- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Electrophysiological Properties of Neurons: Current-Clamp Recordings in Mouse Brain Slices and Firing-Pattern Analysis
Published: Vol 11, Iss 12, Jun 20, 2021 DOI: 10.21769/BioProtoc.4061 Views: 7469
Reviewed by: Chiara AmbrogioJorge Miranda BarrientosSvetlana Molchanova
Original research article
The authors used this protocol in:

Aug 2020
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Characterization of an electrically active cell, such as a neuron, demands measurement of its electrical properties. Due to differences in gene activation, location, innervation patterns, and functions, the millions of neurons in the mammalian brain are tremendously diverse in their membrane characteristics and abilities to generate action potentials. These features can be measured with a patch-clamp technique in whole-cell current-clamp configuration followed by detailed post-hoc analysis of firing patterns. This analysis can be time-consuming, and different laboratories have their own methods to perform it, either manually or with custom-written scripts. Here, we describe in detail a protocol for firing-pattern registration in neurons of the ventral tegmental area (VTA) as an example and introduce a software for its fast and convenient analysis. With the help of this article, other research groups can easily apply this method and generate unified types of data that are comparable between brain regions and various studies.
Graphic abstract:
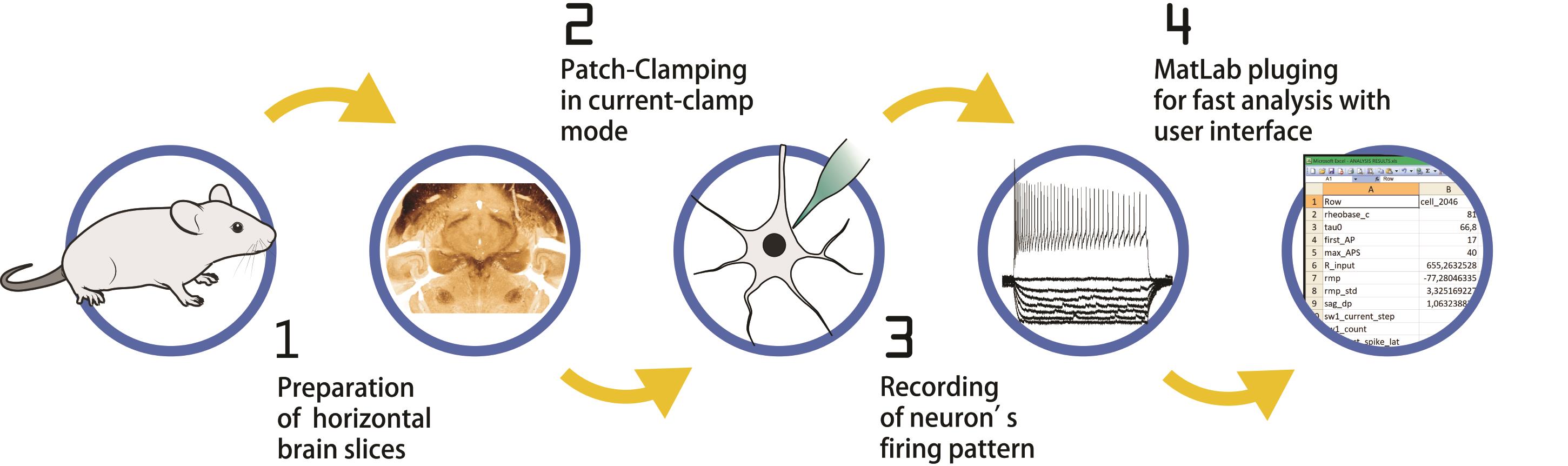
Workflow of the Protocol
Background
The main feature of a neuron is its ability to engage in fast chemo-electrical communication with other cells. The unique neuronal membrane constitution with a high density of ion channels and other specific proteins allows the generation of an action potential within milliseconds after receiving an adequate input. Therefore, characterization of any neuron would be incomplete without description of the electrical properties of its membrane. For this purpose, we can use a convenient “patch-clamp” technique (Neher and Sakmann, 1976), which resulted from centuries of evolution in electrophysiological methods (Verkhratsky and Parpura, 2014). A big advantage of this method is the possibility to combine it with other modern single-cell approaches and collect all necessary data for defining a neuron’s type. Filling of the cell with intracellular dyes through the patch pipette (Marx et al., 2012) and collecting the cell’s aspirate after electrophysiological registration (Sucher and Deitcher, 1995; Fuzik et al., 2016) allow the simultaneous reconstruction of 3D morphology and analysis of mRNA content and firing pattern of the same cell. There are several types of patch-clamp configurations, but we focus here on the whole-cell current-clamp modification, which allows registering changes in the membrane voltage while controlling the electrical current flow; in other words, it allows registering action potentials (AP) in response to specific current injections.
The whole-cell current-clamp configuration of the patch-clamp technique is a well-established method that has been used for decades in the characterization of intrinsic membrane properties of electrically active cells (Neher and Sakmann, 1976; Andrew, 1986; Sanchez-Aguilera et al., 2020). Although most of the researchers are interested in a similar set of parameters and use similar protocols, the experimental details and final data are variable and, therefore, hard to compare (see https://neuroelectro.org/article/index). While preparing to describe somatostatin-expressing neurons in the mouse ventral tegmental area (VTA) for our recent article (Nagaeva et al., 2020), we tried to collect a maximal set of membrane properties for firing pattern analysis based on previous publications (Halabisky, 2006; Ma et al., 2006; Wierenga et al., 2010). The list of these properties can be found in Nagaeva et al., 2020, Appendix Table 1. Similarly, we made a current-stimulation protocol that allowed registering all these properties in one short run. Additionally, we developed a MatLab plugin for fast and convenient extraction of all these parameters.
Our protocol article aims to provide a clear workflow for firing pattern analysis and includes all steps from the preparation of acute brain slices to the final data extraction. It can be applied for electrophysiological studies of previously unknown neurons or as part of the currently popular patch-seq approach (Cadwell et al., 2016; Gouwens et al., 2020). For newly described neurons, the extracted data can be further used for neuron subtyping according to their electrical membrane properties. To do so, one just needs to upload the resulting tables to a clustering algorithm previously published by our group (see further Nagaeva et al., 2020; https://github.com/elifesciences-publications/clustering-for-nagaeva-et.-al.-sst-vta). This protocol will simplify the firing pattern registration and analysis in future studies.
Materials and Reagents
Preparation of acute brain slices
Beakers: 1 L, 2 × 250 ml (VWR, catalog numbers: 213-1128, 213-1124)
Petri dish 60 × 18 mm (VWR, catalog number: 734-2815p)
Tubes for carbogen delivery into solution (Ismatec, catalog number: MF0028)
Paint brush (VWR, catalog number: 470020-430)
Pasteur pipette with a wide neck (you can simply cut off the tip) (Sarstedt, catalog number: NC9891525)
Super glue (Loctite, catalog number: 230992)
Filter paper (Whatman, WHA1001110)
A box full of crushed ice
Medical spatula with smooth ends 150 × 40 × 6 mm (Bochem, VWR catalog number: 231-0601)
Carbogen (95% O2 + 5% CO2)
Floating net for brain slices
A nice one can be 3D-printed from here https://3dprint.nih.gov/discover/3dpx-001623.
Reagents for Artificial Cerebrospinal Fluid (ACSF) and Cutting solution:
NaCl (Fisher BioReagents, catalog number: BP358)
KCl (Amresco, catalog number: 0395)
MgCl2·6H2O (Fisher BioReagents, catalog number: BP214)
NaH2PO4·H2O (Merck, catalog number: 1.0634)
NaHCO3 (Sigma-Aldrich, catalog number: 31437)
D-(+)-Glucose (Alfa Aesar, catalog number: A16828)
Sucrose (Fisher Scientific, catalog number: 10634932)
CaCl2 (Amresco, catalog number: 0556)
Electrophysiology
Beakers: 250 and 25 ml (VWR, catalog numbers: 213-1128, 213-1120)
1-ml micropipette (Thermo Scientific, Finnpipette F2)
1.5-ml Eppendorf tubes for intracellular solution (Eppendorf, catalog number: 0030120086)
Glass capillaries with filament (World Precision Instruments, catalog number: TW150F-4)
Paint brush (VWR, catalog number: 470020-430)
Silver/platinum wire or special net for slice holding in the microscopy chamber (see Figure 9)
1 ml syringe (Terumo, catalog number: SS+01T1)
Syringe PVDF Durapore filter (Merck Millipore, catalog number: SLGV013SL)
Plastic tubing ID 010 × OD 030 (Tygon, catalog number: AAD04091)
Reagents for Intracellular solution (IS):
K-gluconate (Sigma-Aldrich, catalog number: P1847)
HEPES (Alfa Aesar, catalog number: A14777)
EGTA (Sigma-Aldrich, catalog number: E4378)
Na2-ATP (Sigma-Aldrich, catalog number: A6419)
Na-GTP (Sigma-Aldrich, catalog number: G8877)
Na2-phosphocreatine (Sigma-Aldrich, catalog number: P7936)
KOH (Sigma-Aldrich, catalog number: 221473)
ASCF solution (see Recipes)
Cutting solution (see Recipes)
Intracellular solution (see Recipes)
Equipment
Volumetric flask 1 L (Brand, VWR catalog number: 612-5082)
Big scissors (Fiskars, catalog number:1005151)
Small scissors (Bochem, VWR catalog number: 233-2121)
Small tweezers 105 mm (Usbeck, VWR catalog number: 232-0094)
Scalpel (Swann-morton, VWR catalog number: swan0565)
Teaspoon
Razor blade for the vibratome (World Precision Instruments, catalog number: 752-1-SS)
The same can be used for the brain dissection in Step A2b of the Procedure section.
Laboratory scale (0.001-100 g; Mettler PJ360 DeltaRange)
pH-meter (Metrohm, 827 pHlab)
Magnetic stirrer (Merck, IKA big-squid)
Laboratory water bath (Grant Instruments, Bath JB Aqua 12 Plus)
Vibratome (Thermo Scientific, Microm HM650V)
Osmometer (Advanced Instruments Inc., Model 3320)
Micropipette puller (Sutter Instruments, Model P-1000)
Epifluorescent microscope (Olympus, BX51WI)
Fluorescent light source – 100 W mercury arc lamp with a power supply (Olympus, U-RFL-T)
CCD camera (Sony, XC-E150)
Heat controller (Warner Electric, TC-324B single channel)
Laboratory vacuum pump (KNF, N 811 KTP)
Amplifier (Molecular Devices, Axon Instruments, Multiclamp 700B)
Digidata (Molecular Devices, Axon Instruments, Model 1322a)
Micromanipulators (Sensapex, µMp-3)
Software
pClamp 8.2 (or later) package (Molecular Devices, https://www.moleculardevices.com/products/axon-patch-clamp-system/acquisition-and-analysis-software/pclamp-software-suite#Resources)
Software for your microscopy camera
MATLAB R2018b (Mathworks, https://se.mathworks.com/products/matlab.html)
Microsoft Excel 2016 (Microsoft, https://www.microsoft.com/en-ww/microsoft-365/excel)
Procedure
Preparation of acute brain slices
Note: Here, we prepare horizontal slices of the mouse midbrain, aiming to patch dtTomato-positive fluorescent cells from the VTA. The reader can prepare slices from any other brain area using the same reagents and procedure.
Prepare Artificial Cerebrospinal Fluid (ACSF) and Cutting solutions (see Recipes) if you use juvenile mice younger than P30. Store solutions not longer than 3-4 days at 4°C.
Note: If you are using older animals, see Ting et al. (2018).
Put all powders except CaCl2 in 1-L beaker and mix with 800 ml MilliQ water using a magnetic stirrer. Add CaCl2 after all other powders are fully dissolved.
Pour the solution into 1-L volumetric flask and adjust the volume. Adjust pH to 7.3-7.4 by bubbling with carbogen.
Prepare your working space for brain dissection.
Place “dry” instruments in a convenient order to reach them fast: big scissors, small scissors, small tweezers, scalpel, bent piece of filter paper, and waterproof superglue (Figure 1).
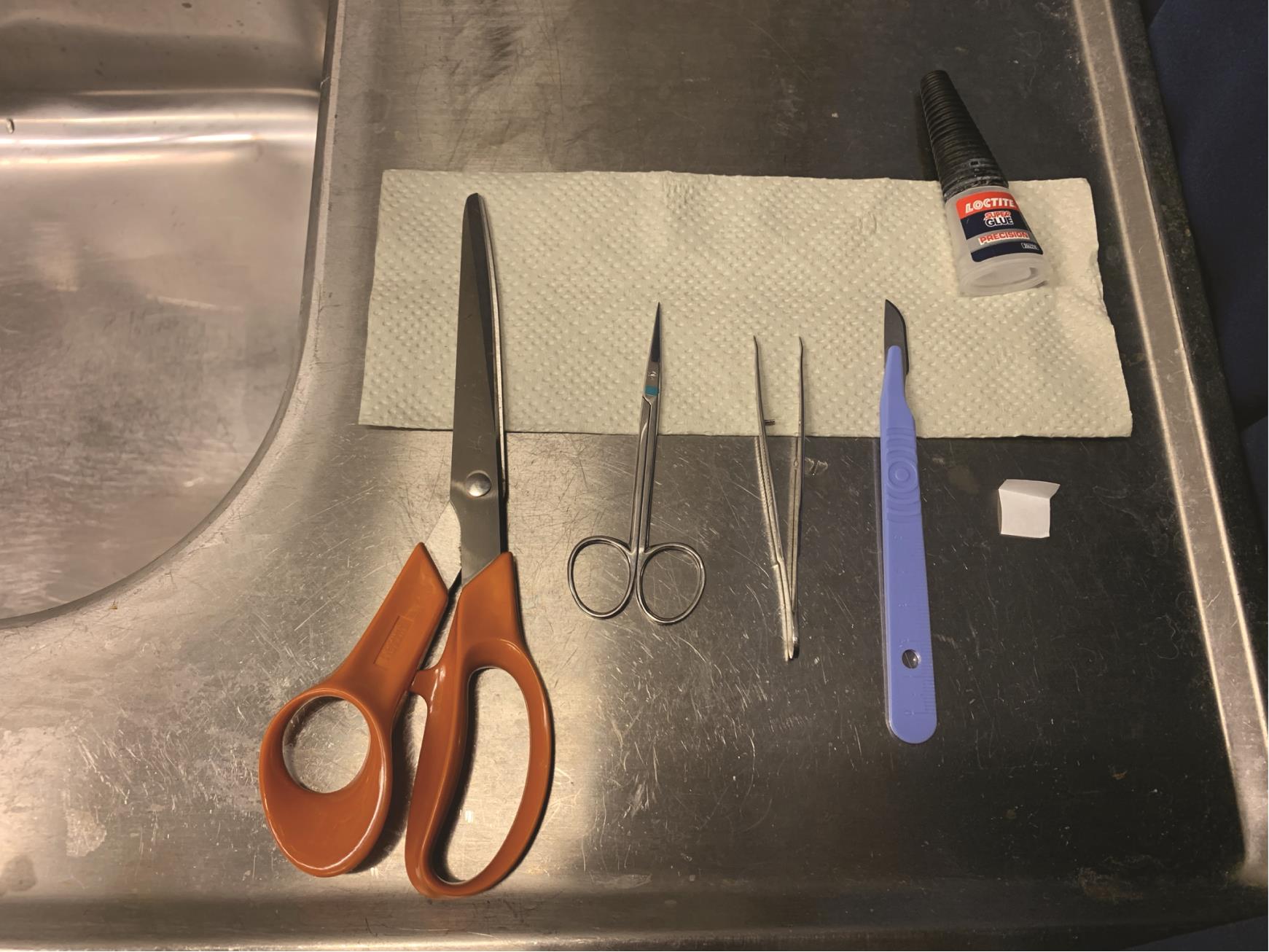
Figure 1. Set of “dry” instrumentsPut the instruments – teaspoon, razor blade, medical spatula with narrow, smooth ends, and 60-mm Petri dish covered by filter paper – and approximately 250 ml of cutting solution on ice (Figure 2).
Note: The ACSF solution should be chilled to 0°C beforehand and have floating pieces of ice. Aerate the solution with carbogen for 5 min before and during surgery (Figure 2).
Tip: Sharp and convenient instruments ensure the success of any surgical operation. All instruments should be cleaned with MilliQ water after use.

Figure 2. Set of “cold” instrumentsPut a 250-ml beaker with constantly aerated 200 ml of ACSF solution and a floating net in a water bath at 33°C (Figure 3).

Figure 3. Beaker filled with ACSF solution and a floating net for incubation of brain slices at 33°CPrepare vibratome: insert the blade and tune the program. It is convenient to have a small paint brush, small tweezers, and a Pasteur pipette with a wide neck near the vibratome (Figure 4).
Note: We have used a vibratome program with the following parameters: feed=225 µm, frequency=88 Hz, amplitude=0.9 mm, and velocity=0.9 mm/s.

Figure 4. Set of instruments for brain slicingWe recommend having all necessary equipment and instruments at hand range as you would need to move between them quickly. When everything is ready, it is time to start the brain dissection.
Brain dissection (approx. 1 min).
Notes:
As the brain cells are very sensitive to hypoxia, and the metabolism is faster in warm environment, it is essential to transfer the brain from a live animal to cold cutting solution as rapidly as possible. We recommend using a stopwatch in the beginning, aiming to finish the whole procedure within 60-80 s.
Important note: Consult your local animal welfare authorities for information on the appropriate anesthesia type to use before animal decapitation.
For brain research purposes, authorities sometimes allow fast physical euthanasia of neonatal/juvenile mice with the method of decapitation, but it should be only performed by well-trained personnel.
Decapitate the mouse with big scissors in one move.
Cut the skin from the neck to between the eyes with small scissors.
Cut the skull from the back to the bregma along the midline with the same small scissors. Be careful not to touch the brain surface with the scissors.
Open the skull half by half with small tweezers from the midline to the side.
Note: It is very important to choose appropriate tweezers and use them carefully, especially if your target is the cerebral cortex.
Use the scalpel to cut out unnecessary parts of the brain within the skull. We cut out half of the cerebellum and frontal pole, as shown in Figure 5.
Note: From here on, all cutting procedures aim at getting midbrain horizontal slices and should be revised for obtaining other brain regions of interest.
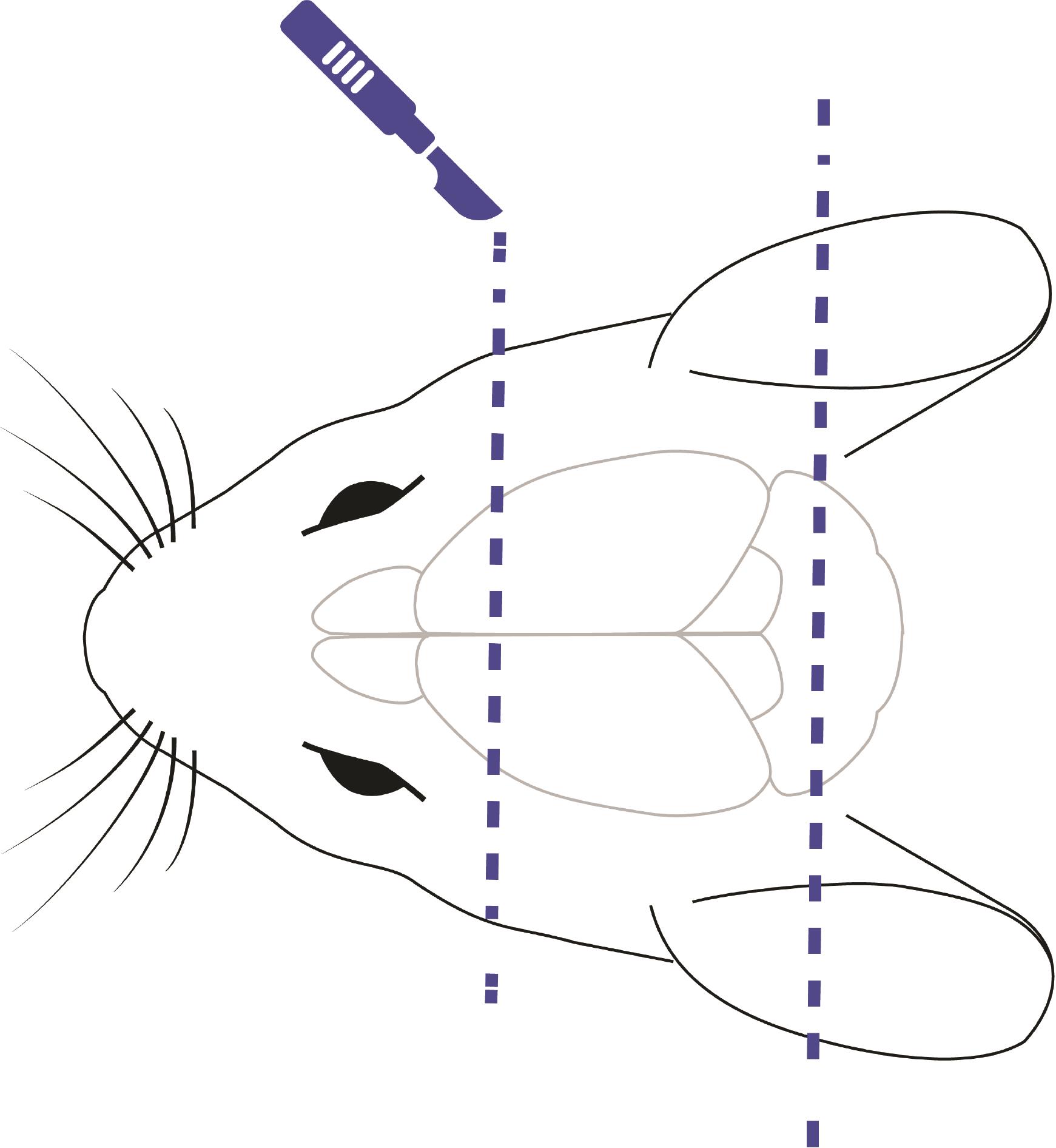
Figure 5. Schematic representation of mouse head with the brain inside. Dashed lines illustrate scalpel cuts.Note: Here, we switch to “cold” instruments and environment to slow down the metabolism.
Take out the brain from the skull with the help of a medical spatula and immediately transfer it into the cold aerated cutting solution. Leave it there for 1 min while preparing the cold vibratome platform for supergluing the brain.
Brain slicing (max 5-10 min).
Note: Background knowledge on the use of the vibratome is required to perform brain slicing.
Take out the brain from the cutting solution with a teaspoon and put it onto the Petri dish with the cortex facing down (that is, “on the cortex”). Then, you can cut out all unnecessary parts with a cold razor blade if you did not do so already before taking the brain out from the skull (see above).
Put a small drop of superglue onto the vibratome platform and immediately transfer the brain onto it with the help of the bent piece of filter paper. Do not change brain orientation (ventral part is up) and glue it so that the cerebellum is facing the vibratome blade.
Place the platform into the buffer tray and fill up the tray with the cold aerated cutting solution (Step A3f).
With the help of small tweezers, carefully take off the filter paper, which still is stuck to the brain.
Note: If you would like to extend the time of slicing, you can aerate the cutting solution also in the buffer tray.
When the brain is placed as shown in Figure 6, it is ready for making horizontal midbrain slices. Carefully cut away unnecessary upper slices with 225-µm steps until the brain and cerebellum are fully connected. At this level [corresponds to -4.72 from Bregma (Franklin and Paxinos, 2008)], you will also see a distinctive “circle” in the center of the slice (see Figure 7). The next slice is the first midbrain slice that contains the VTA.
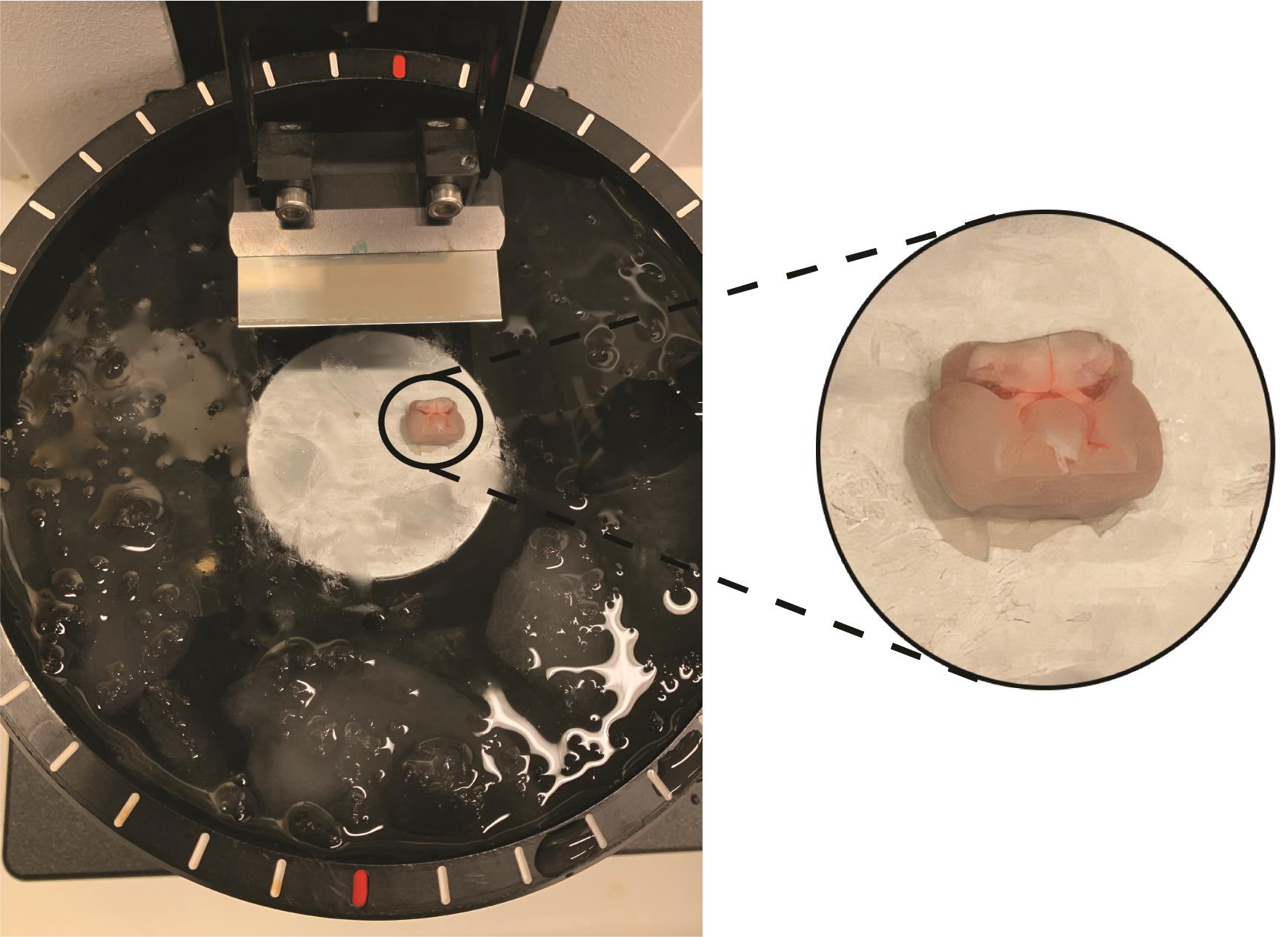
Figure 6. Orientation of the brain inside the vibratome tray. The ventral part up and cerebellum is facing the blade. The right part of Figure 6 shows a close-up of the brain inside the tray.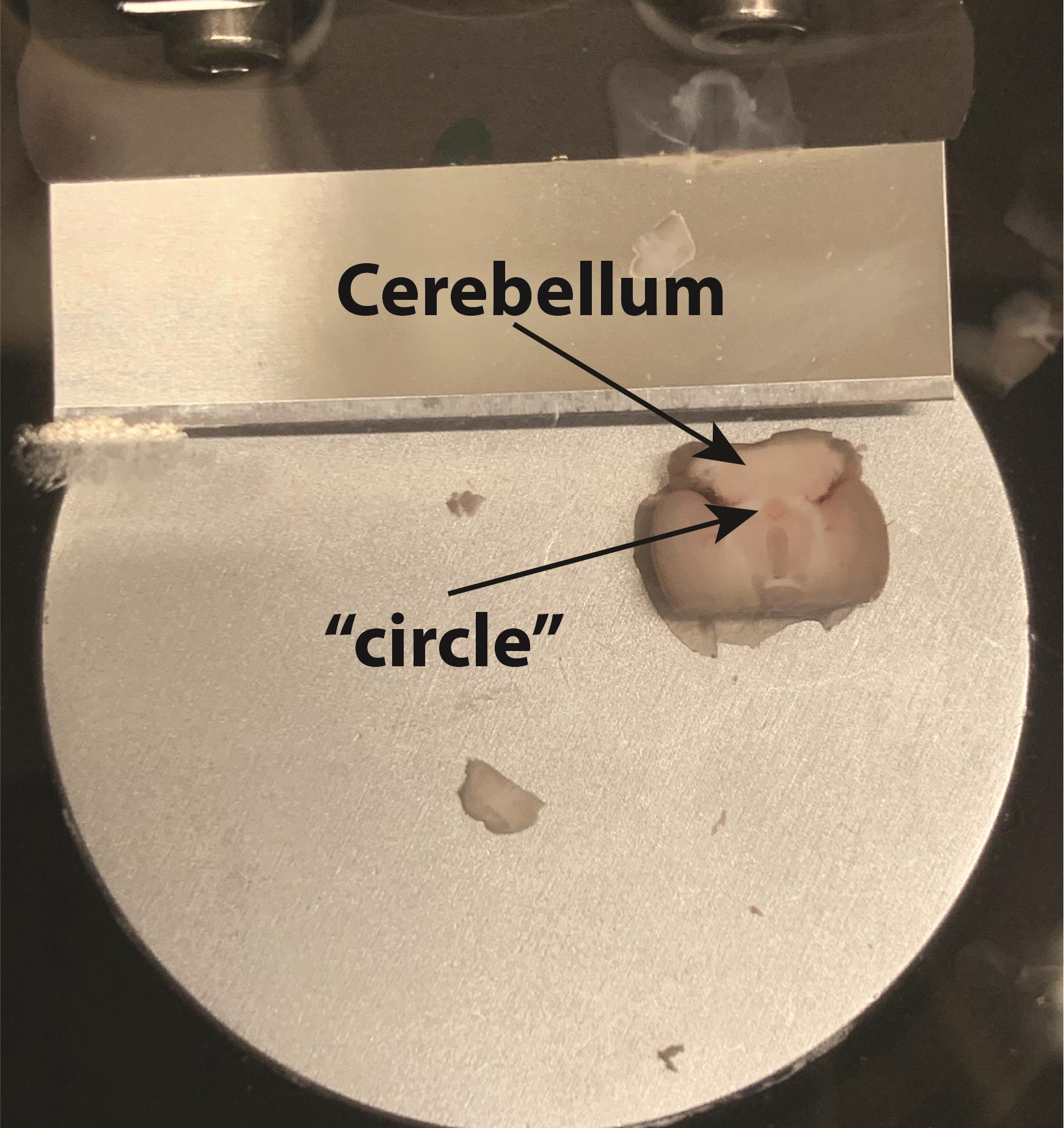
Figure 7. The correct level to start cutting horizontal VTA-containing slices (approximately at -4.72 from Bregma)It is possible to get two or three 225-µm-thick horizontal slices from the mouse VTA, which correspond to Bregma levels of about -4.72 mm, -4.56 mm, and -4.28 mm.
Transfer the resulting slices into 33°C ACSF solution with the help of a Pasteur-pipette immediately after cutting each slice.
Incubate the slices at 33°C for 60 min; then, keep the beaker with slices at room temperature. The slices are viable for at least 4 h. We start electrophysiology right after 60 min of incubation at 33°C.
Note: It is critical that during incubation the slices are continuously aerated but do not float around because of the bubbles.
Electrophysiology
Note: Previous theoretical knowledge on electrophysiology and technical training in cell patching are required to perform further protocol steps.
Prepare K-gluconate-based Intracellular solution (IS).
Mix all reagents with 15 ml of MilliQ water in a 20-ml beaker.
Put the beaker on ice before adding ATP, GTP, and phosphocreatine.
Note: From here on, try to keep the solutions cold all the time, as ATP and GTP are sensitive to temperature.
While mixing with a magnetic stirrer, measure the pH and adjust to 7.2 with KOH.
Measure osmolarity and adjust it to 285 mOsm by adding MilliQ water milliliter by milliliter.
Aliquot the IS solution in 1.5-ml Eppendorfs and store at -20°C for a maximum 2-3 months.
Note: Thaw an Eppendorf tube containing IS every time just prior to the experiment and remember to keep it cold during the experiment (on ice or in the fridge at 4°C).
Prepare 3-5 MΩ glass electrodes from borosilicate capillaries according to puller manufacturer’s instructions. We recommend preparing electrodes with a 4-step program. Figure 8 depicts our program for the Sutter P-1000 puller as an example. Note that parameters might be different depending on the capillaries’ RAMP test result (melting point temperature) and puller type.
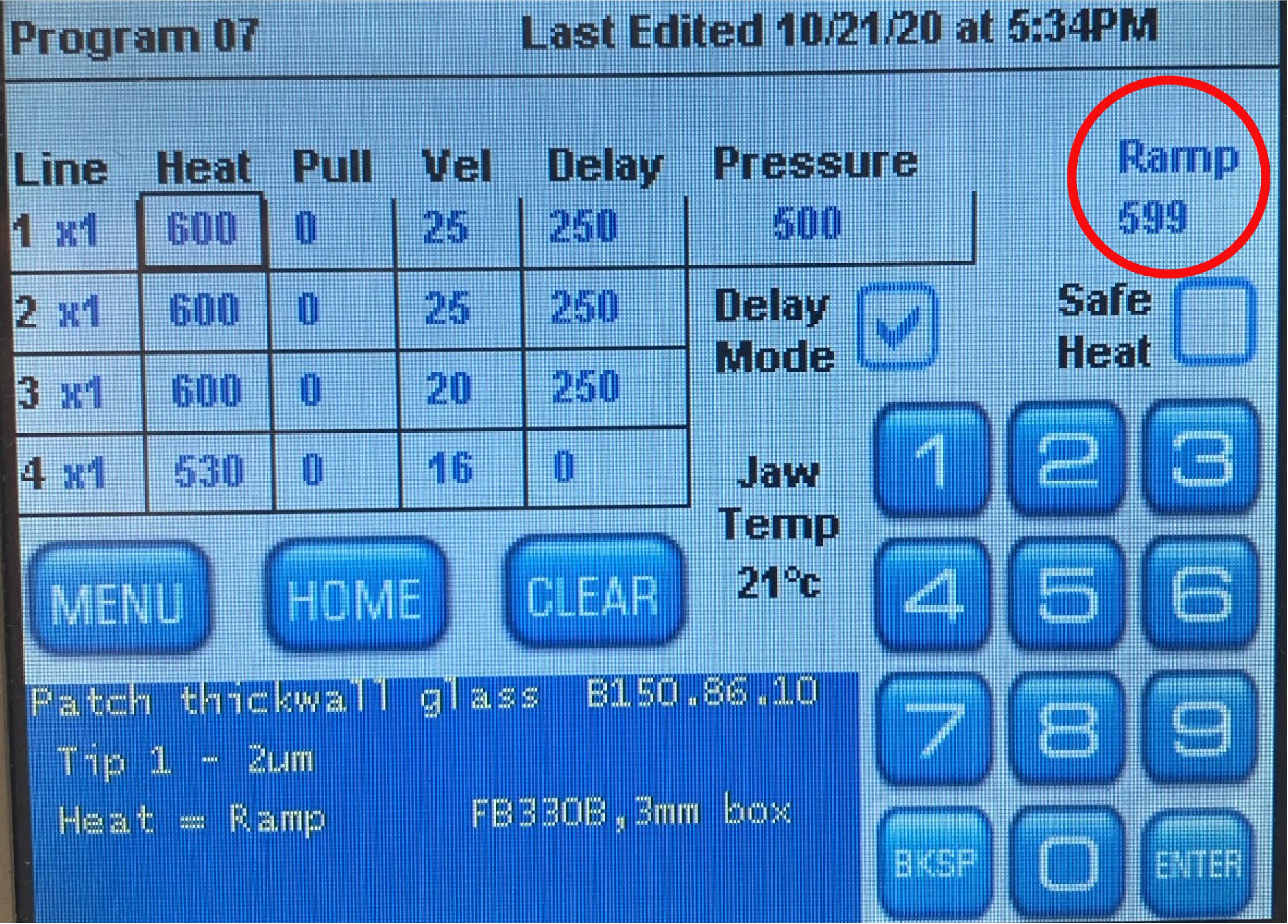
Figure 8. Example of the Sutter P-1000 puller program for suitable glass electrode preparation. Please note the RAMP.Prepare your electrophysiological setup for patching.
Switch on the computer, fluorescent lamp, bright-field lamp, camera, solution heat controller, vacuum pump, amplifier, digidata, and manipulators.
Prepare a 250-ml beaker with a constantly aerated ACSF solution. Tune the perfusion speed to 1-3 ml/min for a 1-ml recording chamber. Tune the heat controller to keep the solution at 33°C.
Patching.
Transfer a slice from the incubation beaker into the recording chamber with a wide-neck Pasteur pipette.
Place the brain slice in the middle of the chamber with the cerebellum in its lower part, as shown in Figure 9. Press the slice down with a silver wire or special net slice holder.
Note: The brain slice should be at the bottom of the recording chamber and remain stationary during the solution flow.
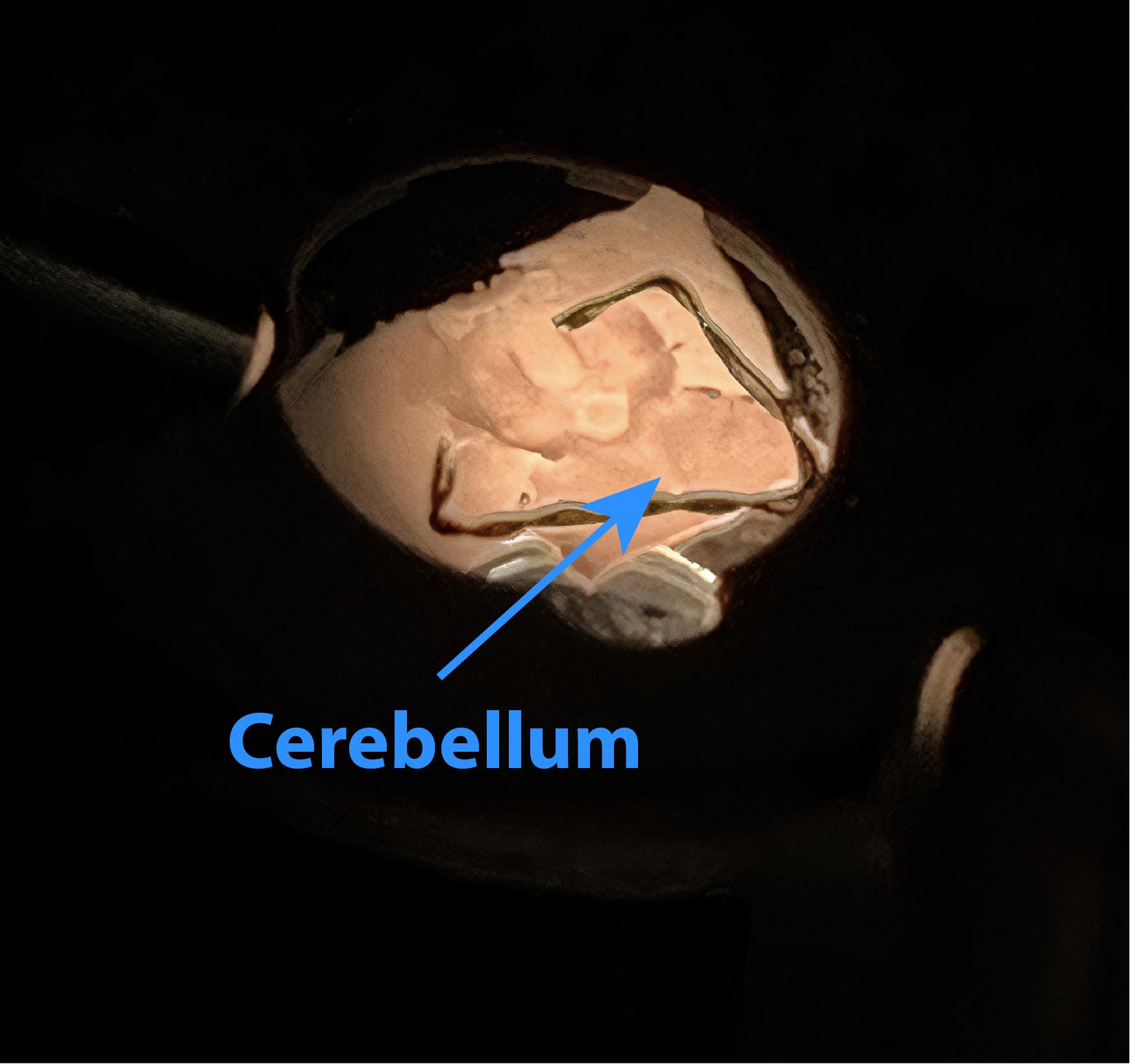
Figure 9. Orientation of the brain slice within the recording chamber with the cerebellum toward the researcher. A silver wire holds the slice at the bottom of the chamber.Fill up the glass electrode with cold IS solution and attach it to the headstage.
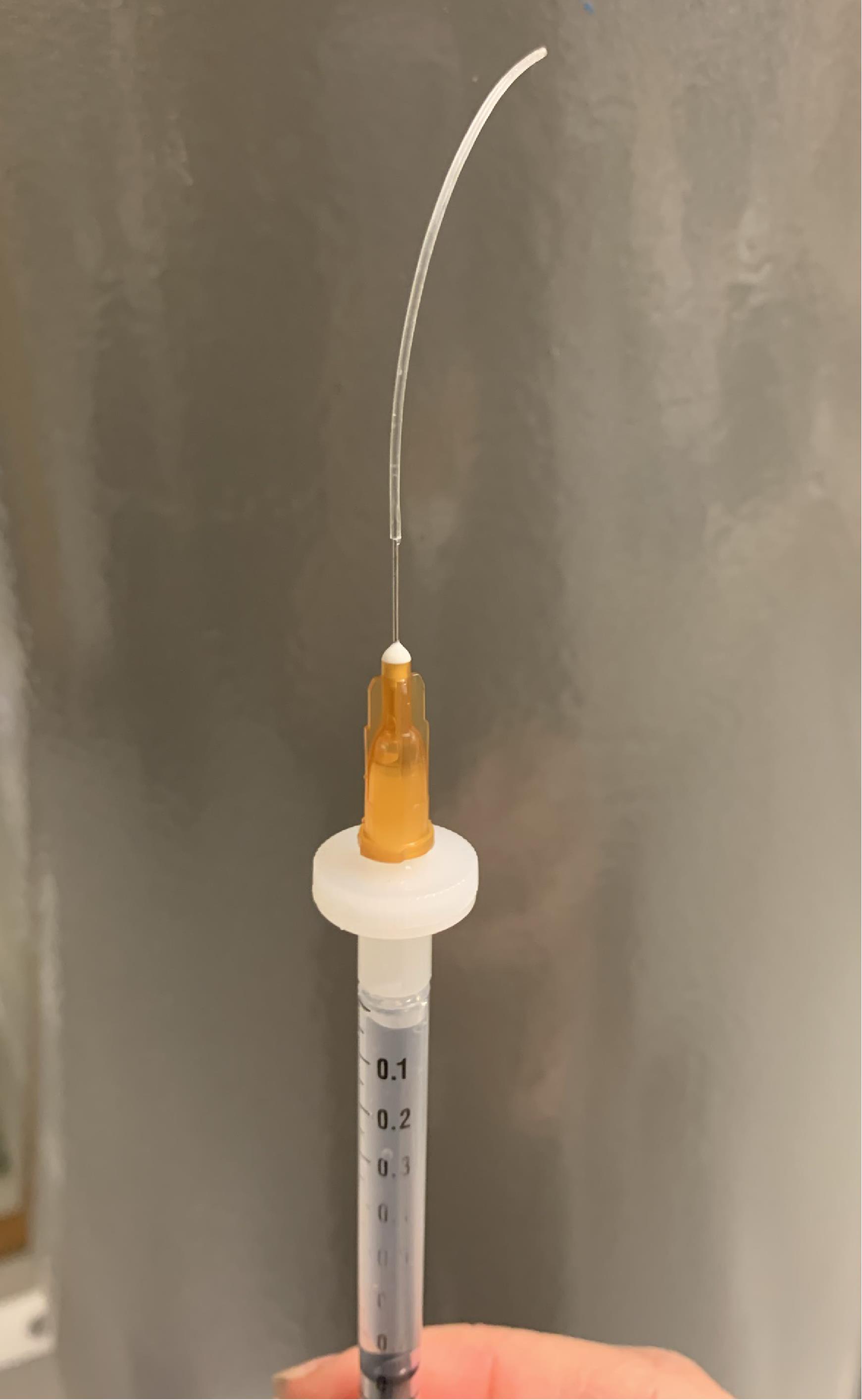
Figure 10. Example of the custom-made syringe for filling the intracellular solutionTip: You can make a very convenient syringe for IS filling (Figure 10) by using a 1 ml syringe, a Merck Millex Durapore filter, and a piece of flexible plastic tubing ID 010 × OD 030.
Make positive pressure in the patch pipette by blowing into the tube and close it with a valve (Figure 11).
Tip: To check whether the pressure is good enough, you can open the valve near your ear. If the pressure is good, you should hear a hollow flop.

Figure 11. The tube and valve set is connected to the headstage for making positive/negative pressure inside the glass electrodePut the electrode down into the solution and check the resistance of the pipette in the Seal Test window (Figure 12). It should be 3-5 MΩ. Reset the pipette offset to 0 mV.
Note: From here to step (v), the amplifier is in Voltage Clamp (VC) mode. While searching for a cell, the voltage is not clamped.
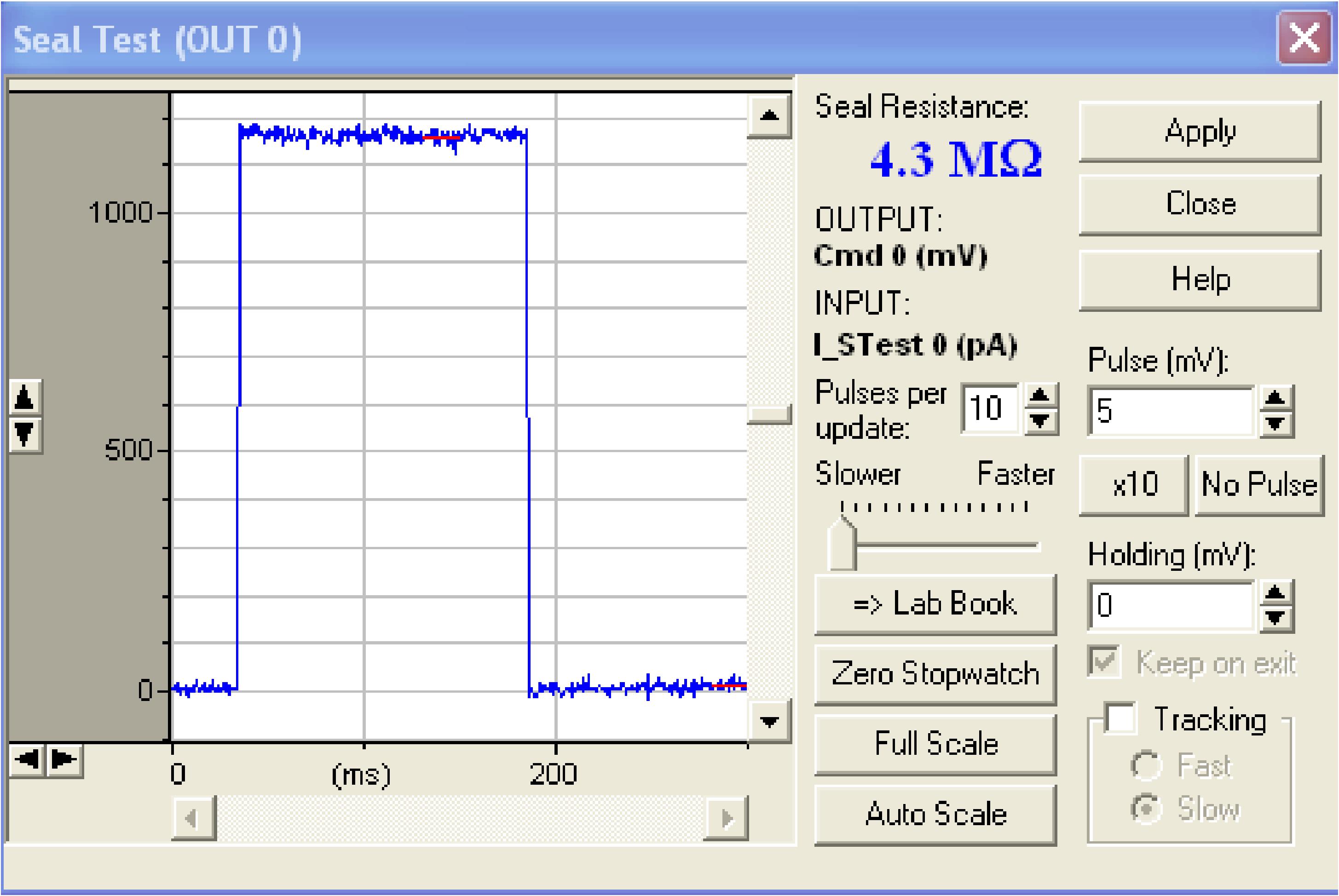
Figure 12. Seal test window showing the pipette resistance of 4.3 MΩLocate the pipette over the region of interest under small magnification, e.g., 5×. To patch cells in the VTA, locate the pipette over the region shown in Figure 13.
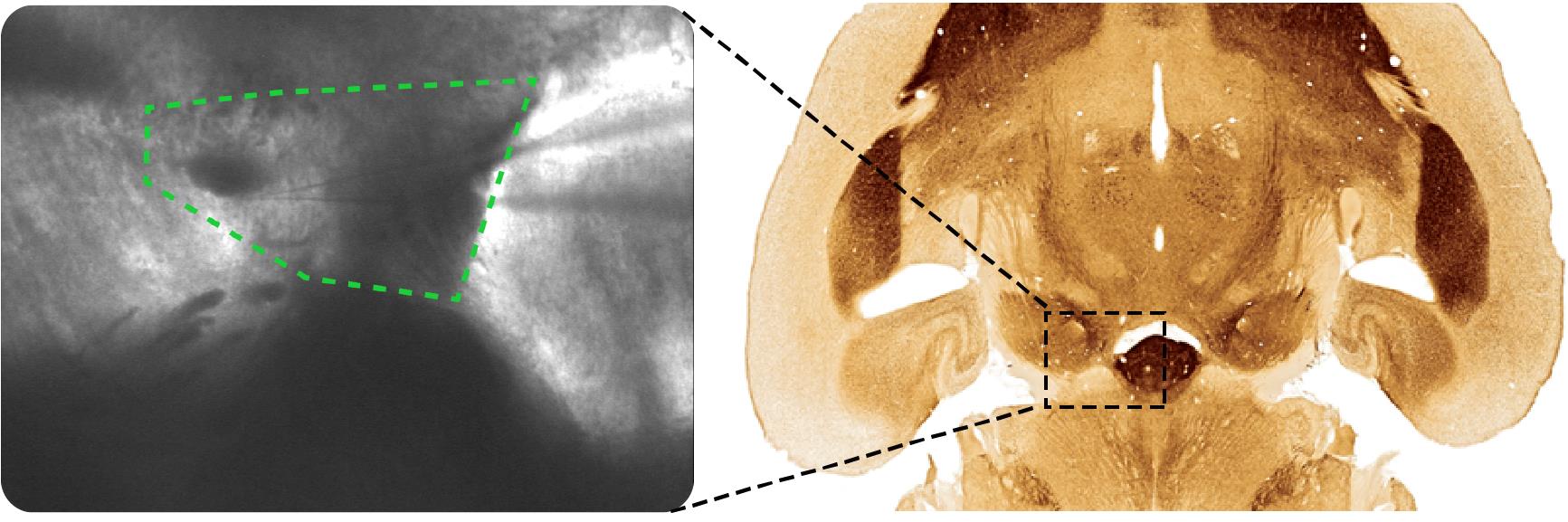
Figure 13. VTA region (marked with green dashed line on the left) under a microscope 5× magnification
Note: For convenience, the right image shows the same midbrain region marked with a dark dashed line within the horizontal brain slice at -4.72 cm from the bregma (Franklin and Paxinos, 2008).Go down with the pipette at 4/5 manipulator speed until the pipette is slightly above the slice focus.
Switch to a higher magnification (e.g., 40×) and find the pipette.
Switch to camera view.
Slow down manipulator speed to 2-3/5 and slowly go down towards the slice surface, first with the objective and then with the pipette.
Stop the pipette right above the slice surface.
Tip: At this level, you can already evaluate the condition of the slice. If there are many bubbled cells on the surface, it might be difficult to find a healthy cell in the slice.
Dim the light and switch to fluorescence. Search a healthy neuron with the objective. Do not move the objective too far; otherwise, it will be difficult to find your pipette again.
Note: An unhealthy neuron is usually round, has clear visible nuclei, and an unclear or, sometimes, very contrasting contour.
After finding a suitable neuron, remember its location. You can put some sign on the screen or mark it otherwise (Figure 14).
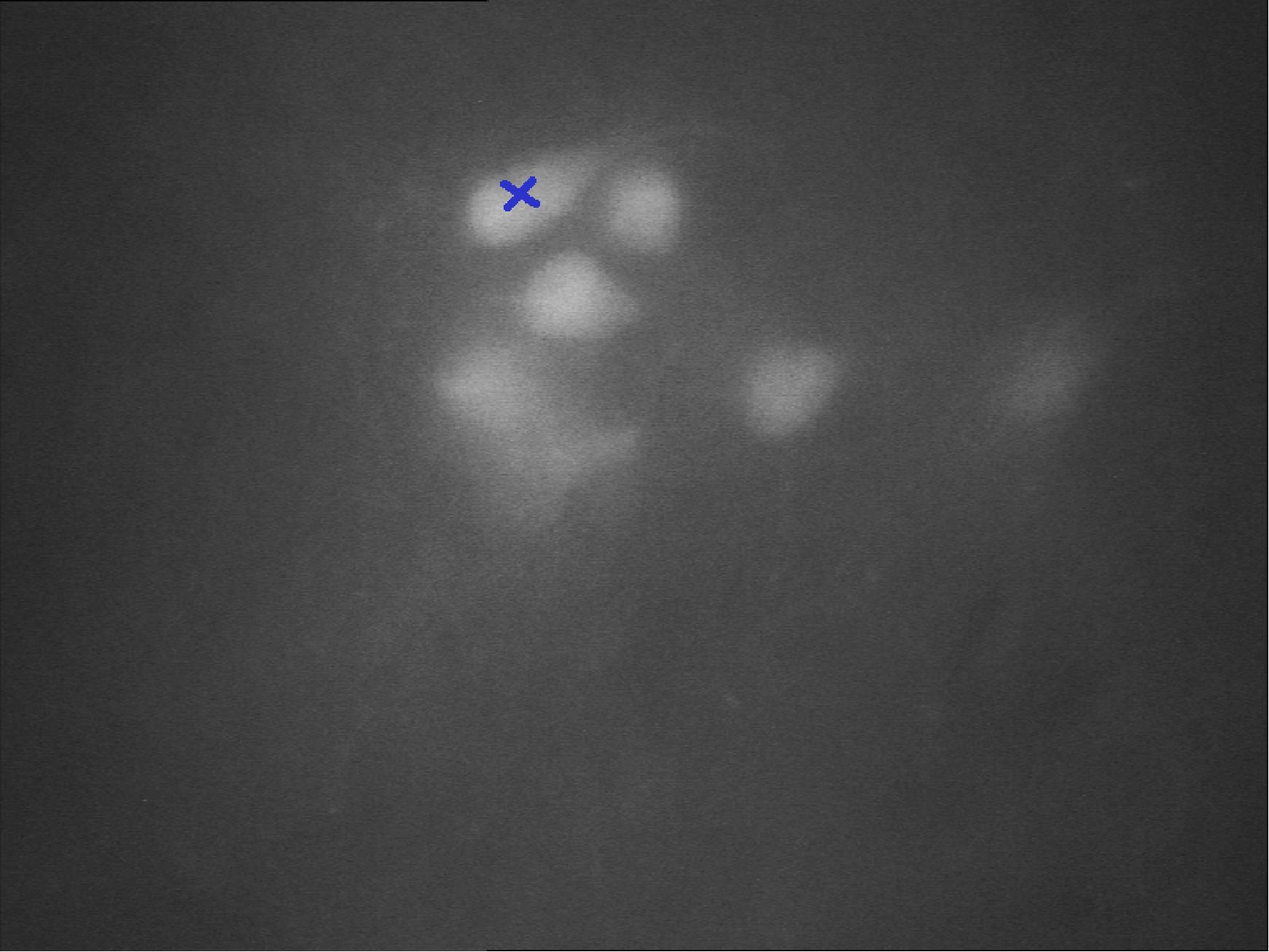
Figure 14. Neurons expressing a fluorescent marker protein under the microscope at 40× magnification. The blue cross indicates a neuron suitable for patching.Go up with the objective and find the pipette. Move it to the neuron’s location above the slice.
Blow into the tube to make sure there is a positive pressure inside the pipette. Reset the pipette offset to 0 mV once more. Pipette resistance must be stable all the time you go down towards the neuron.
Go down to the cell at the slowest manipulator speed. Follow the pipette resistance.
When the resistance rises by 0.1-0.2 MΩ (you might also see a small black dot on the cell, which corresponds to the pipette shadow), release the pipette pressure by opening the valve. Simultaneously, make a slight negative pressure in the pipette by sucking or with the help of a syringe.
At this point, resistance rises to GΩ values (named gigaseal) (Figure 15), which is the first indication of a successful patch.
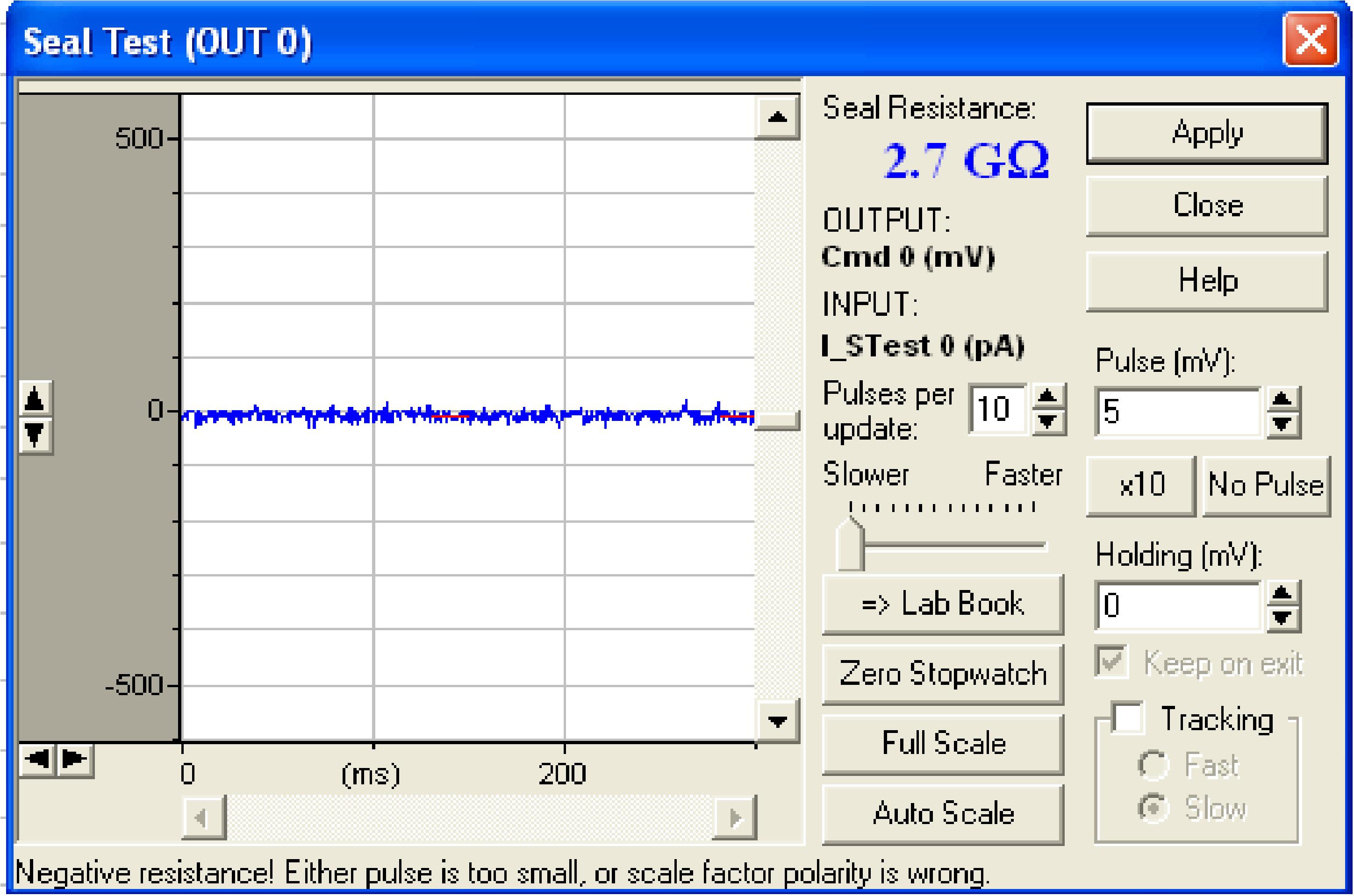
Figure 15. Seal test window showing the gigaseal configuration (Seal Resistance = 2.7 GΩ)Clamp the holding potential at -70 mV; compensate fast and slow capacitances by clicking c-fast and c-slow buttons to get rid of transient currents. Hold the cell at gigaseal for 1-2 min to make the contact more stable.
Break the membrane by making a strong but short suction to establish a whole-cell configuration.
Note: At this point, the electrode and neuron become one electrical unit. This allows registration of the currents flowing through the neuronal channels and/or changes in the whole-cell membrane voltage.
Immediately after the breakage, switch to the Membrane Test window and read the cell parameters (Figure 16).
Note: It is good to know the cell’s capacitance (Cmin pF), access resistance (Rain MΩ), and holding current (Holdin pA).
Cmmay vary depending on the cell size and usually is the 10-100 pF range. You can use this parameter for further characterization of the neuron along with other parameters extracted from the firing pattern analysis.
Rashows the quality of the contact between the glass electrode and cell membrane; it should not exceed 20 MΩ. If it does, just try to suck a bit one more time and re-brake the membrane.
Holdshows the applied current that is required to hold the cell at a certain voltage (-70 mV in our case); this value should be between 0 and -50 pA.
Switch to the I=0 mode and read the neuron’s Resting Membrane Potential (RMP) in the MultiClamp software window V (mV).
Note: Please, make a note of the liquid junction potential (LJP), which appears between two liquids with different ionic compositions. For extracellular ACSF and IS solutions used here, LJP is +12 mV. Usually, we do not correct it during recordings but always mention it in the Method section.
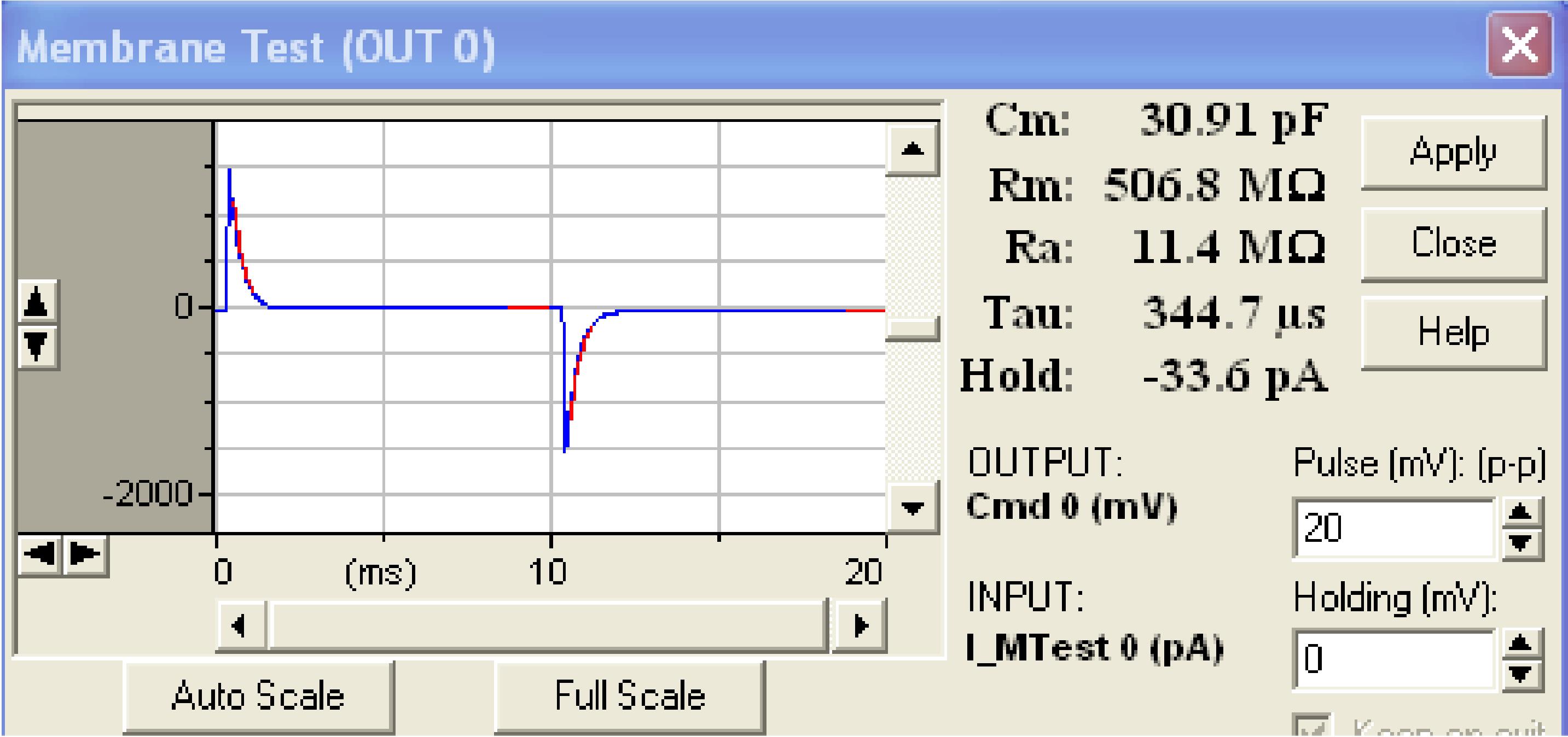
Figure 16. Membrane Test window showing a successful whole-cell configuration in voltage-clamp mode and several cell parametersSwitch to the Current Clamp (IC) mode and run the current-steps protocol provided below (Figure 17).
Notes:
We do not recommend injecting any current in the current-clamp mode unless you are sure of the normal RMP for a certain cell type.
The episodic stimulation protocol lasts 2 s. It includes an 800-ms current step ranging from -60 to 680 pA in 10-pA increments and a 1.2-s recovery pause to allow the cell to get back to its normal RMP. This pause might be longer, depending on cell type and stimulation intensity.
If you want to define AP threshold and rheobase current more precisely, you can additionally run a similar protocol from 0 to 30-50 pA in 1-pA increments.
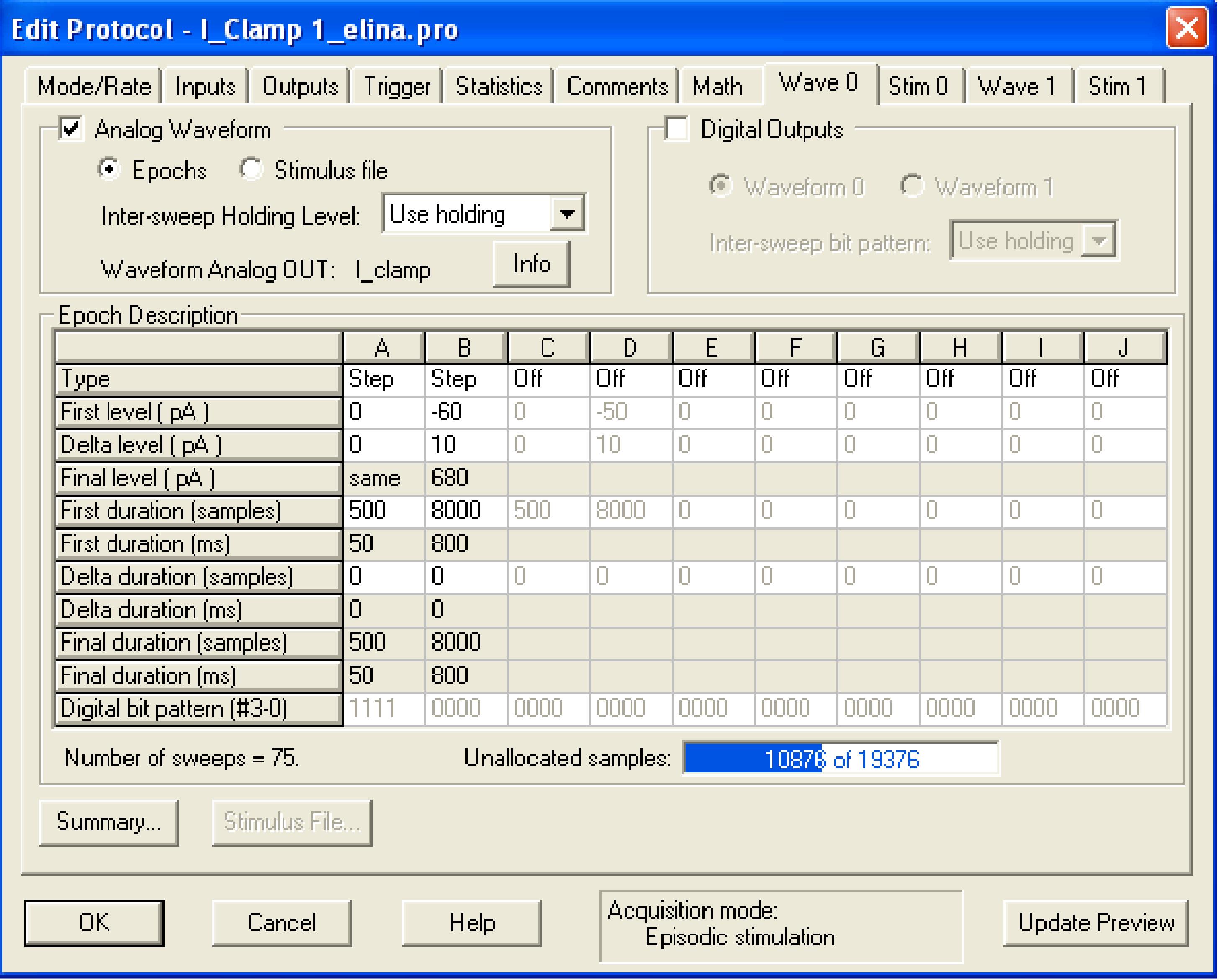
Figure 17. Recommended protocol for current-steps: 800-ms current steps from -60 pA to +680 pA in 10-pA increments
Data analysis
The features from raw *.abf traces are extracted in a semi-automatic way using FFFPA (fast-forward firing pattern analysis), an open-source plugin for MATLAB. FFFPA provides a flexible method to detect the most common parameters of APs (including peaks, thresholds, and AHPs) (Figure 18) and compute the most frequently analyzed features automatically, allowing to process of large batches of files quickly and accurately. Results of the automatic detection are visualized and can be inspected and adjusted manually if needed. Detailed definitions of the extracted features are summarized in Nagaeva et al., 2020, Appendix Table 1.
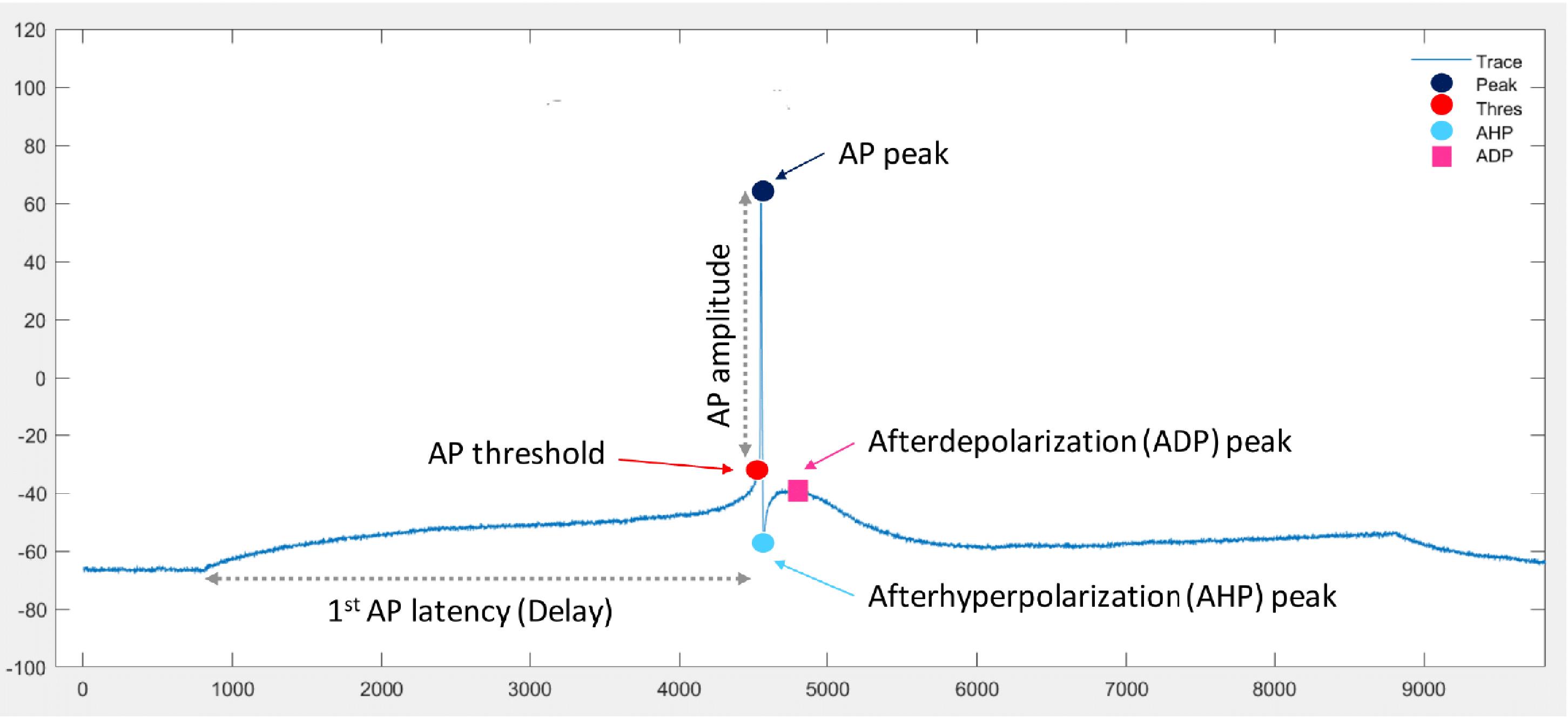
Figure 18. Basic parameters of action potential (AP), which are detected automatically by the FFFPA plugin. For definition of the parameters, see Nagaeva et al., 2020, Appendix Table 1.
Installation
Installation requires MATLAB 2018a or a newer version with Signal Processing and Curve Fitting toolboxes.
Download the FFFPA plugin from https://github.com/zubara/fffpa and unzip it to a local directory.
Double-click on the ‘fffpa.mlappinstall’ file from MATLAB file explorer.
Click “Install” in the opened dialog window.
Once installed, open the app from the MATLAB Apps toolstrip. The graphical user interface for data import will appear (Figure 19).
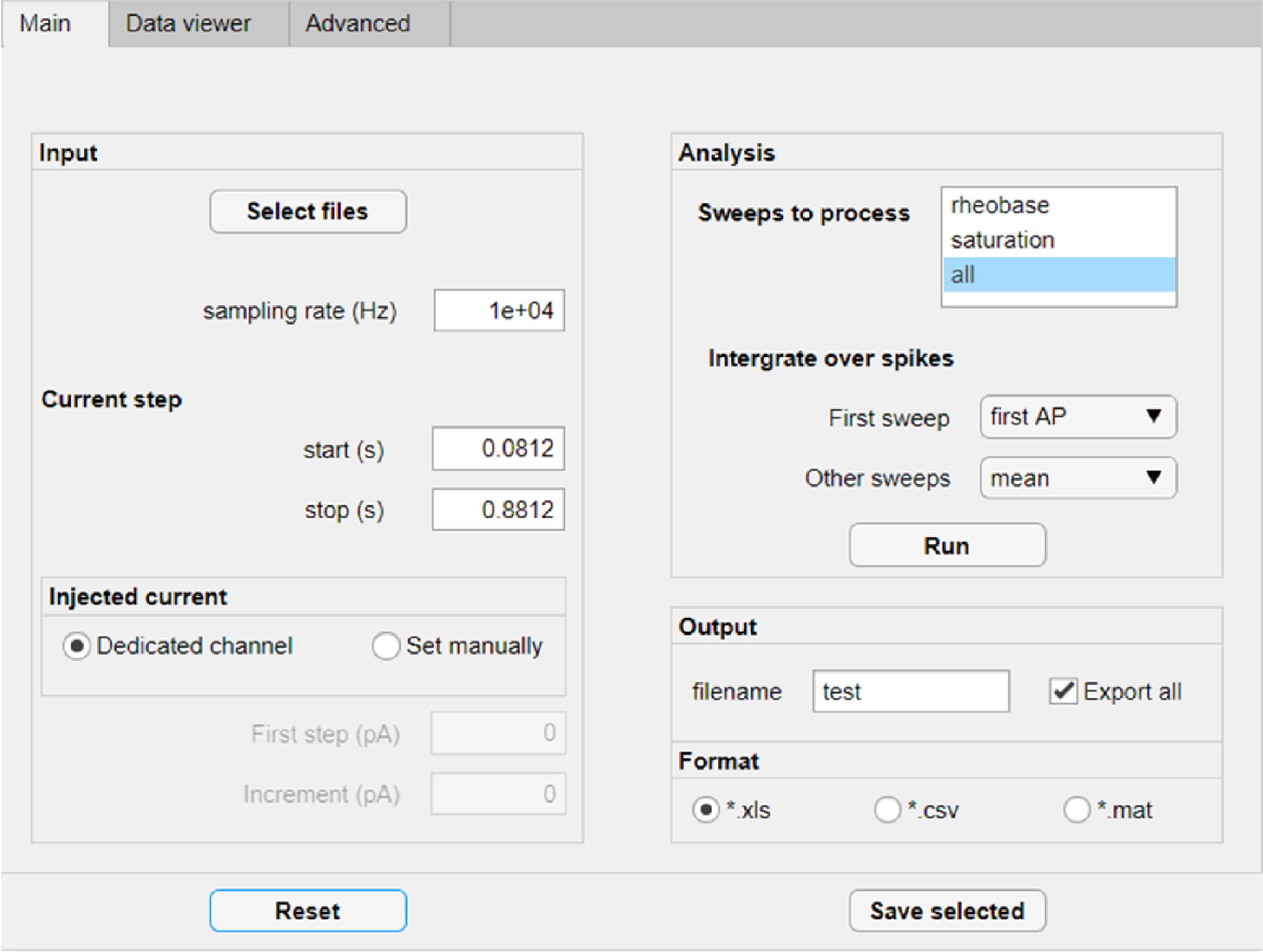
Figure 19. FFFPA graphical user interface for data import
Data import
Click “Select files” and choose one or several ‘.abf’ files to process.
Specify the sampling frequency in Hz and the time of the onset and the offset of the injected current step in seconds.
Choose the sweeps that you wish to analyze. FFFPA can detect, for example, the rheobase current step defined as the first sweep where APs occurred, or the saturation current step defined as the sweep with the maximum number of APs.
Specify the summary statistics for the analyzed APs. If multiple sweeps are analyzed, these options can differ between the first and other sweeps.
Specify the filename and the desired output format.
(Optional) Go to the ‘Advanced’ tab (Figure 20) to specify the features that you wish to extract from the traces. Detailed definitions of the extracted features are summarized in Nagaeva et al., 2020, Appendix Table 1.
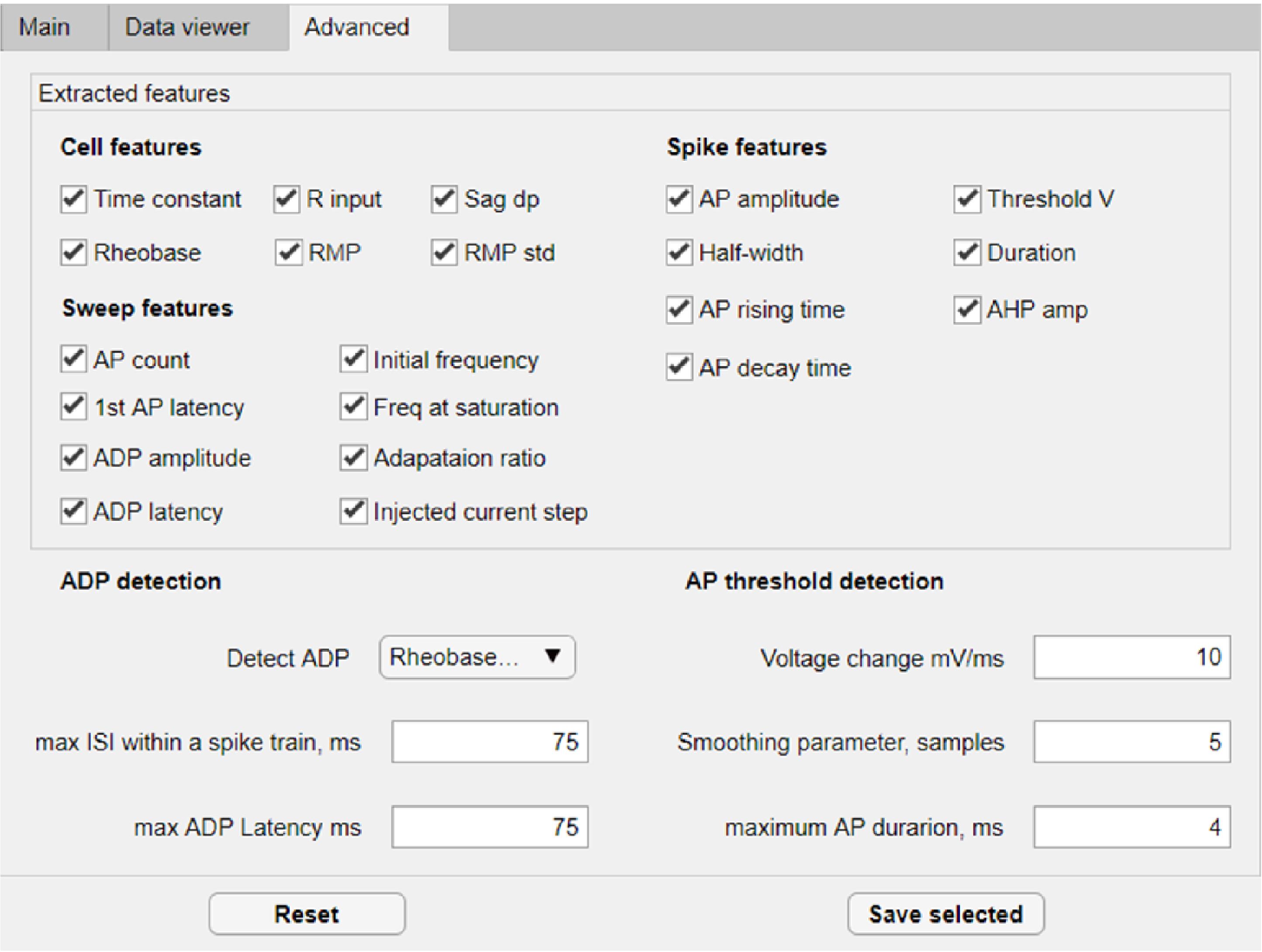
Figure 20. FFFPA feature selection and advanced options tabClick the ‘Run’ button on the ‘Main’ tab (Figure 19).
To inspect the results of automatic feature detection, go to the ‘Data viewer’ tab (Figure 21). This tab opens automatically after clicking ‘Run’ once the data is processed.
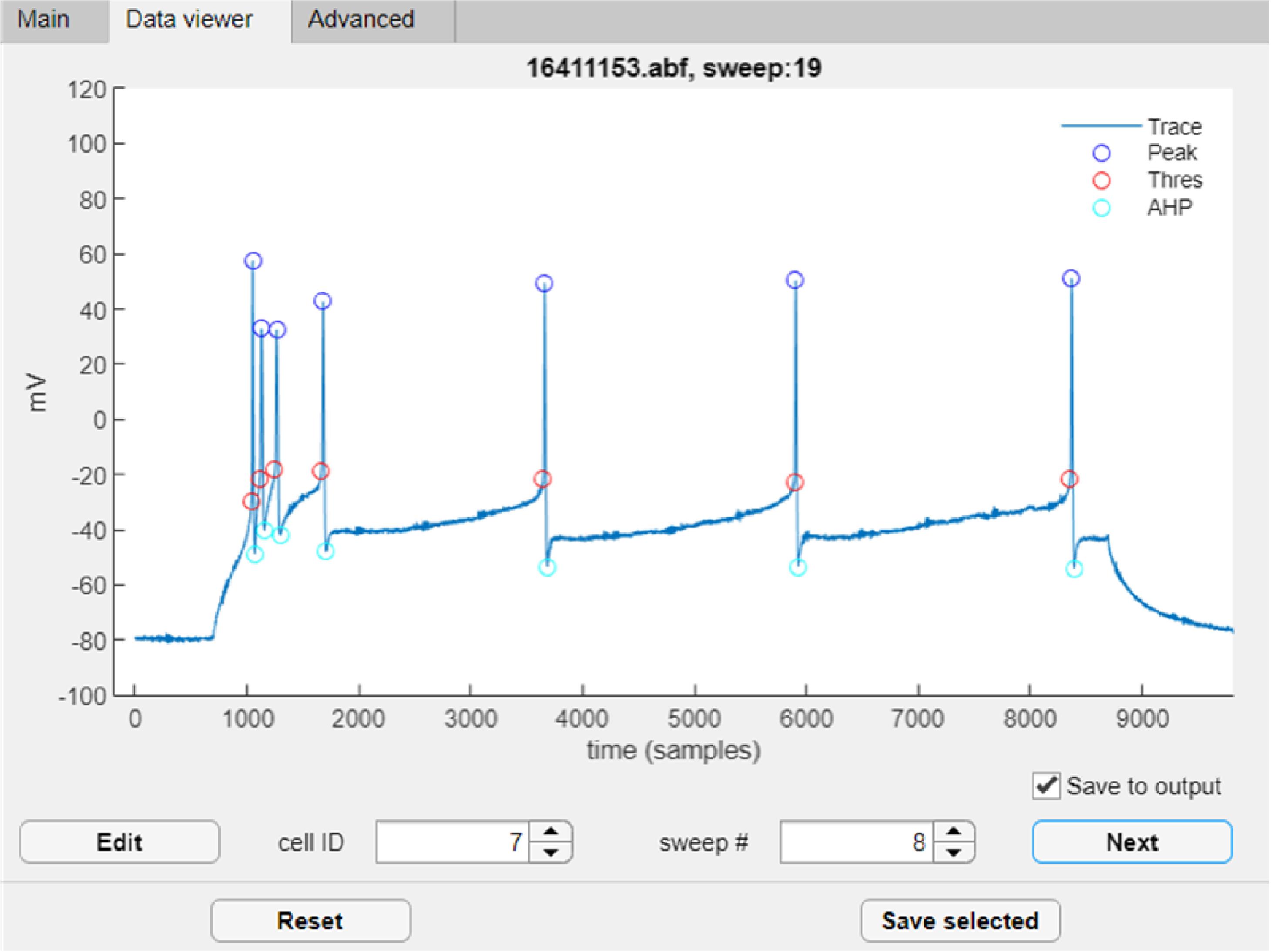
Figure 21. FFFPA Data viewer tabThe plot area displays the raw trace and the detected events AP peaks (blue circles), activation thresholds (red), afterhyperpolarization (AHP, cyan), and afterdepolarization (ADP, not shown). Please note that, by default, the ADP is only detected after the first AP or train of APs at rheobase.
Use the ‘Next’ button to go through all sweeps of all analyzed cells consecutively or use the ‘cell ID’ and ‘sweep #’ fields to navigate to the specific sweep of the specific cell manually.
If needed, the Data viewer allows you to correct the detection results. To do this, first click ‘Edit,’ then click the element that you want to adjust and specify its new position by clicking on the trace. To correct another point, click “Z” to activate the editing option again. After all corrections have been made, press ‘X’ to save the results.
You can also discard the current cell from the output by unchecking the ‘Save to output’ box.
Once satisfied with the results, click ‘Save selected.’ This step will compute features based on the (adjusted) detections and save the output file to disk. The summary file can be found in the same directory as the input data.
Notes
You can watch a short tutorial for the FFFPA plugin here: https://vimeo.com/497798349.
Recipes
Note: All recipes should be made in MilliQ water.
Artificial Cerebral Spinal Fluid (ACSF) solution (Table 1)
Table 1. Artificial Cerebral Spinal Fluid (ACSF) solution recipe
ASCF solution Concentration in mM g/L NaCl 126 7.36 KCl 1.6 0.12 MgCl2·6H2O 1.2 0.243 NaH2PO4·H2O 1.2 0.166 NaHCO3 18 1.5 D-Glucose 11 1.98 CaCl2 2.5 0.37 Carbogen needed for pH adjustment final pH 7.3-7.4
Final osmolarity:
300-310 mOsmSucrose-based cutting solution (Table 2)
Table 2. Sucrose-based cutting solution recipe
Cutting solution Concentration in mM g/L NaCl 60 3.506 KCl 2 0.148 MgCl2·6H2O 8 1.626 NaH2PO4·H2O 1.2 0.172 NaHCO3 30 2.52 D-Glucose 10 1.802 Sucrose 140 47.922 CaCl2 0.3 0.044 Carbogen needed for pH adjustment final pH 7.3-7.4 Final osmolarity:
300-310 mOsmPotassium gluconate-based intracellular solution (Table 3)
Table 3. Potassium gluconate-based intracellular solution recipe
Intracellular solution Concentration in mM g/20 ml K-gluconate 130 0.609 NaCl 6 0.007 HEPES 10 0.047 EGTA 0.5 0.004 Na2-ATP 4 0.044 Na-GTP 0.35 0.004 Na2-phosphocreatine 8 0.041 KOH for pH adjustment final pH 7.2-7.3 Final osmolarity:
280-290 mOsm
Acknowledgments
This work was supported by grants from the Academy of Finland (1317399), The Finnish National Agency for Education EDUFI, and the Sigrid Juselius Foundation.
The protocol described here was adapted from a previous publication: Nagaeva, E., Zubarev, I., Bengtsson Gonzales, C., Forss, M., Nikouei, K., de Miguel, E., Elsila, L., Linden, A. M., Hjerling-Leffler, J., Augustine, G. J. and Korpi, E. R. (2020). Heterogeneous somatostatin-expressing neuron population in mouse ventral tegmental area.Elife 9: e59328. doi: 10.7554 (Nagaeva et al., 2020).
Competing interests
The authors declare no competing interests.
Ethics
Animal procedures described in the current protocol were authorized by the National Animal Experiment Board in Finland (Eläinkoelautakunta, ELLA; Permit Number: ESAVI/1172/04.10.07/2018).
References
- Andrew, R. D. (1986). Intrinsic membrane properties of magnocellular neurosecretory neurons recorded in vitro. Fed Proc 45(9): 2306-2311.
- Cadwell, C. R., Palasantza, A., Jiang, X., Berens, P., Deng, Q., Yilmaz, M., Reimer, J., Shen, S., Bethge, M., Tolias, K. F., Sandberg, R. and Tolias, A. S. (2016). Electrophysiological, transcriptomic and morphologic profiling of single neurons using Patch-seq.Nat Biotechnol 34(2): 199-203.
- Franklin, K.B.J. and Paxinos, G. (2008). The Mouse Brain in Stereotaxic Coordinates, 3rd Ed. Elsevier, Amsterdam.
- Fuzik, J., Zeisel, A., Mate, Z., Calvigioni, D., Yanagawa, Y., Szabo, G., Linnarsson, S. and Harkany, T. (2016). Integration of electrophysiological recordings with single-cell RNA-seq data identifies neuronal subtypes. Nat Biotechnol 34(2): 175-183.
- Gouwens, N. W., Sorensen, S. A., Baftizadeh, F., Budzillo, A., Lee, B. R., Jarsky, T., Alfiler, L., Baker, K., Barkan, E., Berry, K., Bertagnolli, D., Bickley, K., Bomben, J., Braun, T., Brouner, K., et al. (2020). Integrated Morphoelectric and Transcriptomic Classification of Cortical GABAergic Cells. Cell 183(4): 935-953 e919.
- Halabisky, B., Shen, F., Huguenard, J. R. and Prince, D. A. (2006). Electrophysiological classification of somatostatin-positive interneurons in mouse sensorimotor cortex. J Neurophysiol 96(2): 834-845.
- Ma, Y., Hu, H., Berrebi, A. S., Mathers, P. H. and Agmon, A. (2006). Distinct subtypes of somatostatin-containing neocortical interneurons revealed in transgenic mice.J Neurosci 26(19): 5069-5082.
- Marx, M., Gunter, R. H., Hucko, W., Radnikow, G. and Feldmeyer, D. (2012). Improved biocytin labeling and neuronal 3D reconstruction. Nat Protoc 7(2): 394-407.
- Nagaeva, E., Zubarev, I., Bengtsson Gonzales, C., Forss, M., Nikouei, K., de Miguel, E., Elsila, L., Linden, A. M., Hjerling-Leffler, J., Augustine, G. J. and Korpi, E. R. (2020). Heterogeneous somatostatin-expressing neuron population in mouse ventral tegmental area.Elife 9: e59328.
- Neher, E. and Sakmann, B. (1976). Single-channel currents recorded from membrane of denervated frog muscle fibres. Nature 260(5554): 799-802.
- Sanchez-Aguilera, A., Monedero, G., Colino, A. and Vicente-Torres, M. A. (2020). Development of Action Potential Waveform in Hippocampal CA1 Pyramidal Neurons. Neuroscience 442: 151-167.
- Sucher, N. J. and Deitcher, D. L. (1995). PCR and patch-clamp analysis of single neurons. Neuron 14(6): 1095-1100.
- Ting, J. T., Lee, B. R., Chong, P., Soler-Llavina, G., Cobbs, C., Koch, C., Zeng, H. and Lein, E. (2018). Preparation of Acute Brain Slices Using an Optimized N-Methyl-D-glucamine Protective Recovery Method.J Vis Exp(132).
- Verkhratsky, A. and Parpura, V. (2014). History of electrophysiology and the patch clamp. Methods Mol Biol 1183: 1-19.
- Wierenga, C. J., Mullner, F. E., Rinke, I., Keck, T., Stein, V. and Bonhoeffer, T. (2010). Molecular and electrophysiological characterization of GFP-expressing CA1 interneurons in GAD65-GFP mice. PLoS One 5(12): e15915.
Article Information
Copyright
![]() Nagaeva et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Nagaeva et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Nagaeva, E., Zubarev, I. and Korpi, E. R. (2021). Electrophysiological Properties of Neurons: Current-Clamp Recordings in Mouse Brain Slices and Firing-Pattern Analysis. Bio-protocol 11(12): e4061. DOI: 10.21769/BioProtoc.4061.
- Nagaeva, E., Zubarev, I., Bengtsson Gonzales, C., Forss, M., Nikouei, K., de Miguel, E., Elsila, L., Linden, A. M., Hjerling-Leffler, J., Augustine, G. J. and Korpi, E. R. (2020). Heterogeneous somatostatin-expressing neuron population in mouse ventral tegmental area.Elife 9: e59328.
Category
Cell Biology > Cell-based analysis > Electrophysiological technique
Neuroscience > Basic technology > Acute slice preparation
Biophysics > Electrophysiology > Patch-clamp technique
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.