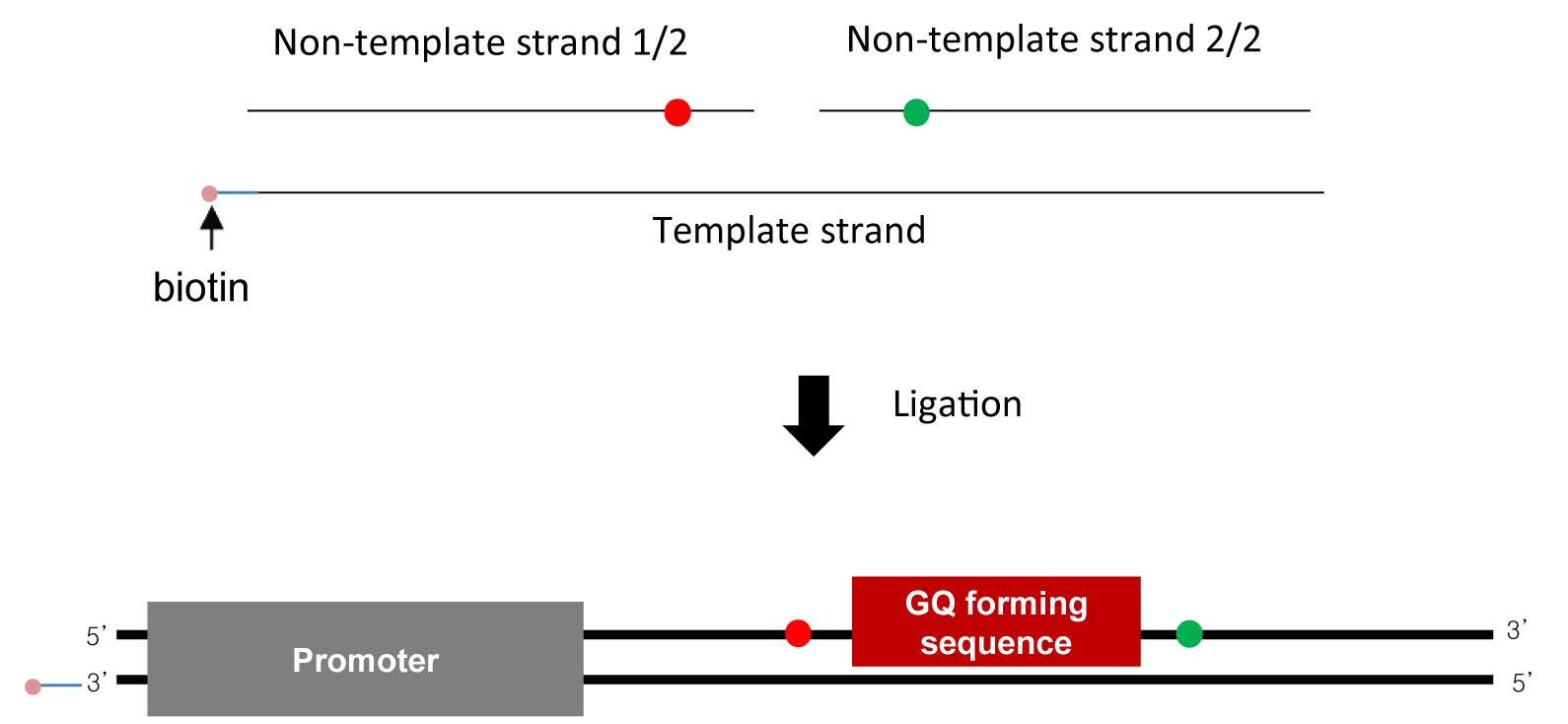
Figure 2. Sample design. DNA substrate is made using annealing and ligation (top). DNA substrate contains a T7 promoter and a GQ-forming sequence. Biotin (pink) is labeled at the upstream end for surface immobilization, and Cy3 (green) and Cy5 (red) are labeled as a FRET pair to detect GQ formation.Distilled water
Liquid nitrogen
Acrylamide:bis=29:1 (30%) (Biosesang, catalog number: A2003-1)
10× TBE (Biosesang, catalog number: TR2004-100-00)
N, N, N’, N’-tetra-methylethylenediamine (TEMED) (Bio-Rad, catalog number: 161-0801)
Ammonium persulfate (APS) (Sigma-Aldrich, catalog number: 1001799334)
Formamide (Sigma-Aldrich, catalog number: 01246)
UREA (Bio-Rad, catalog number: 161-0731)
Ethanol (Carlo Ebra Reagents, catalog number: 528167)
Alexa Fluor 488 C5-maleimide (Thermo Fisher, Invitrogen, catalog number: A10254)
NAP-5-column (GE Healthcare, catalog number: 17-0853-02)
Primary antibody: Anti-DNA-RNA hybrid (S9.6) antibody (Kerafast, catalog number: ENH001)
Note: We divide the stock solution of S9.6 antibody into aliquots, freeze each tube in liquid nitrogen, and store them at -20°C (short term) or -80°C (long term).
Secondary antibody: Anti-Mouse IgG (H+L) (Jackson ImmunoResearch, catalog number: 715-005-151)
Sulfuric acid (Daejung, catalog number: 7664-93-9)
Hydrogen peroxide solution (30%) (DaeJung, catalog number: 7722-84-1)
Biotin-PEG-SC (Laysan Bio Inc., catalog number: 141-63)
mPEG-SVA (Laysan Bio Inc., catalog number: 149-75)
Epoxy glue (LOCTITE, catalog number: 326795)
T4 DNA ligase (New England Bio Labs, catalog number: M0202M)
10× T4 DNA ligase buffer (New England Bio Labs, catalog number: B0202S)
T7 RNA polymerase (New England Bio Labs, catalog number: M0251S)
Trolox (Sigma-Aldrich, catalog number: 238813)
PCD (Oriental Yeast Co., catalog number: 46852004)
PCA (Sigma-Aldrich, catalog number: 37580)
Spermidine (Sgima-Aldrich, catalog number: 85558)
Note: Only a small amount to be used within a week is diluted and stored in a drawer from which light is completely blocked. After using the stock solution, the air in the container should be replaced with nitrogen gas and stored at room temperature.
rNTP set (GE Healthcare, catalog number: 27202501)
1,4-dithiothreitol (Merck, catalog number: 222-468-7)
Note: DTT is known to be easily oxidized in water over time. To prevent degradation, divide 1 M DTT into aliquots and store them at -20°C after dissolving. Moreover, it is recommended not to use DTT solution several months after making it even if it has been stored at -20°C.
Acetic acid (J.T.Baker, Fisher Scientific, catalog number: 14-650-388)
Sodium bicarbonate (Fisher Scientific, catalog number: S233-500)
Sodium borate (Fisher Scientific, catalog number: S248-500)
Aminopropylsilane (UCT SPERCIALTIES, LLC, catalog number: A0700)
Fluorescent beads (FluoSpheres, Invitrogen, catalog number: 1890851)
10× TBE buffer (Biosesang, catalog number: TR2004-100-00)
PCR purification kit (QIAGEN, catalog number: 28106)
Trolox solution (see Recipes)
PCA solution (see Recipes)
T50 buffer (see Recipes)
Denaturing gel solution (see Recipes)
Piranha solution (see Recipes)
Silanization solution (see Recipes)
PEGylation solution (see Recipes)
Imaging buffer (see Recipes)
4× elongation buffer (see Recipes)
Equipment
Home-built TIRF microscope setup
Note: The components used in the home-built total internal reflection fluorescence (TIRF) microscope system are as follows: EMCCD, microscope, lasers (473-nm, 532-nm, 633-nm), shutter controller, prism, dichroic mirrors, piezo stage, and microscope temperature control system. A detailed experimental setup can be found in previously published papers (Roy et al., 2008; Lee et al., 2010).
Microscope temperature control system (Live Cell Instrument, model: CU-109)
Incubator (FINE PCR, model: ALB 6400)
Thermal cycler (Bio-Rad, model: C1000)
Vertical electrophoresis cell (Mini-PROTEAN) (Bio-Rad, model: 1658000 FC)
Gel documentation system (SYNGENE, model: G:box Chemi XT4 system)
Ultrasonic cleaner (used as a water bath) (Branson, model: 3510E-DTH)
Note: The ultrasonic cleaner can serve as a water bath in heating mode. We set the temperature to 69°C, but the actual temperature was roughly 55°C when running the denaturing gel.
Razor (Dorco, South Korea)
Diamond solid thin drill (OD: 0.75 mm) (UKAM Industrial Superhard Tools, catalog number: 2040075)
Glass jar (for Piranha)
Water purification system (Millipore, model: ZRXQ-003-EU)
Syringe pump (HARVARD APPARATUS, model: PHD2000)
Spectrophotometer (Thermo Scientific, model: NANODROP 2000)
Software
Labview (version: 2015) (National Instruments, USA, https://www.ni.com/)
IDL (version: 7.0) (David Stern & ITT Visual Information Solutions, USA, https://www.l3harrisgeospatial.com/Software-Technology/IDL)
Matlab (version: R2015a) (MathWorks, USA, https://www.mathworks.com)
Origin (version: 8.5) (Electronic Arts, USA, https://www.origin.com)
Procedure
Sample preparation
DNA ligation
Annealing
Prepare PCR-size tubes that are compatible with the use of a thermal cycler.
Mix 4 µl each 100 µM DNA oligo in T50 buffer by pipetting (final concentration: 5 µM, volume: 80 µl).
Place the tube containing the oligos in a thermal cycler.
Incubate the solution for 3 min at 95°C.
Cool the solution slowly from 80°C to 25°C. A cooling speed of -1°C per min should be adequate.
Maintain the temperature of the thermal cycler at 4°C after annealing.
Ligation
Transfer the annealed DNA to a 1.7-ml tube.
Add 60 µl distilled water.
Add 16 µl 10× T4 ligase buffer.
Mix the solution by pipetting.
Add 4 µl 5× T4 DNA ligase (Total volume of the solution: 160 µl).
Mix the solution by pipetting and store in an incubator maintained at 16°C for 16 h.
Add 80 µl 5 M NaCl and 560 µl EtOH to the 160 µl solution (after ligation) and mix by pipetting.
Incubate the solution at -20°C for more than 2 h.
Centrifuge the solution at 4°C (rcf: 16,100 × g, and use balancer).
Remove the buffer. Be sure not to suck up the pellet.
Dry the pellet by leaving the lid open inside a drawer at room temperature for 30 min to eliminate any remaining EtOH.
Dissolve the pellet in 4 µl distilled water and add 4 µl 99% formamide.
Purification
Prepare the gel-casting chamber (1-mm thick) and wash with distilled water.
Make the denaturing gel (see Recipes).
Add 7 µl TEMED and 60 µl 20% APS and mix well.
Note: When adding TEMED and APS, the denaturing gel solution should be cool enough to prevent an abrupt solidification before pouring it into the casting chamber.
Pour the solution into the interstice of the gel plate set and insert a comb.
Wait until the gel has completely solidified.
Assemble the inner chamber and mount in the outer chamber.
Make 0.5× TBE buffer and pour into the inner and outer chambers.
Place the outer chamber in a heat bath maintained at 55°C.
Before pre-running, wash the wells by pipetting.
Run the gel at 200 V without any sample loaded for more than 30 min.
Stop the pre-running and wash the wells again by pipetting.
Load 8 µl sample in each well.
Run the gel again for 40 min at 200 V.
Stop the gel running and take a picture of the gel under a red-colored lamp.
Extract only the first band at the top using a razor.
Note: The other bands below the first band correspond to DNA fragments that are not properly ligated.
Prepare the gel-purification kit and place the gel slices in a column.
Pour 200 µl distilled water into the column and incubate for 16 h at room temperature.
Recovery and re-annealing
Centrifuge the purification kit at 16,100 × g for 1 min.
Collect 180 µl purified DNA and mix with 90 µl 5 M NaCl and 630 µl EtOH.
Store at -20°C for 2 h.
Centrifuge at 4°C for 40 min.
Remove the solution.
Dry the open tube in a drawer for 30 min to eliminate any remaining EtOH.
Dissolve the pellet in 10 µl distilled water and mix with 0.2 µl 1 M LiCl and 0.4 µl 1 M Tris-HCl (pH 8.0).
Note: We used LiCl instead of NaCl to prevent G-quadruplex formation during the annealing process.
Transfer to a PCR-size tube.
Place the tube in a thermal cycler.
Incubate for 3 min at 95°C
Cool the sample slowly from 80°C to 25°C. A cooling speed of -1°C per min is sufficient.
Store at -20°C.
Antibody labeling
Dissolve 1 mg maleimide Alexa Fluor 488 dye in 55 µl DMSO.
Mix 50 µl antibody and 2 µl dye in 100 mM sodium bicarbonate solution.
Incubate the mixture for 2 h at room temperature.
Eliminate the unreacted dye by purification using an NAP5-column.
Check the labeling efficiency using a spectrophotometer.
Store the labeled antibody at 4°C.
Flow cell preparation
Hole making
Make holes in a quartz slide using a diamond drill bit (Figure 3).
Note: If the size of the holes is too large, epoxy glue can leak into the inner space of the flow cell. To prevent this, it is important to compare the size of the holes with the diameter of the PE tubing during drilling.
PEG coating
Cleaning
Place cover glasses and quartz slides into glass jars.
Rinse cover glasses and quartz slides with distilled water several times.
Pour Piranha solution (see Recipes) into the jars and incubate for 20-30 min to clean the cover glasses and quartz slides.
Repeat step iii another 3 times.
Rinse the cover glasses and quartz slides with distilled water several times.
Silanization
Rinse the cleaned cover glasses and quartz slides twice with methanol.
Pour silanization solution (see Recipes) into the jars.
Remove the solution after a 30-min reaction.
Rinse the cover glasses and quartz slides with methanol and subsequently with distilled water.
Remove the cover glasses and quartz slides from the jars and dry by blowing nitrogen gas.
PEGylation
Make PEGylation solution (see Recipes).
Drop 70 µl PEGylation solution on a quartz slide.
Place a cover glass on top.
Incubate in a drawer for 4 h.
Store at -20°C after incubation.
Flow cell assembly (Figure 3)
Thaw and rinse the PEGylated cover glasses and quartz slides with distilled water.
Dry the cover glasses and quartz slides by blowing nitrogen gas.
Place the quartz slides on aluminum foil.
Attach double-sided tape to the quartz slides to make channels.
Place a cover glass on each quartz slide.
Block the sides of each channel with epoxy glue.
Insert tubing into the holes and fix them with epoxy glue.
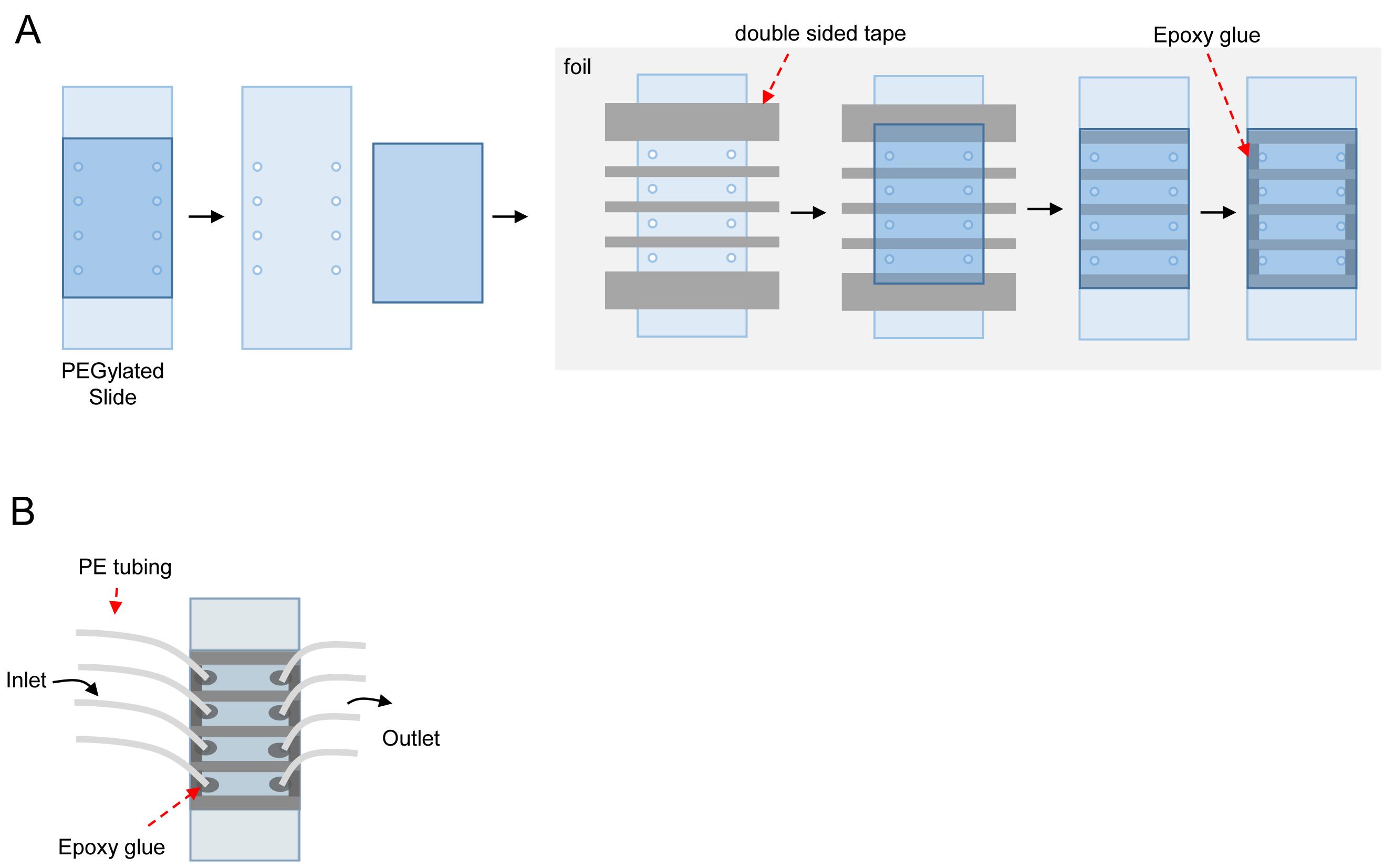
Figure 3. Flow cell assembly. A. Procedure to prepare the flow cell from a PEGylated slide. B. Flow cell after assembly. Inlet and outlet are connected to the injected solution and syringe pump, respectively.
Single-molecule fluorescence measurement
Preparation
Turn on the devices and set the microscope temperature control system to 37°C.
Drop a small amount of water on the objective lens as water immersion.
Place a fluorescent bead slide on the microscope stage.
Drop a small amount of oil on the bead slide and mount a prism.
Illuminate the bead slide, and focus on it by properly adjusting the distance between the bead slide and the objective lens.
Adjust the illumination area.
Turn on the CCD and adjust the dichroic mirrors to divide the imaging area into three different wavelength ranges.
Measure the signals from the fluorescent beads.
To identify fluorescence signals of different channels from the same molecule, make a reference mapping file with a fluorescent bead image.
Note: We use three-color alternating-laser excitation to obtain signals from Cy3, Cy5, and Alexa Fluor 488 dye. To do this, we divide the whole CCD image into three equal areas corresponding to different wavelength ranges. Therefore, we need to know the position information of the same molecules in the different detection channels (see Figure 4). This step is crucial in experiments using a labeled antibody, because only signals emitted by co-localization of the antibody with DNA are considered specific binding. The three dyes were selected due to their superior photostabilities and good quantum yields.
Remove the bead slide.

Figure 4. Fluorescent bead image to obtain the position information of the same molecules. Fluorescent beads are excited by a 473-nm laser, and images are recorded separately according to three different wavelength ranges (Cy3, Cy5, and Alexa Fluor 488 channels). Through Gaussian fitting, the peaks are searched (white circle: co-localized points).Experiments to observe GQ formation during single-round transcription
Preparation of the stalled elongation complex.
Note: 11 nucleotides of the non-template strand from the transcription start site consist of only adenine and guanine.
If T7 RNA polymerase transcribes the DNA without cytosine and thymine, transcription will be stopped with 11-nt-long RNA. This conformation is called the stalled elongation complex. After immobilizing this stalled elongation complex on the surface of the flow cell, we inject rNTP to restart elongation. Using this strategy, we could observe GQ formation under single-round transcription.
Prepare a PCR-size tube with 5 µl distilled water.
Add 2.5 µl 4× elongation buffer, 0.25 µl 1 µM DNA, and 0.25 µl mixture of 2.5 mM GTP and ATP.
Mix by pipetting.
Add 2 µl T7 RNA polymerase.
Mix by pipetting very gently.
Place tube in a thermal cycler and incubate at 37°C.
Note: The incubation time should not exceed 40 min. Due to the error rate of T7 RNA polymerase in transcription, unexpected RNA with the wrong bases could be created and affect the GQ and R-loop formation efficiency.
Make 600 µl imaging buffer (see Recipes).
Mount the flow cell on the microscope stage.
Connect the outlet of the flow cell to a syringe pump.
Using a syringe pump, wash out the channel with 90 µl T50 buffer more than twice.
Inject 90 µl streptavidin (200 ng/ml) into the channel.
Wash out the unbound streptavidin in the channel with T50 buffer 5 min after injection.
Turn on the CCD and adjust the focus and illumination area.
Add 0.5 µl solution containing the stalled elongation complex to 100 µl imaging buffer and gently mix by pipetting.
Inject 90 µl imaging buffer containing the stalled elongation complex into the channel.
Check whether the number of spots is sufficient at Cy3 excitation.
Wash out the unbound stalled elongation complexes in the channel 3 times with 90 µl imaging buffer.
Add rNTP to the imaging buffer (the final concentration of rNTP should be 2 mM).
Move the microscope stage and search for the proper imaging area in the channel.
Turn on the auto-focusing system.
Note: The auto-focus in the z-direction uses a real-time optical astigmatism analysis of single-molecule images. The detailed methods can be found in our previous paper (Hwang et al., 2012).
Start the measurement.
Inject the imaging buffer containing rNTP at 25 s after starting measurement.
Measure for 10 min.
Stop the measurement.
Experiments to observe GQ formation during multiple-round transcription
Add 0.25 µl 1 µM DNA to 9.75 µl distilled water and store in a drawer during the experiment.
Make 600 µl imaging buffer (see Recipes).
Mount the flow cell on the microscope stage.
Connect the outlet of a flow cell to a syringe pump.
Using a syringe pump, wash out the channel more than twice with 90 µl T50 buffer.
Inject 90 µl streptavidin (200 ng/ml) into the channel.
Wash out the unbound streptavidin in the channel with T50 buffer 5 min after injection.
Add 0.4 µl 25 nM DNA to 100 µl imaging buffer and gently mix by pipetting.
Inject 90 µl imaging buffer containing DNA into the channel.
Check whether the number of spots is sufficient at Cy3 excitation.
Wash out the unbound DNA in the chamber 3 times with 90 µl imaging buffer.
Add rNTP and T7 RNA polymerase to the imaging buffer (their final concentration should be 2 mM and 8 nM, respectively) and mix by pipetting.
Incubate at 37°C for 5 min.
Move the microscope stage and search for a proper imaging area in the channel.
Turn on the auto-focusing system.
Start the measurement.
Inject the imaging buffer containing rNTP and T7 RNA polymerase at 25 s after starting measurement.
Measure for 30 min. (In the case of a time-lapse experiment, measure for 3 min at each time point. Do not measure the same area to avoid photobleaching.)
Stop the measurement.
Experiments to observe R-loop formation
Add 0.25 µl 1 µM DNA to 9.75 µl distilled water and store in a drawer during the experiment.
Make 600 µl imaging buffer (see Recipes).
Mount the flow cell on the microscope stage.
Connect the outlet of a flow cell to a syringe pump.
Using a syringe pump, wash out the channel with 90 µl T50 buffer more than twice.
Inject 90 µl streptavidin (200 ng/ml) into the channel.
Wash out the unbound streptavidin in the channel with T50 buffer 5 min after injection.
Add 0.4 µl 25 nM DNA to 100 µl imaging buffer and gently mix by pipetting.
Inject 90 µl imaging buffer containing DNA into the channel.
Check whether the number of spots is sufficient at Cy3 excitation.
Wash out the unbound DNA in the chamber 3 times with 90 µl imaging buffer.
Take the S9.6 antibody out the -80°C freezer and store in the -20°C freezer for 10 min.
Thaw the S9.6 antibody in on ice.
Add rNTP and T7 RNA polymerase to the imaging buffer (Their final concentration should be 2 mM and 8 nM, respectively) and mix by pipetting.
Add the S9.6 antibody and labeled secondary antibody to the imaging buffer (Their final concentration should be 33 nM) and mix by pipetting.
Incubate the imaging buffer containing rNTP, T7 RNA polymerase, and labeled antibody at 37°C for 5 min.
Move the microscope stage and search for a proper imaging area in the channel.
Adjust the incident beam intensity of the blue laser.
Note: It is challenging to know the proper incident beam intensity since there are a few spots (junk) in the Alexa Fluor 488 channel before injecting the labeled antibody into the flow cell. We recommend finding the proper incident beam intensity by injecting antibody into the flow cell where pre-formed DNA-RNA hybrid exists.
Turn on the auto-focusing system.
Start the measurement.
Inject the buffer containing rNTP, T7 RNA polymerase, and labeled antibody at 25 s after starting measurement.
Measure for 30 min (In the case of a time-lapse experiment, measure for 3 min at each time point. Do not measure the same area to avoid photobleaching).
Stop the measurement.
Data analysis
FRET efficiency, E, is defined as the fluorescence intensity of the acceptor (Cy5) relative to the sum of the donor (Cy3) and acceptor (Cy5) intensities at donor excitation. Using the FRET efficiency between Cy3 and Cy5 labeled at the nucleotides flanking the GQ-forming sequence, we could identify DNA conformations formed in the GQ-forming region. Before rNTP and RNAP injection, the low FRET (E = 0.13) indicates that the region remains as a duplex. After the injection of rNTP and RNAP, the FRET efficiency increases to E = 0.82 via an intermediate FRET of E = 0.37 (Figure 5A, top and middle panels). The high FRET state is identified as GQ, but the exact conformation corresponding to the intermediate FRET is not yet known. On the other hand, the R-loop formation is detected by monitoring the binding of the fluorescently labeled S9.6 antibody, which specifically binds to DNA–RNA hybrids (Figure 5A, bottom panel). Note: Non-specific binding of the S9.6 antibody was screened by SNR and binding lifetime criteria).
From a time-lapse experiment, the evolution of the population of each state can be investigated (Figure 5B). Note: Overlapping molecules with a high intensity that deviates from the average intensity of all molecules by 30% are excluded. It is evident that GQ accumulates over time, whereas the intermediate state vanishes. The time delay from GQ formation to the first antibody binding can also be measured. The time delay is mostly negative (Figure 5C), indicating that R-loop formation precedes GQ formation. R-loop formation efficiency can be measured by counting the DNA substrate to which the S9.6 antibody binds. We found that the R-loop formation efficiency is increased up to 5 times when GQ exists on the non-template strand of DNA (Figure 5D), indicating the existence of a positive feedback loop between GQ formation and R-loop formation.
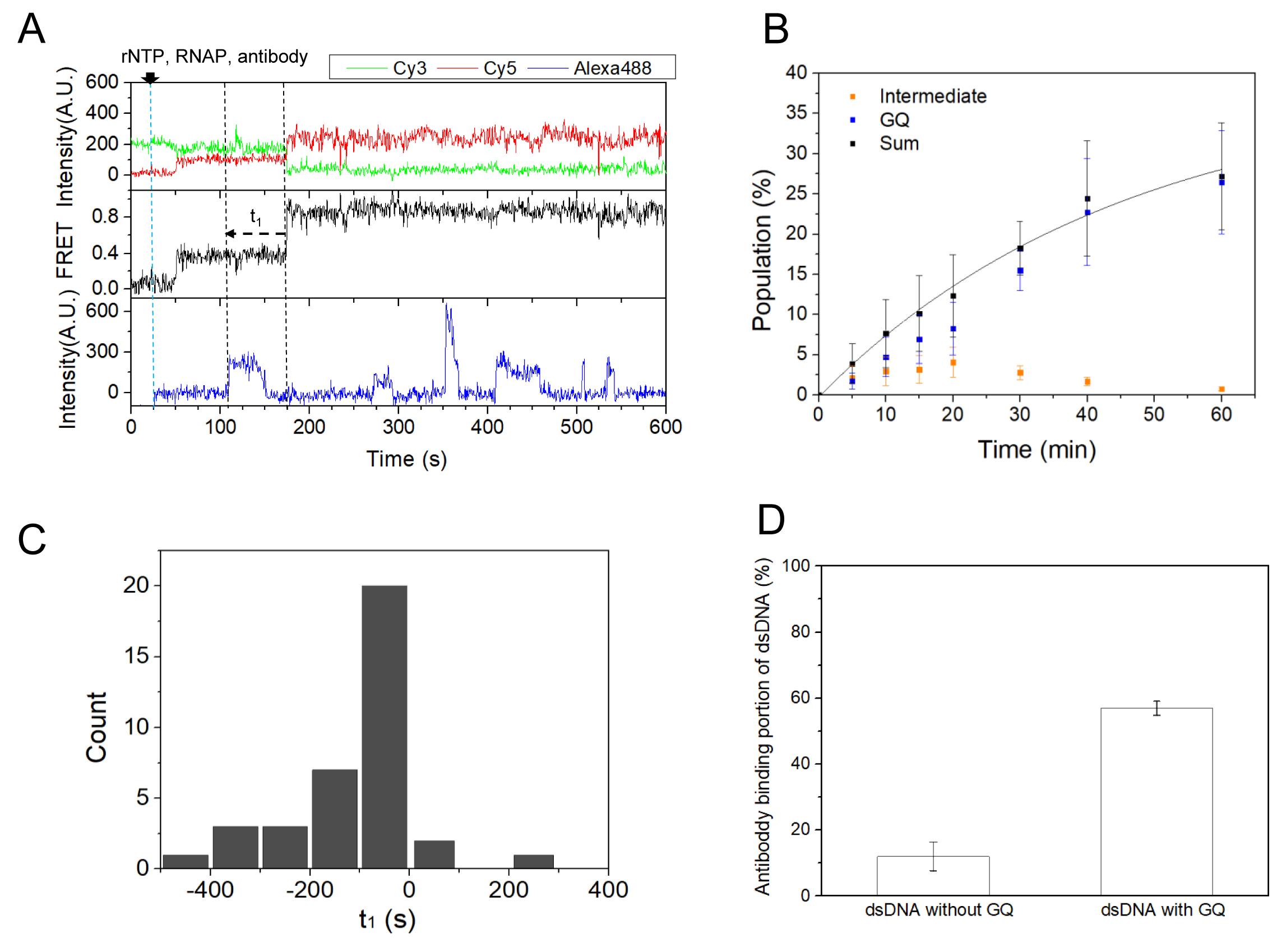
Figure 5. Co-transcriptional formation of GQ coupled with R-loops. A. Representative time traces showing GQ and R-loop formation. GQ: Cy3 (top, green) and Cy5 (top, red) fluorescence intensities at Cy3 excitation, and the corresponding FRET (middle). R-loop: a sudden increase in Alexa Fluor 488 fluorescence intensity at Alexa Fluor 488 excitation (bottom). B. Relative populations of the intermediate state (orange) and GQ state (blue) over time. The population sum of the intermediate state and GQ state (black) is fitted to a single-exponential function with a time constant of 45.3 ± 5.6 min (black lines). C. A histogram of the time difference between GQ formation and the first antibody binding [t1 is defined in the middle panel of A]. D. The portion of dsDNA that is coupled with antibody binding for 20 min after the start of transcription. The R-loop formation efficiency significantly increases when GQ exists on the non-template strand. Figure 5 was adapted from the original paper (Lim and Hohng, 2020).
Recipes
Trolox
Dissolve 50 mg Trolox in 50 ml distilled water
Add 50 µl 3 M NaOH and vortex
Rotate the tube for one day at room temperature (RPM: 30)
Filter the solution with a 0.2-µm membrane filter
Store at 4°C
PCA
Dissolve 154 mg PCA in 20 ml distilled water
Add 20 µl 3M NaOH and vortex
Filter the solution and store at 4°C
T50 buffer
Add 500 µl Tris-HCl (pH 8.0) and 500 µl 5 M NaCl to 49 ml distilled water
Vortex vigorously
Denaturing gel solution
Pour 2.5 ml distilled water into a 15-ml tube containing 4.8 g UREA
Add 0.5 ml 10× TBE buffer
Add 3 ml Acry:Bis=29:1 solution
Vortex vigorously
Warm the solution in a microwave to dissolve the UREA
Cool at 4°C for 15 min
Piranha solution
Pour 90 ml sulfuric acid into a heat-resistant container
Add 30 ml hydrogen peroxide solution (30%) (final ratio of hydrogen peroxide solution to sulfuric acid should be 1:3)
Gently shake
Note: The solution will boil and be hot after mixing.
Silanization solution
1 ml aminoprophysilane
5 ml acetic acid
100 ml methanol
PEGylation solution
Mix 2 mg biotin-PEG-NHS ester, 80 mg PEG-NHS ester, and 640 µl 100 mM sodium bicarbonate solution
Vortex and centrifuge
Note: After dissolving, promptly use to avoid degradation.
Imaging buffer
Mix 65 µl Trolox, 10 µl distilled water, 10 µl PCA, 3 µl PCD, 4 µl 1M Tris-HCl (pH 8.0), 2.5 µl 2 M KCl, 2 µl 1 M MgCl2, and 2 µl 100 mM spermidine
Note: According to the type of experiment, a portion of distilled water can be replaced with rNTP, RNAP, and antibody.
4× elongation buffer
Mix 160 mM Tris-HCl (pH 8.0), 200 mM KCl, 80 mM MgCl2, and 4 mM DTT
Store at -20°C
Acknowledgments
The National Research Foundation of Korea [NRF-2019R1A2C2005209 to S.H.]. This protocol was derived from the original paper “Single-molecule fluorescence studies on cotranscriptional G-quadruplex formation coupled with R-loop formation,” published in Nucleic Acids Research in 2020.
Competing interests
Nothing to declare.
References
- Aguilera, A. and Garcia-Muse, T. (2012). R loops: from transcription byproducts to threats to genome stability. Mol Cell 46(2): 115-124.
- Biffi, G., Tannahill, D., McCafferty, J. and Balasubramanian, S. (2013). Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem 5(3): 182-186.
- Biffi, G., Tannahill, D., Miller, J., Howat, W. J. and Balasubramanian, S. (2014). Elevated levels of G-quadruplex formation in human stomach and liver cancer tissues. PLoS One 9(7): e102711.
- Chan, Y. A., Aristizabal, M. J., Lu, P. Y., Luo, Z., Hamza, A., Kobor, M. S., Stirling, P. C. and Hieter, P. (2014). Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-chip. PLoS Genet 10(4): e1004288.
- De Magis, A., Manzo, S. G., Russo, M., Marinello, J., Morigi, R., Sordet, O. and Capranico, G. (2019). DNA damage and genome instability by G-quadruplex ligands are mediated by R loops in human cancer cells. Proc Natl Acad Sci U S A 116(3): 816-825.
- Duquette, M. L., Handa, P., Vincent, J. A., Taylor, A. F. and Maizels, N. (2004). Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev 18(13): 1618-1629.
- Falabella, M., Kolesar, J. E., Wallace, C., de Jesus, D., Sun, L., Taguchi, Y. V., Wang, C., Wang, T., Xiang, I. M., Alder, J. K., Maheshan, R., Horne, W., Turek-Herman, J., Pagano, P. J., St Croix, C. M., Sondheimer, N., Yatsunyk, L. A., Johnson, F. B. and Kaufman, B. A. (2019). G-quadruplex dynamics contribute to regulation of mitochondrial gene expression. Sci Rep 9(1): 5605.
- Gellert, M., Lipsett, M. N. and Davies, D. R. (1962). Helix formation by guanylic acid. Proc Natl Acad Sci U S A 48: 2013-2018.
- Hansel-Hertsch, R., Spiegel, J., Marsico, G., Tannahill, D. and Balasubramanian, S. (2018). Genome-wide mapping of endogenous G-quadruplex DNA structures by chromatin immunoprecipitation and high-throughput sequencing. Nat Protoc 13(3): 551-564.
- Hwang, W., Bae, S. and Hohng, S. (2012). Autofocusing system based on optical astigmatism analysis of single-molecule images. Opt Express 20(28): 29353-29360.
- Kreig, A., Calvert, J., Sanoica, J., Cullum, E., Tipanna, R. and Myong, S. (2015). G-quadruplex formation in double strand DNA probed by NMM and CV fluorescence. Nucleic Acids Res 43(16): 7961-7970.
- Lee, J., Lee, S., Ragunathan, K., Joo, C., Ha, T. and Hohng, S. (2010). Single-molecule four-color FRET. Angew Chem Int Ed Engl 49(51): 9922-9925.
- Lim, G. and Hohng, S. (2020). Single-molecule fluorescence studies on cotranscriptional G-quadruplex formation coupled with R-loop formation. Nucleic Acids Res 48(16): 9195-9203.
- Marsico, G., Chambers, V. S., Sahakyan, A. B., McCauley, P., Boutell, J. M., Antonio, M. D. and Balasubramanian, S. (2019). Whole genome experimental maps of DNA G-quadruplexes in multiple species. Nucleic Acids Res 47(8): 3862-3874.
- Masse, E. and Drolet, M. (1999). Escherichia coli DNA topoisomerase I inhibits R-loop formation by relaxing transcription-induced negative supercoiling. J Biol Chem 274(23): 16659-16664.
- Roy, D. and Lieber, M. R. (2009). G clustering is important for the initiation of transcription-induced R-loops in vitro, whereas high G density without clustering is sufficient thereafter. Mol Cell Biol 29(11): 3124-3133.
- Roy, R., Hohng, S. and Ha, T. (2008). A practical guide to single-molecule FRET. Nat Methods 5(6): 507-516.
- Santos-Pereira, J. M. and Aguilera, A. (2015). R loops: new modulators of genome dynamics and function. Nat Rev Genet 16(10): 583-597.
- Sanz, L. A., Hartono, S. R., Lim, Y. W., Steyaert, S., Rajpurkar, A., Ginno, P. A., Xu, X. and Chedin, F. (2016). Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol Cell 63(1): 167-178.
- Siddiqui-Jain, A., Grand, C. L., Bearss, D. J. and Hurley, L. H. (2002). Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc Natl Acad Sci U S A 99(18): 11593-11598.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Lim, G. and Hohng, S. (2021). Single-molecule Fluorescence Technique to Monitor the Co-transcriptional Formation of G-quadruplex and R-loop Structures. Bio-protocol 11(13): e4069. DOI: 10.21769/BioProtoc.4069.
Category
Biophysics > Microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.