- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Transcriptional Run-on: Measuring Nascent Transcription at Specific Genomic Sites in Yeast
Published: Vol 11, Iss 12, Jun 20, 2021 DOI: 10.21769/BioProtoc.4064 Views: 3139
Reviewed by: Julie WeidnerLaia ArmengotAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
DNA transcription by RNA polymerases has always interested the scientific community as it is one of the most important processes involved in genome expression. This has led scientists to come up with different protocols allowing analysis of this process in specific locations across the genome by quantitating the amount of RNA polymerases transcribing that genomic site in a cell population. This can be achieved by either detecting the total number of polymerases in contact with that region (i.e., by chromatin immunoprecipitation (ChIP) with anti-RNA polymerase antibodies) or by measuring the number of polymerases that are effectively engaged in transcription in that position. This latter strategy is followed using transcription run-on (TRO), also known as nuclear run-on (NRO), which was first developed in mammalian cells over 40 years ago and has since been adapted to many other different organisms and high-throughput methods. Here, we detail the procedure for performing TRO in Saccharomyces cerevisiae for single genomic regions to study active transcription on a single gene scale. To do so, we wash the cells in the detergent sarkosyl, which prevents new initiations at the promoter level, and then perform an in situ reaction, leading to the radiolabeling of transcripts by RNA polymerases that were already engaged in transcription at the moment of harvesting. By subsequently quantitating the signal of these transcripts, we can determine the level of active transcription in a single gene. This presents a major advantage over other forms of transcription quantitation such as RNA polymerase ChIP, since in the latter, both active and inactive polymerases are measured. By combining both ChIP and TRO, the amount of inactive or paused polymerases on a particular gene can be estimated.
Graphic abstract:
Transcriptional run-on scheme
Background
Transcription is an essential step in gene expression. It is a highly regulated process during which an RNA molecule is produced from a template DNA sequence by the action of RNA polymerases. Many scientists are interested in the complex process of transcription, which still remains to be fully understood and many others need to quantitate gene transcription in a specific region of the genome for practical purposes. Thus, several methods have been developed over the years to study eukaryotic transcription (reviewed in Pérez-Ortín et al., 2012).
Each developed method reveals different aspects of the transcription process. In this method article, we detail the protocol for transcription run-on (TRO), also known as nuclear run-on (NRO), which is particularly useful for detecting RNA polymerases that are engaged in active transcription at the moment of the experiment (Smale, 2009). The addition of sarkosyl detergent permeabilizes cells and prevents new initiations of transcription at the promoter level. Therefore, once new substrates (NTPs) are provided for the transcription reactions, only previously fully engaged RNA polymerases will act, incorporating a radiolabeled analog of UTP into the newly produced nascent RNA. This radiolabeling then allows for the detection of elongating RNA polymerases by hybridizing the nascent RNA onto nylon membranes that contain immobilized DNA fragments corresponding to the genomic sites of interest.
TRO was introduced over 40 years ago and has since experienced many upgrades for its use in different organisms and high-throughput sequencing (Greenberg and Ziff, 2004). The first attempt to take TRO genome-wide in yeast was the Genomic Run-On (GRO) method developed in 2004 (Garcia-Martinez et al., 2004), in which the labeled RNAs are hybridized to arrays. The protocol was then adapted to become high-resolution (Bio-GRO) and high-throughput (GRO-seq) (Core et al., 2008; Jordán-Pla et al., 2016 and 2019).
Here, we describe in detail the TRO method developed for Saccharomyces cerevisiae used in one of our recent publications (Corzo et al., 2019), which detects active RNA polymerases in single genes.
Materials and Reagents
Whatmann filter paper, 180 µm thickness (Macherey-Nagel, catalog number: 742213)
1.5 ml tubes (Eppendorf, catalog number: 22 36 411-1)
50 ml and 15 ml Falcon tubes (Eppendorf, catalog numbers: 0030122178 and 0030122151)
FastPrep 2 ml Lysing Matrix tubes (MP Biomedicals, catalog number: 115076200-CF)
ProbeQuantTM G-50 Micro Column (GE28-9034-08)
Saran wrap
Acetate film
Glass beads (Sigma, catalog number: G9268) washed in acid following the manufacturer’s protocol
HCl 37% (Sigma, catalog number: 320331)
Pfu DNA polymerase (Promega, catalog number: M7741)
QIAquick PCR Purification Kit (Qiagen, catalog number: 28104)
QIAquick Gel Extraction Kit (Qiagen, catalog number: 28115)
Yeast DNA Extraction Kit (Thermo Fisher, catalog number: 78870)
E. coli gDNA extraction (see He, 2011)
Hybond-N membrane (Amersham, catalog number: RPN303N)
NTP Set 100 mM (Invitrogen, catalog number: R0481)
Sarkosyl N-lauroylsarcosina (Sigma, catalog number: L5125)
DTT (Roche, catalog number: 11583786001)
α-32P UTP (Perkin-Elmer, catalog number: NEG507H001MC, 10 µCi/µl)
Cold MilliQ water
Acid phenol pH 4.3 (Sigma, catalog number: P4682)
Sodium acetate (Sigma, catalog number: 127-09-3)
100% ethanol (JT Baker, catalog number: 8025)
Chloroform (Sigma, catalog number: C2432)
NaOH pellets (Sigma, catalog number: 221465)
Random hexamers 50 µM (Invitrogen, catalog number: N8080127)
dNTP set (100 mM) (Invitrogen, catalog number: 10297018)
Klenow fragment (NEB, catalog number: M0212S)
α-32P dCTP (Perkin-Elmer, catalog number: NEG513H250UC, 10 µCi/µl)
NaCl (Labotaq, catalog number: SO0227005P)
Sodium citrate (Prolabo, catalog number: 27833.363)
MgCl2 (Prolabo, catalog number: 25108295)
EDTA (Sigma, catalog number: E5134-1KG)
SDS (Amresco, catalog number: 0227)
Sodium dihydrogen phosphate monohydrate (Sigma, catalog number: 10049-21-5)
Di-sodium hydrogen phosphate (Sigma, catalog number: 7558-79-4)
Potassium phosphate (Merck, catalog number: 1048731000)
Tris base (Sigma, catalog number: T1503-10KG)
Boric acid (Panreac, catalog number:131015.1211)
Yeast extract (Pronadisa, catalog number: 1702)
Glucose (Prolabo, catalog number: 24379.363)
Agarose low EEO (Sigma, catalog number: A0576-100G)
Red safe (Labotaq, catalog number: 8014)
DNA 1 Kb ladder (Invitrogen, catalog number: 10787-026)
DNA gel loading dye 6× (Thermo Scientific, catalog number: R0611)
Denaturalization buffer (see Recipes)
Neutralization buffer (see Recipes)
SCC 20× (see Recipes)
Transcription buffer 2.5× (see Recipes)
TES (see Recipes)
Hybridization solution (see Recipes)
Wash solution I (see Recipes)
Wash solution II (see Recipes)
Neutralization solution (stripping) (see Recipes)
Stripping solution (see Recipes)
10× TBE buffer (see Recipes)
Tris-HCl, pH 7 (see Recipes)
Phosphate buffer, pH 7 (see Recipes)
Equipment
Slot Blot blotting manifold (Hoefer Scientific, catalog number: PR648)
Thermocycler (Bio-Rad, catalog number: T100)
NanoDrop (Thermo Scientific, catalog number: 840274100)
Thermoblock heater (Labnet)
Vacuum pump
Spectrophotometer (Eppendorf, catalog number: EP6135000923)
Falcon centrifuge fixed rotor (Eppendorf, model: 5810R), max speed 3,220 × g
Microcentrifuge fixed rotor (Eppendorf, model: 5424), max speed 17,000 × g
UV crosslinker (Stratagene, model: UV Stratalinker 1800)
FastPrep-24TM 5G instrument (MP Biomedicals, catalog number: 116005500)
Hybridization oven (UVP, model: HB-1000 Hybridizer)
PhosphoImager screen and cassette (FUJIFILM BAS)
STORM-840 imaging system (GE Healthcare)
Vortex mixer (Jencons, model: VX100), use at max speed
Geiger counter (Thermo, model: Mini 900)
Microwave oven
Orbital shaker (Appleton Woods Stuart), slow shaking at about 50 rpm
Heat-resistant gloves
Electrophoresis chamber and power supply (Bio-Rad, catalog number: 1640300)
Gel casting tray and comb (Bio-Rad)
Software
GelQuant.NET software (http://biochemlabsolutions.com/GelQuantNET.html)
Procedure
Notes:
The procedure uses 32P; appropriate equipment, facilities, and training for radioactive work is needed.
Filter tips should be used when manipulating radioactivity or RNA.
The whole protocol takes 10 days to complete without stopping. The membranes and DNA fragments of interest, yeast and E. coli gDNA, and yeast cell pellets can be made and stored in advance. Mayor stopping points would be after membrane preparation (A); after labeled RNA hybridization, exposition, and scanning (B-D); and after labeled gDNA hybridization, exposition, and scanning (F).
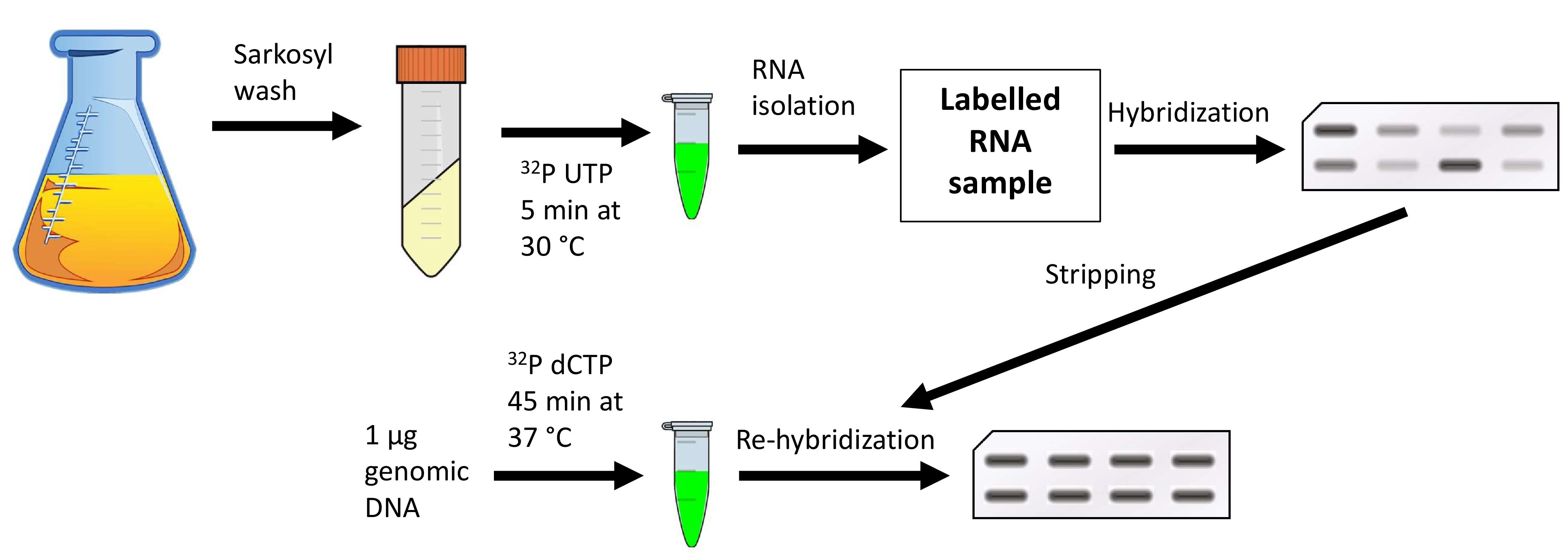
Preparing the membranes
Treat the Slot Blot (SB) blotting manifold with 0.1 M HCl for 1 h to eliminate any contaminants. Wash with distilled H2O abundantly and leave to dry in a fume hood on top of a clean filter paper to avoid contamination.
Prepare the DNA fragments corresponding to the genes of interest (i.e., the genes you would like to check are being actively transcribed and would like to quantitate) to be loaded onto the membrane:
Design PCR oligos to obtain fragments of DNA of around 300 bp to be analyzed.
Perform PCR reactions using Pfu DNA polymerase to ensure fidelity of the DNA product (following the manufacturer’s protocol) and to obtain 100 ng DNA per membrane (usually 3-4 50 µl PCR reactions for 12 membranes). Template yeast gDNA can be isolated using a kit.
Combine the PCR reactions of the same DNA fragment, purify using QIAquick PCR purification columns according to the manufacturer’s protocol, and measure the DNA concentration using a NanoDrop. Check the sizes on a 1% agarose gel by loading approximately 500 ng DNA measured by the NanoDrop to ensure there are no other non-specific bands at a size different from that expected (~300 bp). If there are other bands, you will need to perform an extra step to purify the 300 bp band from the gel by loading all your DNA on a 1% agarose gel, cutting the 300 bp band, and purifying using QIAquick Gel extraction columns. DNA can be stored for up to 1 year at -20°C.
Apart from your PCR DNA of interest, we recommend that you include a negative control consisting of DNA from another organism (e.g., E. coli genomic DNA) and a positive control consisting of genomic DNA from the organism you are using (e.g., yeast genomic DNA).
Mix 100 ng DNA and 60 μl SSC 20× (final concentration of 6×), and add MilliQ H2O up to a final volume of 200 μl per sample and membrane.
Boil DNA at 95°C on a thermoblock for 10 min and immediately place on ice.
Mount the SB blotting manifold (see Figure 1):
Connect the lower fitting to the vacuum pump to draw samples through the SB.
Place the middle block of the SB on top of the bottom block with the red rubber facing downward.
Dampen an 11 × 3 cm Whatman in 6× SSC and place it on top of the SB.
Carefully place an 11 × 3 cm Hybond N membrane on top, with the top left corner cut in order to know the orientation of the membrane.
Place the top block of the SB on top of this, making sure that the number 1 and letter A of the SB are also on the top left, and screw in the red screws bit by bit, making sure all screws are tightened at the same time. Be careful not to tighten too hard so as not to make indentations in the membrane.
Turn on the vacuum pump (use at maximum setting).
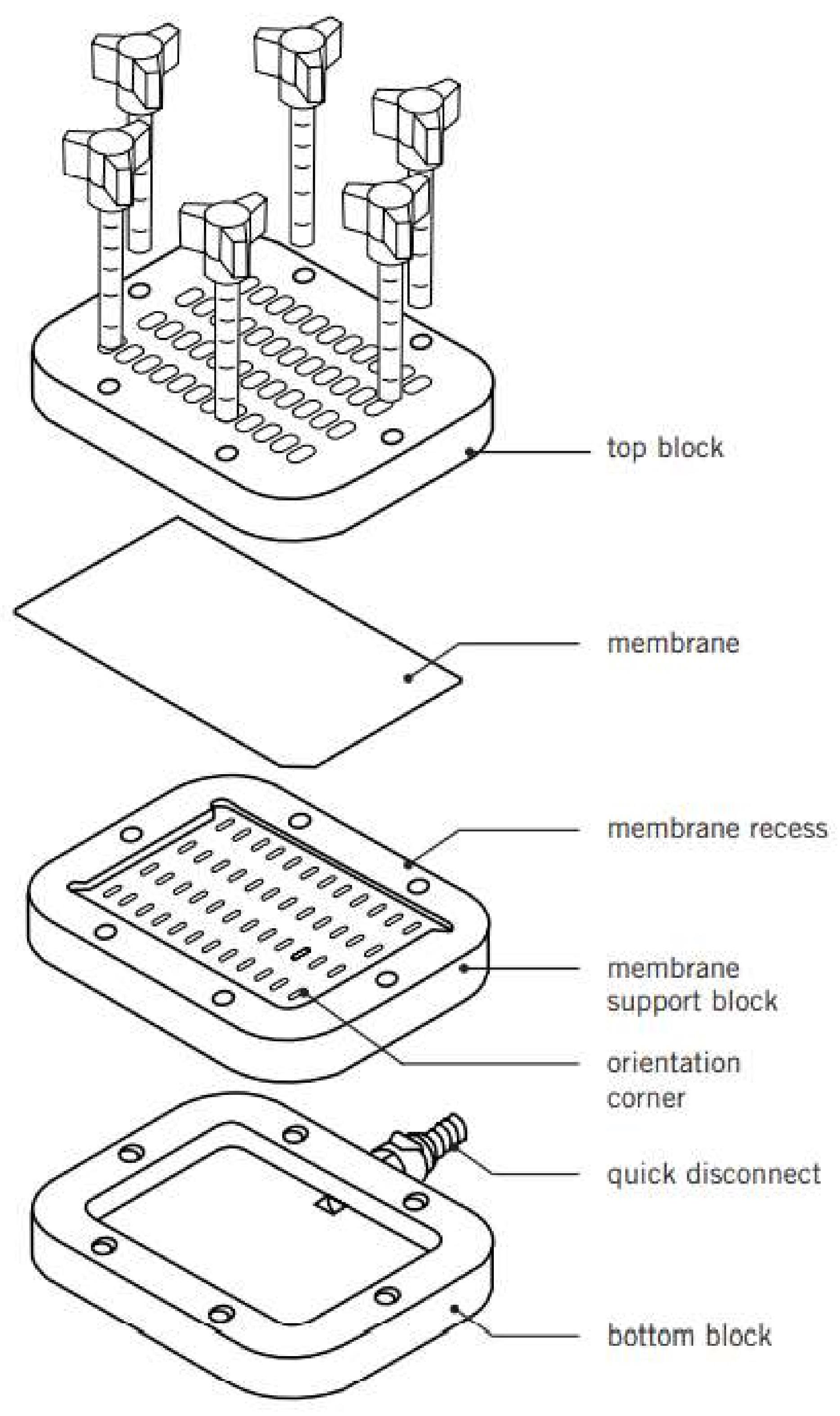
Figure 1. The Hoefer PR648 Slot Blot (SB) set up for standard use (taken from the Hoefer PR648 manual)Blotting:
Add 500 μl 6× SSC to each well and make sure the liquid is being correctly sucked through the membrane and Whatmann paper.
Wait until the wells are dry.
Add each boiled DNA sample, previously placed on ice, into the assigned well, being careful not to touch the sides of the well, and wait until it dries.
Once all the DNA has been sucked through the SB, turn the vacuum pump off and unscrew the SB.
Dampen a 12 × 4 cm Whatmann in denaturalization buffer and carefully place the membrane on top, with the DNA facing upward. Incubate at room temperature for 10 min.
Repeat the previous process (4e) using neutralization buffer, but incubate for 5 min.
Place the membrane on a dry Whatmann and label it with a pencil.
Fix the DNA to the membrane by UV-crosslinking at 70000 μJ/cm2. Membranes can now be stored for up to a year after fully drying them out between Whatmann filter paper.
Note: From Step B9 onward, you need to work in a radioactive facility.
Run-on reaction
Grow your yeast strains in 25-ml YPD cultures or similar until they reach an OD600nm of 0.5 from a dilution of a small 5-ml culture grown overnight. Make sure you have the same volume of cells across samples.
Centrifuge at 3,220 × g for 3 min at RT and discard the supernatant. At this point, you can flash freeze your cells in liquid nitrogen and store the pellets in a -80°C freezer for up to 1 month.
Prepare the transcription mix: 60 μl transcription buffer, 8 μl NTPs (A,C,G) at 10 mM each, and 3 μl 0.1 M DTT per sample. Do not add UTP. Place on ice until use.
Resuspend the cell pellets in 5 ml 0.5% sarkosyl.
Centrifuge at 1,800 × g for 3 min at RT and discard the supernatant carefully.
Resuspend the pellet in 1 ml 0.5% sarkosyl and move to a 1.5-ml Eppendorf tube.
Centrifuge at 700 × g for 4 min at RT and discard the supernatant using a pipette.
Centrifuge the samples again at 700 × g and RT for 1 min to remove any remaining supernatant.
Resuspend in 60 μl MilliQ H2O and incubate at 30°C until the samples are warm.
Add 8 μl α-32P UTP (Perkin-Elmer NEG507H001MC, 10 µCi/µl) per sample to the transcription mix and place at 30°C to warm up.
Once the samples and transcription mix are warm (around 5 min at 30°C), add 79 μl transcription mix to each sample and incubate for exactly 5 min at 30°C. To achieve this, it is best to leave 30 s between samples.
After the 5 min incubation, quickly add 800 μl cold MilliQ H2O and place on ice to stop the reaction.
Centrifuge at 3,500 × g for 1 min at 4°C if possible (if not, at RT) and discard the supernatant. At this point, you can stop and freeze the samples at -20°C before continuing. Samples should be used as soon as possible since the 32P will decay over time. Use within a few days.
RNA isolation
Note: A commercially available RNA isolation kit could be used instead.
Resuspend the pellet in 400 μl TES and transfer to a FastPrep tube containing 400 μl glass beads and 400 μl acid phenol.
Break the cells using the FastPrep (3 cycles of 30 s at 5 m/s) at RT.
Centrifuge for 5 min at 16,200 × g (if possible at 4°C; if not, at RT).
Transfer the supernatant to an Eppendorf tube and add 400 μl chloroform.
Centrifuge for 5 min at 16,200 × g (if possible at 4°C; if not, at RT).
Transfer the supernatant to a fresh Eppendorf tube and add 1 ml 100% ethanol and 40 μl sodium acetate 3M pH 5.5.
Precipitate RNA overnight at -20°C.
The next day, centrifuge at 16,200 × g for 15 min at 4°C if possible (if not, at RT). In the meantime, start pre-hybridizing the prepared membranes (step D1) so that the labeled RNA can be used immediately.
Discard the supernatant.
Wash with 1 ml 70% ethanol and centrifuge at 16,200 × g for 5 min at 4°C if possible (if not, at RT).
Discard the supernatant, then centrifuge again and eliminate any leftover ethanol.
Dry the pellet at room temperature for 5-10 min.
Add 100 μl MilliQ H2O and incubate at 65°C (for approximately 5 min), vortexing the sample every 30 s until all the RNA has been resuspended.
Add 25 μl NaOH 0.2 N and incubate for 5 min on ice. This step fragments the RNA and helps it to hybridize with the DNA on the membranes.
Neutralize by adding 25 μl HCl 0.2 N.
Hybridization
Pre-hybridize each membrane by incubating at 65°C inside a 15-ml Falcon tube containing 5 ml hybridization solution for at least 1 h (the longer, the better). Make sure you place the membrane with the fixed DNA toward the inside of the tube and try to minimize overlapping of the membrane. A 50-ml Falcon tube can be used for larger membranes so that they do not overlap. For this and the following steps, a hybridization oven was used; the Falcon tubes where inserted into a glass tube and rotated. If you have only a few membranes, you can use the glass tube directly. Make sure you screw the lids on well to prevent leakage.
Once the RNA is ready, discard the hybridization solution and add 3 ml fresh hybridization solution and the labeled RNA.
Incubate at 65°C for 48 h while rotating.
Reuse or save the labeled RNA by storing at 4°C. Reuse within 1 week since you will lose the signal over time. When reusing, heat the solution in the hybridization oven to ensure that it liquifies.
Rinse the membrane in 5 ml wash solution I. Discard the solution in an appropriate radioactive recipient.
Add 5 ml fresh wash solution I and incubate for 20 min at 65°C while rotating.
Discard the solution, add 5 ml wash solution II, and incubate for 10 min at 65°C.
Repeat the previous step.
Carefully take each membrane out of the Falcon tube and remove the excess liquid using tissue paper before placing it with the fixed DNA side facing down on a piece of Saran wrap. Place a previously cut 12 × 4 cm acetate film on top and wrap the Saran wrap around the membrane, making sure that no creases are formed.
Place the wrapped membrane in a cassette with a Fuji screen and expose for around 72 h at RT (the Geiger should indicate 2-5 cps per membrane). Make sure that the DNA-fixed side of the membrane is facing up and is in contact with the screen. We recommend placing the membranes with the cut edge at the top left-hand side.
After scanning the Fuji screen, the membranes must be stripped and hybridized a second time with genomic DNA to normalize the signal.
Stripping
Incubate each membrane in 5 ml 50 mM NaOH in a 15-ml Falcon tube for 30 min at 45°C inside a hybridization oven while rotating.
Discard the NaOH and neutralize the membranes by adding 5 ml neutralization solution for an extra 15 min under the same conditions.
Transfer the membranes to a single glass tray that can hold 100 ml stripping solution previously boiled in a microwave. Caution when manipulating the boiling solution; use heat resistant gloves.
Incubate the membranes in boiling stripping solution at room temperature for 10 min on an orbital shaker at slow speed to ensure no loss of liquid.
Wash the membranes once with distilled H2O and leave to dry at RT on a clean filter paper; save them or proceed to the next step. Measure the membranes with a Geiger counter to make sure the radioactive signal has disappeared.
When they are ready to be re-hybridized, transfer the stripped membranes into a fresh 15-ml Falcon tube and pre-hybridize (Step D1).
Radioactive labeling of genomic DNA
Extract yeast genomic DNA using your preferred method (a kit can be used) and dilute 1 μg in a final volume of 37 μl MilliQ H2O.
Boil for 5 min at 95°C on a thermoblock and immediately transfer to ice. Add 5 μl random hexamers, 5 μl each 10 mM dNTP (A, T, G), 1 μl Klenow fragment, and 5 μl α-32P dCTP (Perkin-Elmer NEG513H250UC, 10 µCi/µl).
Incubate at 37°C for 45 min on a thermoblock.
Purify the labeled genomic DNA using a ProbeQuantTM G-50 Micro Column following manufacturer’s protocol.
If to be used immediately, boil the labeled DNA at 95°C for 5 min; and if not, freeze at -20°C. Labeled DNA should be used as soon as possible since 32P decays over time. Use within a few days.
The labeled DNA obtained will be sufficient for 12 membranes. Dilute the obtained DNA with MilliQ H2O until 20 μl per membrane is obtained. For example, if you have 12 membranes, you need to add 190 μl MilliQ H2O to the resulting 50 μl labeled DNA to obtain 20 μl DNA per membrane.
Add 3 ml fresh hybridization solution and 20 μl diluted labeled DNA to a 15-ml Falcon containing the membrane, and hybridize for 24 h at 65°C.
Follow the same steps as above (steps in Procedure D).
Expose the membrane overnight or longer depending on the signal obtained (48 h if the Geiger indicates 5-10 cps).
Scanning the membranes and quantitation
Scan the Fuji screens using a STORM-840 imaging system or similar.
Be careful to remember the order of the membranes and to not let the Fuji screen be exposed to light for long, since light erases the screen.
If the signal observed is not high enough, you can re-expose the screen for longer.
Quantitate the radioactive signal using the free GelQuant.NET software (http://biochemlabsolutions.com/GelQuantNET.html) or similar.
Normalize the RNA radioactivity intensity to the DNA radioactivity intensity.
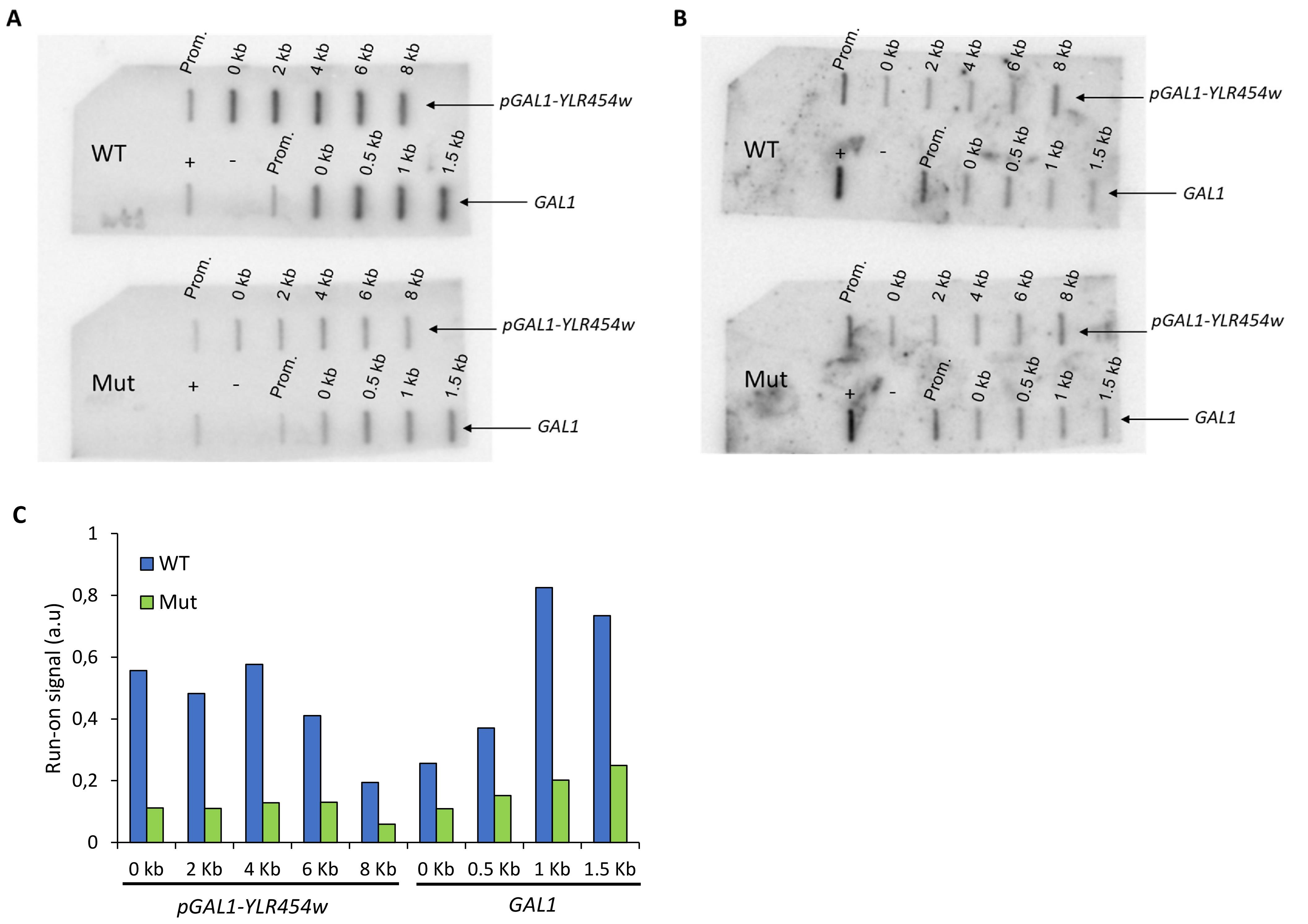
Figure 2. Results from a typical TRO experiment. A. Membrane after labeled RNA hybridization. Both top and bottom membranes have the same DNA fragments crosslinked on them: several fragments of the pGAL1-YLR454w gene construct, positive (yeast gDNA) and negative (E. coli gDNA) controls, and several fragments of the GAL1 gene. The top membrane was hybridized with labeled RNA from WT yeast cells and the bottom from mutant (Mut) yeast cells. The mutant in this example corresponds to pdf1Δ. B. The same membranes shown in A were stripped and re-hybridized with labeled yeast gDNA. C. Graph showing the quantitation of the results shown in A and B.
Data analysis
The phosphoimages taken by STORM or a similar imaging system should be in .gel format.
First, we need to quantitate the labeled RNA membranes (as seen in Figure 2A), load the image into the GelQuant.NET program (see Figure 3), and adjust the contrast by pressing the .GEL scale button. Afterwards, rotate and center the image using the “Rotate 90” and zoom buttons.
Next, proceed to quantitate each line by drawing a small red square around the line in question. Then, press the control button on the bottom panel to ensure that you use the same sized square for all the lines, and press the quantify button. A number should appear on the right-hand side, which corresponds to the quantitation of the square minus the background. We usually use the automatic local background option (bottom right), but you can determine your own background by clicking on the manual background option.
Repeat this on the next line by dragging the fixed red square over it and pressing quantify; repeat until you have quantitated every relevant line (the positive and negative controls do not need to be quantitated, they just need to be visible or absent, respectively).
On the top right, you will see a series of numbers corresponding to the volume quantitation of your lines. You can copy these numbers into a clipboard by selecting the box under the numbers, highlighting the numbers that you want to copy, and pressing the clipboard button at the bottom. Then, copy these numbers into an Excel sheet.
Repeat the steps 2-5 for the DNA-labeled membranes (as seen in Figure 2B). These numbers should be more similar to each other.
Once you have all the numbers for each fragment and each membrane in Excel, you can now finish analyzing them. First, normalize the RNA membranes by the number of uridines (or thymines) in each DNA fragment loaded onto the membranes, as this can affect the amount of labeled signal obtained. Then, normalize the RNA numbers to the DNA membranes. Finally, normalize them to the number of cells harvested in each sample using the O.D. numbers.
We repeat each experiment 3 times and perform a Student’s t-test on each fragment in the WT with respect to the mutant to identify significant changes in active RNA polymerases on each DNA fragment studied.
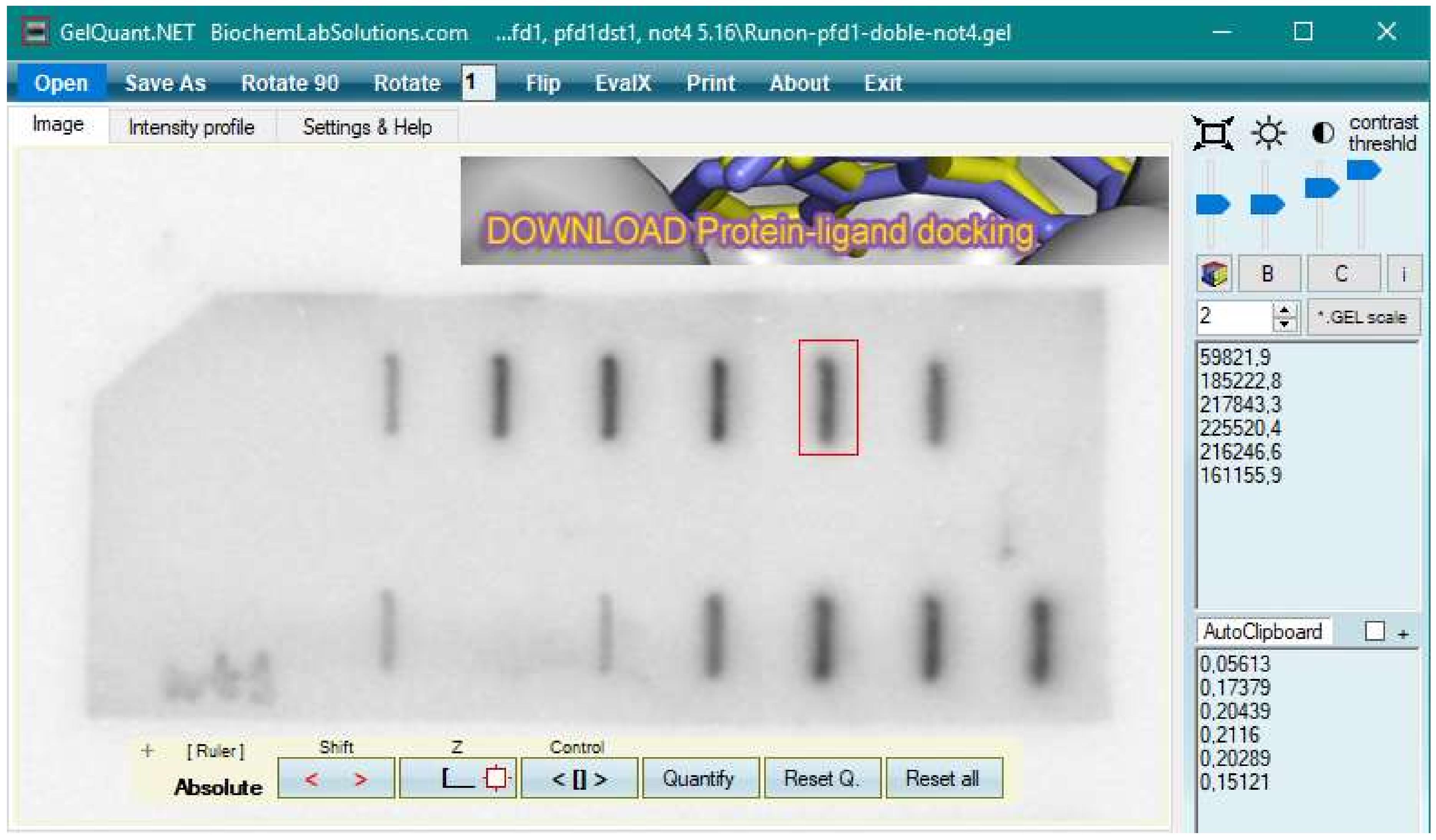
Figure 3. Screenshot of the GelQuant program used to quantitate the hybridized labeled RNA signal. At the top-right, the .GEL scale, zoom, brightness, and contrast buttons can be found. At the top, the open, save as, rotate, etc buttons can be found. At the bottom, buttons to draw the red quantitation square (z) can be found, and next to it, the control button that is selected to allow the same red square to be dragged over the different bands for quantitation. The quantitate button is next to it, which when selected, will give numbers on the right-hand side of the page. At the bottom-right, the auto local background option can be found, and above this is the clipboard option.
Recipes
Note: Autoclave all stock solutions to sterilize.
Denaturalization buffer
1.5 M NaCl
0.5 M NaOH
Neutralization buffer
1 M NaCl
0.5 M Tris-HCl, pH 7
SSC 20×
150 mM NaCl
20 mM Na citrate
Add HCl until pH 7
Transcription buffer 2.5×
550 mM Tris-HCl, pH 7
500 mM KCl
80 mM MgCl2
TES
10 mM Tris-HC,l pH 7.5
10 mM EDTA, pH 8
0.5% SDS
Hybridization solution
0.5 M phosphate buffer, pH 7
7% SDS, 0.5 M EDTA pH 8
Wash solution I
1× SSC
0.1% SDS
Wash solution II
0.5× SSC
0.1% SDS
Neutralization solution (stripping)
550 mM Tris-HCl, pH 7
0.1× SSC
0.1% SDS
Stripping solution
55 mM potassium phosphate, pH 70.1% SDS
10× TBE buffer
13 M Tris base
450 mM boric acid
25 mM EDTA, pH 8
Tris-HCl 1M, pH 7
1 M Tris base
add HCl until pH 7
Phosphate buffer 1 M, pH 7
99.4 g Na2HPO4
41.4 g NaH2PO4·H2O
Check pH is at 7
Acknowledgments
This work was funded by the Spanish Ministry of Economy and Competitiveness, and European Union funds (FEDER) [BFU2016-77728-C3-1-P].
The original research article in which this protocol was used is Begley et al. (2019).
Competing interests
None declared.
References
- Begley, V., Corzo, D., Jordan-Pla, A., Cuevas-Bermudez, A., Miguel-Jimenez, L., Perez-Aguado, D., Machuca-Ostos, M., Navarro, F., Chavez, M. J., Perez-Ortin, J. E. and Chavez, S. (2019). The mRNA degradation factor Xrn1 regulates transcription elongation in parallel to Ccr4. Nucleic Acids Res 47(18): 9524-9541.
- Core, L. J., Waterfall, J. J. and Lis, J. T. (2008). Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters.Science 322(5909): 1845-1848.
- Garcia-Martinez, J., Aranda, A. and Perez-Ortin, J. E. (2004). Genomic run-on evaluates transcription rates for all yeast genes and identifies gene regulatory mechanisms. Mol Cell 15(2): 303-313.
- Greenberg, M. E. and Ziff, E. B. (1984). Stimulation of 3T3 cells induces transcription of the c-fos proto-oncogene. Nature 311(5985): 433-438.
- He, F. (2011). E. coli Genomic DNA Extraction. Bio-protocol 1: e97.
- Jordan-Pla, A., Miguel, A., Serna, E., Pelechano, V. and Perez-Ortin, J. E. (2016). Biotin-Genomic Run-On (Bio-GRO): A High-Resolution Method for the Analysis of Nascent Transcription in Yeast. Methods Mol Biol 1361: 125-139.
- Jordan-Pla, A., Perez-Martinez, M. E. and Perez-Ortin, J. E. (2019). Measuring RNA polymerase activity genome-wide with high-resolution run-on-based methods. Methods 159-160: 177-182.
- Pérez-Ortín, J. E., de Miguel-Jimenez, L. and Chavez, S. (2012). Genome-wide studies of mRNA synthesis and degradation in eukaryotes. Biochim Biophys Acta 1819(6): 604-615
- Smale, S. T. (2009). Nuclear run-on assay.Cold Spring Harb Protoc 2009(11): pdb prot5329.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Begley, V., de Miguel-Jiménez, L. and Chávez, S. (2021). Transcriptional Run-on: Measuring Nascent Transcription at Specific Genomic Sites in Yeast. Bio-protocol 11(12): e4064. DOI: 10.21769/BioProtoc.4064.
Category
Microbiology > Microbial genetics > RNA > Transcription
Molecular Biology > RNA > Transcription
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.