- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Dual Color, Live Imaging of Vesicular Transport in Axons of Cultured Sensory Neurons
Published: Vol 11, Iss 12, Jun 20, 2021 DOI: 10.21769/BioProtoc.4067 Views: 4043
Reviewed by: Gal HaimovichKatrin DeinhardtMarzia Di Donato
Original research article
The authors used this protocol in:

Jun 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The function of neurons in afferent reception, integration, and generation of electrical activity relies on their strikingly polarized organization, characterized by distinct membrane domains. These domains have different compositions resulting from a combination of selective targeting and retention of membrane proteins. In neurons, most proteins are delivered from their site of synthesis in the soma to the axon via anterograde vesicular transport and undergo retrograde transport for redistribution and/or lysosomal degradation. A key question is whether proteins destined for the same domain are transported in separate vesicles for local assembly or whether these proteins are pre-assembled and co-transported in the same vesicles for delivery to their cognate domains. To assess the content of transport vesicles, one strategy relies on staining of sciatic nerves after ligation, which drives the accumulation of anterogradely and retrogradely transported vesicles on the proximal and distal side of the ligature, respectively. This approach may not permit confident assessment of the nature of the intracellular vesicles identified by staining, and analysis is limited to the availability of suitable antibodies. Here, we use dual color live imaging of proteins labeled with different fluorescent tags, visualizing anterograde and retrograde axonal transport of several proteins simultaneously. These proteins were expressed in rat dorsal root ganglion (DRG) neurons cultured alone or with Schwann cells under myelinating conditions to assess whether glial cells modify the patterns of axonal transport. Advantages of this protocol are the dynamic identification of transport vesicles and characterization of their content for various proteins that is not limited by available antibodies.
Keywords: Axonal transportBackground
Neurons are highly polarized cells. This polarity is critical for neuronal function, i.e., integrating pre-synaptic inputs on the somatodendritic compartment and initiating and propagating action potentials along axons. Myelinated axons are further subdivided into a series of sub-domains, notably including the nodes of Ranvier, the gaps located between adjacent myelin sheaths where action potentials regenerate during saltatory conduction. Nodes are highly enriched in voltage gated Na+ channels, their associated beta subunits, and several neuronal cell adhesion molecules (CAMs). Among the latter is neurofascin (NF) 186, which binds to cognate receptors on Schwann cells, the glial cells that myelinate axons in the peripheral nervous system. Sodium channels and CAMs at the node are tethered to and stabilized by interactions with ankyrin G (AnkG), which forms a specialized submembranous cytoskelon with βIV spectrin. Together, these components form a multimeric nodal complex that differs strikingly from that of other multimeric protein complexes present in other domains of myelinated axons (Salzer et al., 2008). These latter domains include the paranodes, which flank the node, and the juxtaparanodal and internodal domains, which lie underneath the compact myelin sheath.
A key question is whether the components of the node, and of other domains, are transported from the soma to the axon separately to be assembled locally or whether they are transported to their respective domains as pre-assembled complexes. Transport vesicles that shuttle transmembrane proteins from the cell body to the axon (i.e., anterogradely) would predominantly contain a single protein cargo in the former case, whereas they would contain a mixture of protein cargoes in the latter case. A related question is whether vesicles retrogradely transported from the axon to the soma also contain single or multiple cargoes.
One strategy to assess prospective co-expression of proteins in transport vesicles relies on immunofluorescence of proteins in sections of fixed ligated nerves (Cavalli et al., 2005). This method depends on the availability of suitable antibodies against the proteins of interest. In addition, the precise nature of the vesicles being visualized can be potentially ambiguous in the absence of active transport.
As an alternative strategy, we have dynamically imaged vesicles during active transport using multi-color live imaging, a powerful tool to characterize the transport of membrane proteins in neurons (Kaether et al., 2000). To examine whether components of the nodes are transported separately from each other, and from components of other domains, we first infected cultured rat dorsal root ganglia (DRG) neurons with a doxycycline-inducible lentiviral vector to drive simultaneous expression of various tagged proteins. These lentiviral-infected neurons were grown on poly-L-lysine (PLL)/laminin-coated glass bottom dishes either alone or with Schwann cells (SCs) under myelinating conditions. We then carried out pair-wise comparisons of these different cargoes in anterogradely and retrogradely transported vesicles by dual color live imaging and assessed whether vesicles contained single or multiple cargoes. Dynamic imaging allows for the unambiguous identification of vesicles undergoing active transport. Analyzing the transport of multiple proteins relies on distinct fluorescent tags of sufficient brightness to live image over extended time periods and does not require corresponding antibodies. Here, we provide a detailed protocol for dual-color live imaging of the transport of transmembrane proteins in cultured rat DRG neurons.
Materials and Reagents
35 mm glass bottom dishes (MatTek Corporation, catalog number: P35G-1.5-14-C)
100 mm tissue culture dishes (TPP, catalog number: 93100)
Syringe filter unit, 0.45 μm (Millipore Sigma, catalog number: SLHV033RS)
Timed pregnant Sprague Dawley rats (for E15-16 DRG dissection)
293FT cells (Thermo Fisher, catalog number: R7007)
Modified pSLIK (Addgene #25737; Shin et al., 2006) (storage at -20°C)
Modified pFUGW lentiviral vector (Addgene #14883; Lois et al., 2002) (storage at -20°C)
pCMV-VSV-G (gift from J. Milbrandt, Washington University) (storage at -20°C)
pCMV-Δ8.9 (gift from J. Milbrandt, Washington University) (storage at -20°C)
Note: pSLIK was engineered to remove hygromycin resistance and IRES sequences to provide additional space to accommodate larger cDNAs for various cargoes (Zhang et al., 2012). pFUGW was modified to lack the GFP reporter and to add a unique cloning site (Dzhashiashivili et al., 2007).
Gateway LR Clonase II Enzyme Mix (Thermo Fisher, catalog number: 11791020) (storage at -80°C)
Poly-L-lysine (Sigma-Aldrich, catalog number: P5899) (storage at -80°C)
Note: Used for 293FT cell culture.
Poly-L-lysine (Sigma-Aldrich, catalog number: P1274) (storage at -80°C)
Note: Used to coat glass bottom dishes.
Natural mouse laminin (Invitrogen, catalog number: 23017-015) (storage at -80°C)
Dulbecco’s phosphate-buffered saline (Lonza, catalog number: 17-512Q) (store at room temperature)
LipoD293 DNA transfection reagent (Signagen, catalog number: SL100668) (storage at 4°C)
0.25% trypsin (Thermo Fisher, catalog number: 15050065) (storage at -20°C)
Fetal bovine serum (Thermo Fisher, catalog number: 16000-044) (storage at -80°C)
MEM (Thermo Fisher, catalog number: 11090-073) (storage at 4°C)
MEM, minus phenol red (Thermo Fisher, catalog number: 51200-038) (storage at 4°C)
Neurobasal medium (Thermo Fisher, catalog number: 21103-049) (storage at 4°C)
Neurobasal medium, minus phenol red (Thermo Fisher, catalog number: 12348-017) (storage at 4°C)
DMEM (Lonza, catalog number: 12-614F) (storage at 4°C)
MEM non-essential amino acid solution (Thermo Fisher, catalog number: 11140050) (storage at 4°C)
Sodium pyruvate (Thermo Fisher, catalog number: 11360070) (storage at 4°C)
Glucose (Sigma-Aldrich, catalog number: G5146) (storage at 4°C)
L-Glutamine (Thermo Fisher, catalog number: 25030-081) (storage at -80°C)
B27 supplement (ThermoFisher, catalog number: 17504-054) (storage at -20°C)
2.5S nerve growth factor (AbD Serotec, catalog number: PMP04Z) (storage at -80°C)
Doxycycline (Sigma-Aldrich, catalog number: D9891) (storage at -80°C)
HEPES (Thermo Fisher, catalog number: 15630106) (storage at 4°C)
Penicillin-streptomycin (Thermo Fisher, catalog number: 15140122) (storage at -80°C)
Vitamin C (Sigma-Aldrich, catalog number: A0278) (storage at room temperature)
Fluorodexyuridine (FdU) (Sigma-Aldrich, catalog number: F0503) (storage at room temperature)
Uridine (Sigma-Aldrich, catalog number: U3003) (storage at room temperature)
HEK293 cell medium (see Recipes)
NB medium (see Recipes)
NBF medium (see Recipes)
C medium (see Recipes)
CF medium (see Recipes)
Live imaging medium (see Recipes)
Equipment
CO2 incubator (Thermo Fisher, model: Heracell 240)
Microscope and requirements:
Inverted epifluorescence microscope with plan Apo Lambda 100×/1.45 NA objective (Nikon, model: Eclipse Ti-E) equipped with a motorized Epi-fl rotating filter turret.
CCD cameras (Andor Technology, model: Clara; Nikon, model: DS-Qi2)
Heating Insert P (PECON, catalog number: 130-800 207)
Tempcontrol 37-2 digital (PECON, catalog number: 0503.000)
GFP filter cube (Nikon, catalog number: 96362)
594-nm laser bandpass set filter cube (Chroma, catalog number: 49911)
Software
NIS-Elements Advanced Research Software (Nikon, version: 4.30.01)
ImageJ Manual Tracking (Fiji, https://imagej.net/Manual_Tracking)
Excel (Microsoft, version: 2013)
Prism Software (GraphPad, version: 8)
Procedure
Generation of fluorescently tagged proteins
Fuse either EGFP or mKate2 (far red) (see Notes 1, 4, 5, 6) to the C-terminus or other suitable sites of the protein of interest using unique restriction enzyme sites present in the expression vectors. Add restriction enzyme sites as needed via PCR or by first transferring to other vectors with multiple restriction sites prior to subcloning into pSLIK or FUGW. If suitable sites for tags are in between domains, add restriction enzyme sites, linker sequences, and tags by patch PCR (Squinto et al., 1990).
Subclone the resultant fusion protein cDNA into a doxycycline inducible-lentiviral vector, e.g., modified pSLIK (Addgene #25737) using the Gateway LR Clonase II or the constitutively expressing modified pFUGW lentiviral vector (Addgene #14883) using the unique restriction sites present in this vector. Details of the constructs used for live imaging have been described previously (Dzhashiashivili et al., 2007; Zhang et al., 2012; Bekku and Salzer, 2020).
Lentivirus production
Coat 100-mm dishes with 5 ml poly-L-lysine (10 μg/ml; P5899, No.11 in the reagent list) in PBS for 20 min at room temperature; rinse 3 times with PBS at room temperature.
Seed dishes with 6 × 106 293FT cells 24 h prior to transfection and allow cell density to reach ~90% confluence at the time of transfection.
Add fresh HEK293 cell medium (5 ml) 30-60 min before transfection.
Transfect 293FT cells with 5 μg pSLIK or FUGW lentiviral constructs together with helper plasmids Δ8.9 (6.25 μg) and VSVg (3.3 μg) using the LipoD293TM in vitro DNA transfection reagent.
Replace the DNA/LipoD293 complex containing medium with 7 ml HEK293 cell medium 5 h after transfection.
Collect media from the cultures 48 h after transfection and transfer to 50-ml centrifuge tubes. Centrifuge for 15 min at 1,700 × g at room temperature and filter the supernatants through a 0.45-μm filter unit. Filtered supernatants are aliquoted as 0.6-ml samples and stored at -80°C until use.
Preparation of dissociated rat DRG neuron cultures and myelinating co-cultures for live imaging
Coat the glass coverslip at the bottom of each MatTek dish with 0.5 mg/ml PLL (P1274, No.12 in the reagent list) in 300 μl PBS. Incubate at 37°C for 30 min, then wash 3 times with PBS.
Next, coat the glass coverslips with 10 μg/ml laminin in 200 μl PBS. Incubate at 37°C for 30 min.
Prepare dissociated E15-E16 rat DRG neuron cultures for growth as neuron-only cultures or myelinating cocultures as per previously published protocols (Taveggia and Bolino, 2018). Briefly, remove DRGs from rat embryos, incubate them with 1.5 ml trypsin at 37°C for 45 min, add 1 ml C medium, dissociate DRG neurons by repetitive pipetting to triturate ganglia, and centrifuge cells at 193 × g at room temperature for 5 min.
Resuspend the pellet by gentle trituration in ~200 μl CF medium containing penicillin-streptomycin (PS) per DRG. Seed dissociated rat DRG neurons, corresponding to the equivalent of 1 ganglion (approx. 2.1 × 104 cells), onto the Matek PLL/laminin-coated coverslip and incubate for 24 h.
Cultures are cycled on NBF and NB media every other day for a total of 12 days to deplete all non-neuronal cells, leaving neuron-only cultures.
To establish myelinating co-cultures, add 3 × 105 post-natal rat SCs to neuron-only cultures. SCs are prepared from sciatic nerves, and individual aliquots can be stored in liquid N2 as previously described (Kim and Maurel, 2009). To initiate myelination, after 5 days, add vitamin C to the C media to a final concentration of 50 μg/ml; this concentration of vitamin C does not affect the pH of the medium. Myelination typically ensues several days after adding vitamin C.
Dual viral infection and induction of fluorescently tagged proteins in neuron-only and myelinating cocultures
Dilute the first viral supernatant in C medium containing PS. For neuron-only cultures, add 1:1 (virus: C medium) diluted mKate2-tagged pSLIK virus to one-day-old DRG neuron cultures. For myelinating co-cultures, add 1:1 (virus: C medium) diluted EGFP-tagged pSLIK virus to one-day-old neuron cultures. Incubate cultures with viral supernatants for 24 h.
The next day, dilute the second viral supernatant in C medium containing PS. For neuron-only cultures, change the viral medium to freshly diluted EGFP-tagged pSLIK virus (1:1 dilution). For co-cultures, replace the viral medium with new diluted mKate2-tagged FUGW virus (dilution 1:15 of virus: C medium) onto the neuron culture (see Note 9). Incubate for 24 h.
Remove the viral medium, add NBF, and culture the infected neurons for an additional 1-2 days until expression is induced. If longer incubation times are desirable to allow further neuron growth, change the medium to NB for an additional 2 days.
Add 2 µg/ml doxycycline (final concentration) to either NB or NBF in neuron-only cultures depending on the medium cycle on the day doxycycline is added, or into C plus vitamin C in co-cultures to induce protein expression 24-48 h before live imaging (see Note 8).
Note: The schematic time courses of the experiment for neuron only cultures and myelinating cultures (procedures C and D) are shown in Figure 1.
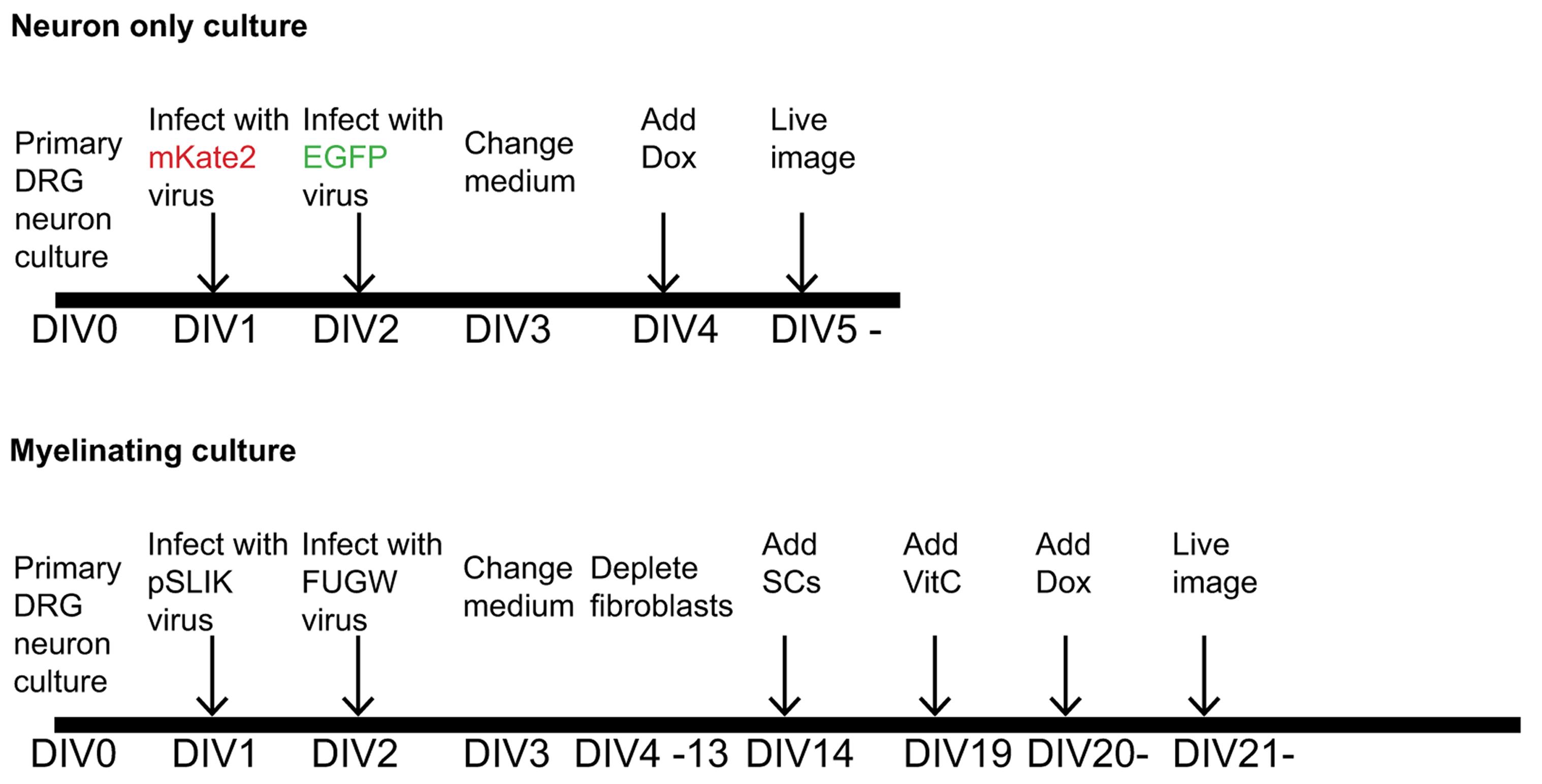
Figure 1. Time course of the dual-color live imaging
Live imaging
Change medium to live imaging media immediately prior to live imaging.
Place cultures growing on a MatTek dish onto a 37°C heated microscope stage (Heat Insert P) connected to a temperature control unit (Tempcontrol 37-2).
Determine the direction of axonal transport for each vesicle relative to the position of the neuronal soma before or at the onset of live imaging.
Capture images at 2 s intervals for ≥2 min with CCD cameras and the 100× objective (see Notes 2, 3, 8) using the perfect focus setting of the NIS-Elements Advanced Research Software (Nikon) to correct focus drift. Process images using real time deconvolution of the NIS-Elements Advanced Research Software. EGFP-tagged proteins were captured with a GFP filter cube (96362, Nikon); those for mKate2 were captured with a 594-nm laser bandpass set filter (49911, Chroma) cube. See the representative still images of dual color live imaging in Figure 2.
Take images of more than 20 neurons for analysis. Loss of focus due to microscope stage drift may require refocusing and may also reduce image numbers available for generating kymographs.
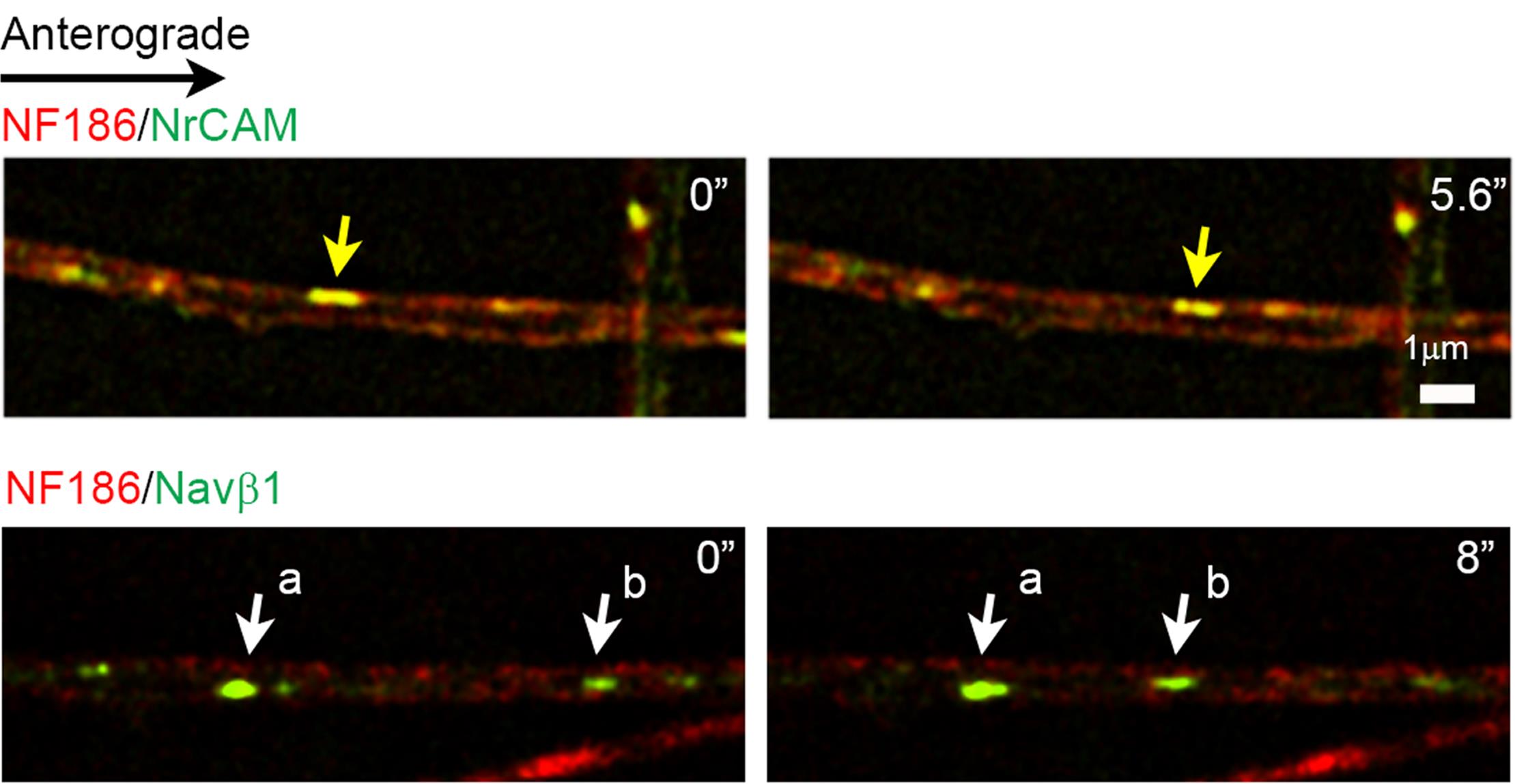
Figure 2. Representative still images of vesicular co-transport and independent transport. Upper panels: Vesicles containing NF186-mKate2 and NrCAM-EGFP are anterogradely co-transported in the axon as evidenced by the yellow color from the overlap of mKate2-labeled NF186 and EGFP-labeled NrCAM in the same vesicles; an example of co-transported vesicles is highlighted by the yellow arrow. Lower panels: Vesicles containing NF186-mKate2 and Navβ1-EGFP are transported separately in the retrograde direction along the axon; examples of vesicles containing Navβ1-EGFP only are highlighted by the white arrows.
Data analysis
Build kymographs by live imaging 15-µm axon segments for 2 min. One option for building kymographs is to use the NIS-Elements Advanced Research Software, which displays pixel intensity changes along a defined linear section over time (Figure 3). ImageJ or other microscopy analysis software is another option for building kymographs. Extract trajectories of individual vesicles from the kymographs and manually count vesicle numbers in each direction. Analyze ≥ 10 neurons for each combination of transfected proteins and compare the rates of co-transport for each protein. For statistical analysis, analyze ≥ 100 vesicles (≥ 10 neurons); sample size may vary depending on the experimental design. Sample size can be calculated by software, e.g., G power.
Confirm quantitative results using ImageJ Manual Tracking, particularly for some anterograde vesicles that can be of limited brightness.
Measure the average velocities of vesicles using ImageJ Manual Tracking.
Compare the cotransport rates by averaging the results from each imaged axon.
Assess statistical significance with either the two-tailed Welch’s t-test or the two-tailed Student’s t-test for unpaired data using the Excel or GraphPad Prism software.

Figure 3. Representative kymograph of co-transported vesicles. This kymograph was generated from live imaging of neurons expressing NF186-EGFP and NF186-mKate2 by the NIS-Elements Advanced Research Software. The trajectory of a vesicle is highlighted by blue circles, which are numbered in temporal sequence. In the kymograph, two retrogradely co-transported vesicles are detectable. Additional representative live images and movies, kymographs, and data analysis are provided in J Cell Biol (Bekku and Salzer, 2020).
Notes
This protocol describes dual color imaging but can be adapted for three or more fluorophores, depending on the microscope’s ability to distinguish the different fluorophores used as tags.
A limitation of live imaging by single-laser epifluorescence microscopy is the time lag that occurs during switching between filters to monitor the different fluorophores. During this lag, if the vesicles are actively being transported, the vesicle will continue to move and exhibit an artefactual spatial separation of different fluorophores demarcating the same vesicle.
Spinning disk microscopy with two cameras is an excellent alternative to simultaneously live image multi-colored vesicles with no time lag as there is no filter switching. In our study, we also used spinning disk microscopy to confirm the results. In terms of image quality, the epifluorescence microscope takes clear images of individual vesicles.
In our studies, proteins were tagged with either EGFP or mKate2 at their C-termini, and their trafficking was analyzed by pairwise comparisons. Unlike mKate2, all other red-fluorophore tags that were tried, i.e. mRFP, mCherry, and DsRed, induced some degree of protein aggregation that impacted trafficking, which is in agreement with Katayama et al. (2008).
It is important to confirm that protein trafficking is not affected by the addition of the fluorescent protein tags. For example, dual expression of a protein tagged with EGFP and the same protein tagged with mKate2 showed that these proteins exhibited complete overlap during intracellular trafficking in neurons (Bekku and Salzer, 2020).
It is also important to show that the addition of the fluorescent tag does not interfere with targeting of the tagged protein to its normal plasma membrane destination (e.g., to the node, synapse, etc.). For a description of how to design fusion proteins see Snapp (2005).
To express two proteins simultaneously in neurons, tagged proteins were subcloned into the inducible viral construct pSLIK, and their expression was simultaneously induced by doxycycline.
For longer live imaging (> 5 min), it may be necessary to refocus on the region of interest as the microscope stage may drift out of focus despite installation of autofocus software.
For myelinating cultures, which require prolonged periods of culture, co-expression of two constructs was best achieved by driving the expression of one construct in the FUGW vector and pSLIK for the other. Co-infection of neurons with two pSLIK constructs adversely affected expression in older cultures, potentially by either impairing the survival of dually infected neurons or dual expression.
Recipes
HEK293 cell medium
DMEM
10% FBS
2 mM L-glutamine
2 mM NEAA
1 mM sodium pyruvate
1× penicillin-streptomycin
NB medium
Neurobasal medium (prepared with or without phenol red)
0.4% glucose
2% B27 supplement
2 mM L-glutamine
50 ng/ml 2.5S NGF
NBF medium
NB medium
FUDR (final concentration: 10 μM FdU + 10 μM Uridin)
C medium
MEM (prepared with or withour phenol red)
0.4% glucose
10% FBS
2 mM L-glutamine
50 ng/ml 2.5S NGF
CF medium
C medium
FUDR (final concentration: 10 μM FdU + 10 μM Uridine)
Live imaging medium
Phenol red-free NB (for neuron-only culture) or C (for co-culture) media
10 mM HEPES
Acknowledgments
We thank Erik Snapp for advice on the use of fluorescent protein tags for live imaging and Michael Cammer for supporting the spinning disk microscopy live imaging. This work was supported by an NIH grant (Grant Number NS043474) to JLS. The protocol is based on work published in J Cell Biology (Bekku and Salzer, 2020; doi: 10.1083/jcb.201906071).
Competing interests
The authors declare no competing interests.
Ethics
All animal experiments were performed in compliance with the relevant policies and institutional guidelines issued by the New York University School of Medicine Institutional Animal Care and Use Committee.
References
- Bekku, Y. and Salzer, J. L. (2020). Independent anterograde transport and retrograde cotransport of domain components of myelinated axons. J Cell Biol 219(6).
- Cavalli, V., Kujala, P., Klumperman, J. and Goldstein, L. S. (2005). Sunday Driver links axonal transport to damage signaling. J Cell Biol 168(5): 775-787.
- Dzhashiashvili, Y., Zhang, Y., Galinska, J., Lam, I., Grumet, M. and Salzer, J. L. (2007). Nodes of Ranvier and axon initial segments are ankyrin G-dependent domains that assemble by distinct mechanisms. J Cell Biol 177(5): 857-870.
- Kaether, C., Skehel, P. and Dotti, C. G. (2000). Axonal membrane proteins are transported in distinct carriers: a two-color video microscopy study in cultured hippocampal neurons. Mol Biol Cell 11(4): 1213-1224.
- Katayama, H., Yamamoto, A., Mizushima, N., Yoshimori, T and Miyawaki, A. (2008). GFP-like proteins stability accumulate in lysosome. Cell Struc Funct 33 (1): 1-12.
- Kim, H. A. and Maurel, P. (2009). Primary Schwann Cell Cultures. In: Doering, L. (Ed). Protocols for Neural Cell Culture. Springer Protocols Handbooks. Humana Press.
- Lois, C., Hong, E. J., Pease, S., Brown, E. J. and Baltimore, D. (2002). Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295(5556): 868-872.
- Salzer, J. L., Brophy, P. J. and Peles, E. (2008). Molecular domains of myelinated axons in the peripheral nervous system. Glia 56(14): 1532-1540.
- Shin, K. J., Wall, E. A., Zavzavadjian, J. R., Santat, L. A., Liu, J., Hwang, J. I., Rebres, R., Roach, T., Seaman, W., Simon, M. I. and Fraser, I. D. (2006). A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc Natl Acad Sci U S A 103(37): 13759-13764.
- Snapp, E. (2005). Design and use of fluorescent fusion proteins in cell biology. Curr Protoc Cell Biol 21.4.1-21.4.13.
- Squinto, S. P., Aldrich, T. H., Lindsay, R. M., Morrissey, D. M., Panayotatos, N., Bianco,. S. M., Furth, M. E. and Yancopoulos, G. D. (1990). Identification of functional receptors for ciliary neurotrophic factor on neuronal cell lines and primary neurons. Neuron 5(6):757-766.
- Taveggia, C. and Bolino, A. (2018). DRG Neuron/Schwann Cells Myelinating Cocultures. In: Woodhoo, A. (Ed). Myelin. Methods in Molecular Biology, vol 1791. Humana Press, New York, NY.
- Zhang, Y., Bekku, Y., Dzhashiashvili, Y., Armenti, S., Meng, X., Sasaki, Y., Milbrandt, J. and Salzer, J. L. (2012). Assembly and maintenance of nodes of ranvier rely on distinct sources of proteins and targeting mechanisms. Neuron 73(1):92-107.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Bekku, Y. and Salzer, J. L. (2021). Dual Color, Live Imaging of Vesicular Transport in Axons of Cultured Sensory Neurons. Bio-protocol 11(12): e4067. DOI: 10.21769/BioProtoc.4067.
Category
Neuroscience > Cellular mechanisms > Cell isolation and culture
Neuroscience > Peripheral nervous system > Schwann cell
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.