- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Differential Analysis of N-glycopeptide Abundance and N-glycosylation Site Occupancy for Studying Protein N-glycosylation Dysregulation in Human Disease
Published: Vol 11, Iss 12, Jun 20, 2021 DOI: 10.21769/BioProtoc.4059 Views: 4328
Reviewed by: Oneil Girish BhalalaPia GiovannelliAmberley D. Stephens

Original research article
The authors used this protocol in:

Oct 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Protein N-glycosylation plays a vital role in diverse cellular processes, and dysregulated N-glycosylation is implicated in a variety of human diseases including neurodegenerative disorders and cancer. With recent advances in high-resolution mass spectrometry-based glycoproteomics technologies enabling large-scale N-glycoproteome profiling of disease and control samples, analysis of the large datasets has become a challenge. Here, we provide a protocol for the systems-level analysis of in vivo N-glycosylation sites on N-glycosylated proteins and their changes in human disease, such as Alzheimer's disease. The protocol includes quantitation and differential analysis of N-glycopeptide abundance, in addition to integrative N-glycoproteome and proteome data analyses, to determine disease-associated changes in N-glycosylation site occupancy and identify differentially N-glycosylated proteins in human disease versus control samples. This protocol can be modified and applied to study proteome-wide N-glycosylation alterations in response to different cellular stresses or pathophysiological states in other organisms or model systems.
Keywords: Protein N-glycosylationBackground
Protein N-glycosylation – the attachment of glycans to asparagine residues – is a major posttranslational modification for regulating many key biological processes from membrane trafficking and protein degradation to immune response and cell-cell communication (Moremen et al., 2012). The importance of proper N-glycosylation in human health is underscored by the finding that mutations in the protein N-glycosylation machinery components cause congenital disorders of glycosylation with multi-system abnormalities (Freeze et al., 2015). Furthermore, increasing evidence links aberrant protein N-glycosylation to various human diseases including cancer, diabetes, and neurodegenerative diseases (Schedin-Weiss et al., 2014; Pinho and Reis, 2015; Reily et al., 2019).
A mechanistic understanding of the roles of protein N-glycosylation in biology and pathophysiology requires the system-wide elucidation of in vivo N-glycosylation sites (N-glycosites) on N-glycosylated proteins (N-glycoproteins) in health and disease. Protein N-glycosylation sites typically have a canonical sequon N-X-S|T, where X represents any amino acid except proline. However, whether a sequon-containing site can be N-glycosylated in vivo is dependent on protein folding and localization, site availability, and accessibility to N-glycosylation enzymes (Moremen et al., 2012; Cherepanova et al., 2016); thus, the in vivo N-glycosylation sites on glycoproteins have to be determined experimentally. Furthermore, since stress and pathophysiological conditions can alter N-glycosylation site occupancy and expose the cryptic N-glycosites (i.e., normally unutilized N-glycosylation sequons) for N-glycosylation (Pinho and Reis, 2015; Cherepanova et al., 2016), it is important to determine disease-associated, site-specific changes in protein N-glycosylation site occupancy to gain mechanistic insights into N-glycosylation dysregulation and its roles in disease pathogenesis.
High-resolution mass spectrometry-based glycoproteomics technologies provide powerful tools for unbiased, large-scale, site-specific N-glycoproteome profiling analyses of complex biological samples. For example, in our recently published study (Zhang et al., 2020), we used an N-glycoproteomics workflow consisting of SDS-mediated protein extraction and filter-aided sample preparation (FASP) (Wisniewski, 2017), zwitterionic chromatography-hydrophilic interaction chromatography (ZIC-HILIC)-based glycopeptide enrichment (Ma et al., 2015), 18O-labeling of in vivo N-glycosylation sites (Kuster and Mann, 1999), and liquid chromatography-tandem mass spectrometry (LC-MS/MS) to characterize protein N-glycosylation in human Alzheimer's disease (AD) and control brains. We identified 4730 18O-labeled N-glycosite-containing peptides (hereafter referred to as N-glycopeptides) and mapped 2294 in vivo N-glycosylation sites on 1132 brain N-glycoproteins (Zhang et al., 2020).
With advanced N-glycoproteomics technologies enabling simultaneous, quantitative measurement of abundance profiles for thousands of N-glycopeptides and in vivo N-glycosylation sites, analysis of such large datasets has become a challenge. In our recent study (Zhang et al., 2020), we performed differential analysis of N-glycopeptide abundance and identified 118 N-glycopeptides with >1.3-fold change in N-glycopeptide abundance in AD as compared with control cases. By integrated analysis of our N-glycoproteome and proteome profiling data from the same brain samples (Zhang et al., 2018), we identified 77 N-glycosites on 60 N-glycoproteins with >1.3-fold changes in N-glycosylation site occupancy in AD versus control brains (Zhang et al., 2020). Furthermore, we performed qualitative assessment of whether an N-glycosite was exclusively occupied in either AD or control brains and identified 89 N-glycosites on 76 glycoproteins with a gain of N-glycosylation in AD and 12 N-glycosites on 11 glycoproteins with a complete loss of N-glycosylation in AD. In total, we identified 137 differentially N-glycosylated proteins in AD versus control brains, including 92 hyperglycosylated proteins containing N-glycosites with increased N-glycosylation site occupancy and/or a gain of N-glycosylation in AD, 39 hypoglycosylated proteins containing N-glycosites with decreased N-glycosylation site occupancy or a loss of N-glycosylation in AD, and 6 aberrantly glycosylated proteins containing both hyperglycosylated and hypoglycosylated N-glycosites. These analyses have identified disease signatures of altered N-glycopeptides, N-glycoproteins, and N-glycosylation site occupancy in AD and suggest new targets for AD biomarker development (Zhang et al., 2020).
Here, we provide a protocol that describes how to perform mass spectrometry-based, systems-level analysis of in vivo N-glycosylation sites on N-glycosylated proteins and their changes in human disease (Figure 1). The protocol also includes quantitation and differential analysis of N-glycopeptide abundance, in addition to integrative N-glycoproteome and proteome data analyses, to determine disease-associated changes in N-glycosylation site occupancy and identify differentially N-glycosylated proteins in human disease versus control samples. For more details on the use and execution of this protocol, please refer to our research article (Zhang et al., 2020). Similar strategies as described in this protocol should be broadly applicable to study proteome-wide N-glycosylation alterations in response to different cellular stresses or pathophysiological conditions in other organisms or model systems.
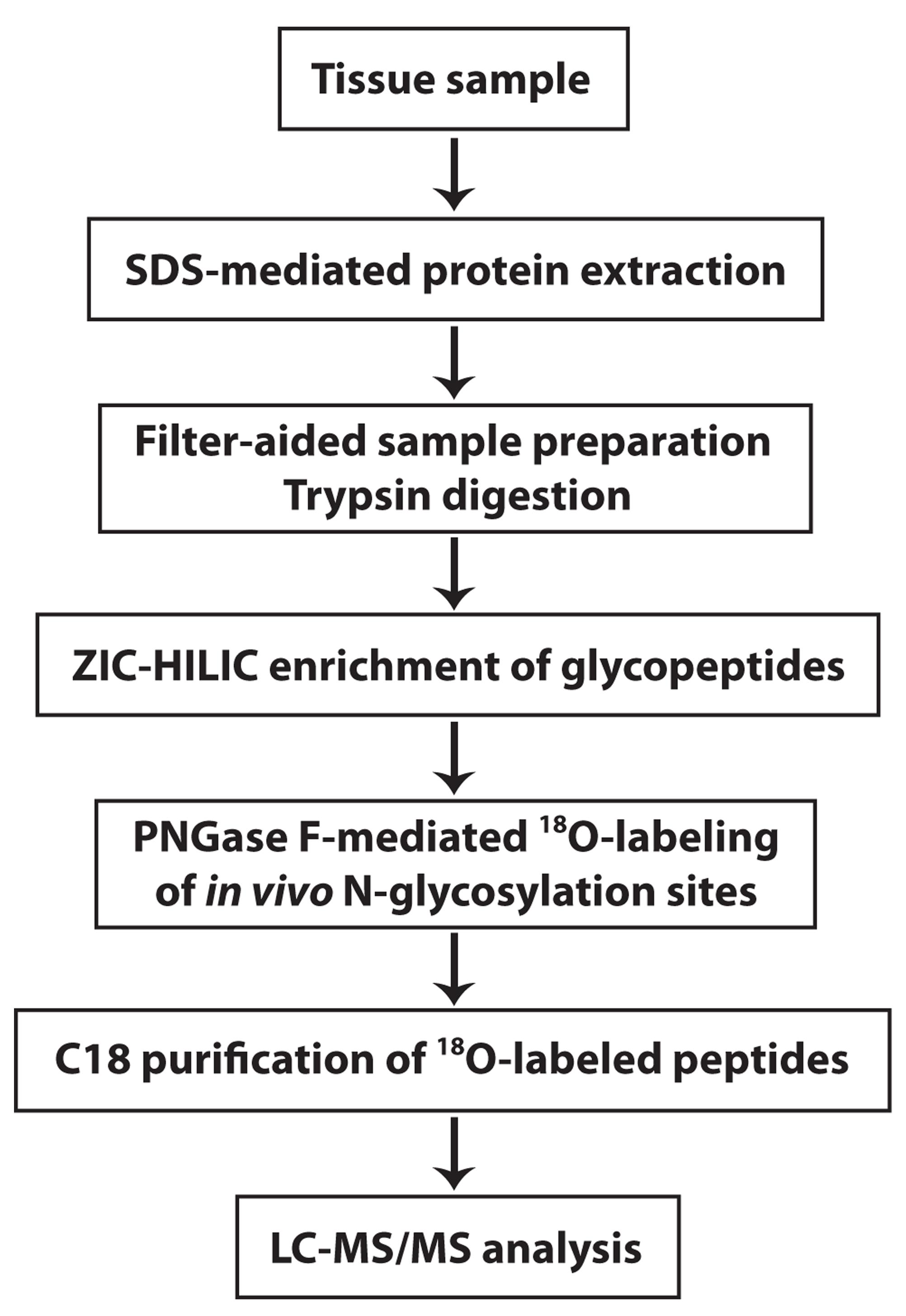
Figure 1. Overview of the experimental workflow. The experimental procedures for these steps are described in this protocol.
Materials and Reagents
Microcon 30-kDa centrifugal filter device (Merck, MRCF0R030)
Syringe, 3 ml (BD syringe)
VWR universal low-retention pipet tips, 200 μl (VWR, catalog number: 76322-150)
Bovine α2-HS-glycoprotein (fetuin) (Sigma, Millipore, catalog number: F3004-25MG)
ZIC-HILIC resin (particle size 10 μm, pore size 200 Å; Merck SeQuant, UmeÅ, Sweden)
H218O (Sigma-Aldrich, catalog number: 487090)
PNGase F (New England Biolabs, catalog number: P0704S)
Urea (Sigma, Millipore, catalog number: U1250)
Sodium dodecyl sulfate (Sigma, Millipore, catalog number: 71725)
DL-dithiothreitol (Sigma, Millipore, catalog number: D0632)
Iodoacetamide (Thermo Scientific, catalog number: A39271)
Ammonium bicarbonate (Sigma, Millipore, catalog number: 09830)
Formic acid (Sigma, Millipore, catalog number: 5330020050)
Sequencing grade modified trypsin (Promega, catalog number: V5111)
Equipment
Benchmark heat block (Benchmark Scientific)
Eppendorf 5424 microcentrifuge (Eppendorf)
Mortar and pestle, 150 ml capacity, porcelain (Grainger)
NanoDrop spectrophotometer and Quartz cuvettes (Thermo Fisher Scientific)
3MTM EmporeTM C8 extraction disk (Thermo Fisher Scientific)
PierceTM C18 tips, 100 μl bed (Thermo Fisher Scientific, catalog number: 87784)
Nano-LC UltiMate 3000 high-performance liquid chromatography system (Thermo Fisher Scientific)
LTQ-Orbitrap Elite mass spectrometer (Thermo Fisher Scientific)
EASY-Spray PepMap C18 column (length, 50 cm; particle size, 2 μm; pore size, 100 Å; Thermo Fisher Scientific)
Savant SpeedVac SC210A Plus centrifuge (Thermo Fisher Scientific)
Precision general purpose water bath (Thermo Fisher Scientific)
Software
Proteome Discoverer version 1.4 or higher (Thermo Fisher Scientific, www.thermofisher.com)
Microsoft Excel (https://www.microsoft.com)
GraphPad Prism 7 (GraphPad, https://www.graphpad.com/scientific-software/prism/)
MetaCore bioinformatics software (https://portal.genego.com)
Procedure
Protein extraction and filter-aided sample preparation
Homogenize frozen tissues by grinding with a mortar and pestle under liquid nitrogen. Lyse ground tissues (25 mg per human disease or control case) in 150 µl lysis buffer (4% SDS, 100 mM DTT, and 100 mM Tris-HCl, pH 7.6) at room temperature, followed by incubation at 95°C for 5 min as described (Wisniewski et al., 2009; Wisniewski, 2017). No protease inhibitors are added to the lysis buffer because the presence of SDS efficiently inactivates protease functions (Wisniewski et al., 2009).
After cooling the homogenates to room temperature, perform centrifugation at 16,000 × g for 5 min to obtain supernatants containing extracted proteins.
Determine the concentrations of total protein in protein extracts by UV spectrometry at 280 nm in a cuvette with a 10-mm pathlength using a NanoDrop spectrophotometer with an extinction coefficient of 1.1 for 0.1% (g/L) solution (Wisniewski et al., 2009).
Spike each protein extract by pipetting a small volume of bovine α2-HS-glycoprotein (fetuin) stock solution (0.4 µg/µl bovine fetuin in lysis buffer) to reach a final concentration of 0.1% fetuin (µg/µg total protein). The spiked-in bovine fetuin protein serves as an internal control for technical variations during sample processing and analysis.
Process the protein extracts as described (Zhang et al., 2018) according to the filter-aided sample preparation (FASP) protocol (Wisniewski et al., 2009; Wisniewski, 2017).
Mix 30 µl protein extract with 200 µl UA solution (8 M urea in 100 mM Tris-HCl, pH 8.5).
Transfer the mixture into a Microcon 30-kDa centrifugal filter unit (MRCF0R030, Merck) and centrifuge at 14,000 × g for 15 min. Discard the flow-through.
Add 200 µl UA solution to the filter unit and repeat the centrifugation. Discard the flow-through.
Add 100 μl UA solution containing 50 mM iodoacetamide to the filter unit and incubate in the dark for 30 min at room temperature.
Centrifuge the filter unit at 14,000 × g for 10 min. Discard the flow-through.
Add 100 μl UA solution to the filter unit and centrifuge again at 14,000 × g for 10 min. Repeat this step twice.
Add 100 μl 50 mM NH4HCO3 to the filter unit and centrifuge again. Repeat this step twice. Discard the flow-through.
Add 40 μl 50 mM NH4HCO3 solution containing sequencing grade trypsin (enzyme to protein ratio 1:100) in the filter unit and incubate at 37°C for 12 h.
Elute the trypsin-digested peptides by adding 100 μl 50 mM NH4HCO3 followed by centrifugation at 14,000 × g for 10 min. Collect the flow-through. Repeat this step five times and combine the eluates.
Add trifluoroacetic acid (TFA) to the peptide solution to a final concentration of 0.5% TFA.
Desalt the peptide samples using PierceTM C18 tips following the manufacturer’s protocol.
Determine the concentration of the purified peptides in each sample by UV spectrometry at 280 nm in a cuvette with a 10-mm pathlength using a NanoDrop spectrophotometer with an extinction coefficient of 1.1 for 0.1% (g/L) solution at 280 nm (Wisniewski et al., 2009).
Dry the peptide samples completely in a SpeedVac vacuum concentrator at room temperature for 3-6 h.
ZIC-HILIC-based glycopeptide enrichment
Prepare ZIC-HILIC microcolumns as described (Ma et al., 2015) by adding a slurry of ZIC-HILIC resin (10 mg) in 100 µl acetonitrile (ACN) to 200-µl tips plugged with a 3MTM EmporeTM C8 extraction disk at the bottom of each tip (Figure 2A).
Use a 3-ml syringe to generate backpressure inside the tip (Figure 2B). Slowly push down the plunger to allow the solvent to flow through the ZIC-HILIC column without causing resin compression.
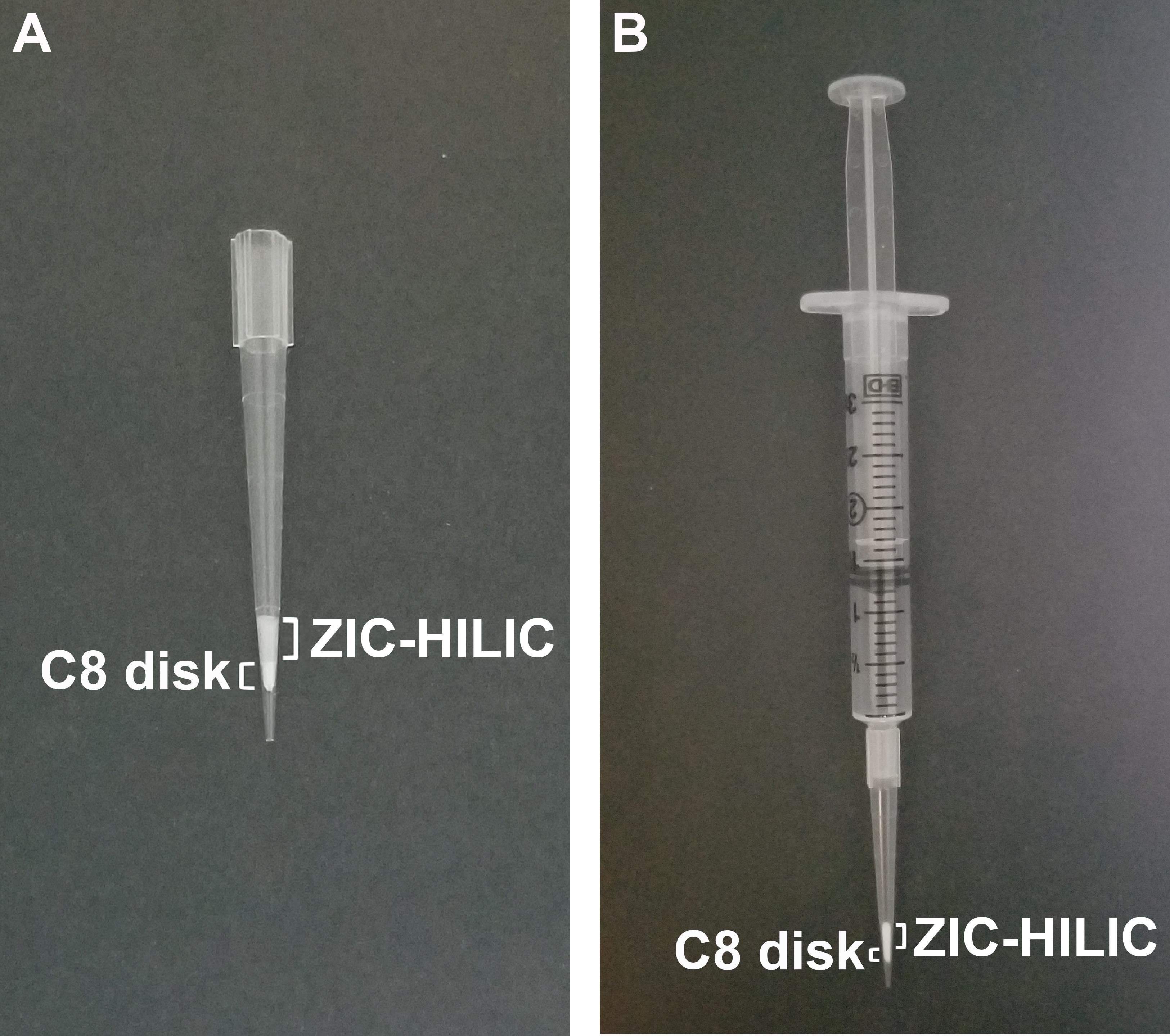
Figure 2. ZIC-HILIC microcolumn. A. ZIC-HILIC microcolumn in a 200-µl tip plugged with a 3MTM EmporeTM C8 extraction disk. B. Use of a 3-ml syringe to aid the flow through the ZIC-HILIC column.Equilibrate the ZIC-HILIC microcolumn by adding 100 µl binding buffer [80% ACN and 5% formic acid (FA)] and slowly pushing the solution down using a 3-ml syringe as described in Step B2.
Reconstitute dry peptides (100 µg peptides per sample) in 200 µl binding buffer.
Load the reconstituted peptide samples onto pre-equilibrated ZIC-HILIC microcolumns. Slowly push the solution down using a 3-ml syringe as described in Step B2. Discard the flow-through.
Wash each ZIC-HILIC microcolumn five times with 100 µl binding buffer, using the syringe to slowly push the solution down as described in Step B2. Discard the flow-through.
Add 80 µl elution buffer (99.5% H2O and 0.5% FA) to the ZIC-HILIC microcolumn, and slowly push down using the syringe to elute the bound glycopeptides into a 1.5-ml tube. Repeat this step three times and collect the eluates in the same tube.
Dry the glycopeptide samples completely in the SpeedVac concentrator at room temperature for 3-6 h.
18O-labeling of in vivo N-glycosylation sites
Reconstitute dry glycopeptides in 50 µl 50 mM NH4HCO3 prepared with H218O.
Add 0.5 µl (250 units) PNGase F to each sample tube and incubate in a water bath at 37°C overnight to allow 18O-labeling of the asparagine residues at in vivo N-glycosylation sites as described in Kuster and Mann (1999).
Desalt the 18O-labeled peptide samples using self-packed C18 ZipTips or PierceTM C18 tips following the manufacturer’s protocol.
Dry the 18O-labeled peptides samples completely in the SpeedVac concentrator at room temperature for 3-6 h.
LC-MS/MS analysis of 18O-labeled N-glycosite-containing peptides
Reconstitute dry 18O-labeled peptides (2 µg per sample) in 5 µl 0.1% fomic acid (FA).
Separate peptides of each sample as described in Zhang et al. (2018) by online reversed phase-HPLC fractionation on an EASY-Spray PepMap C18 column, using a 240-min gradient from 2% to 50% solvent B at a flow rate of 300 nl/min (mobile phase A, 1.95% ACN, 97.95% H2O, 0.1% FA; mobile phase B, 79.95% ACN, 19.95% H2O, 0.1% FA).
Perform mass spectrometric analysis as described in Zhang et al. (2018) using data-dependent acquisition with full MS scans (m/z range, 375-1600; automatic gain control target, 1,000,000 ions; resolution at 400 m/z, 60,000; maximum ion accumulation time, 50 ms) in the Orbitrap mass analyzer. The ten most intense ions in each full scan are fragmented by collision-induced dissociation with a maximum ion accumulation time of 100 ms in the LTQ mass spectrometer (automatic gain control target value, 10,000) to acquire MS/MS spectra.
Data analysis
Database search and quantitation of N-glycopeptide abundance
Analyze LC-MS/MS raw data files using Proteome Discoverer and search the data against the human UniProt TrEMBL database (2016_02 Release, 20,198 reviewed entries) plus the bovine α2-HS-glycoprotein (fetuin).
Perform the database search using the following parameters: 20-ppm precursor ion mass tolerance; 0.5-Da fragment ion mass tolerance; trypsin digestion with up to two missed cleavages; fixed modification: cysteine carbamidomethylation (+57.0215 Da); variable modifications: asparagine deamidation in H218O (18O tag of Asn, +2.9890 Da), asparagine and glutamine deamidation (+0.9840 Da), methionine oxidation (+15.9949 Da), and N-terminal acetylation (+42.0106 Da). Set the false discovery rate (FDR) to 1%.
Select and accept the peptides with an 18O-tagged asparagine residue within the N-glycosylation sequon N-X-S|T|C (X ≠ P) as the in vivo N-glycosite-containing peptides (referred to as N-glycopeptides).
Perform quantitative analysis of N-glycopeptide abundance using Proteome Discoverer to quantitate the peak area (i.e., area under the curve) of each 18O-labeled N-glycosite-containing peptide.
Determine the normalized N-glycopeptide abundance by normalizing the peak area of each 18O-labeled N-glycosite-containing peptide to the corresponding peak area of the 18O-labeled internal standard N-glycopeptide KLCPDCPLLAPLN(18O)DSR derived from the spiked-in bovine fetuin protein in each sample.
Differential analysis of N-glycopeptide abundance
Since a limitation of quantitative N-glycoproteomics is that glycopeptide identifications or abundance values can be missing from some samples (Karpievitch et al., 2012), only the N-glycopeptides with valid abundance values detected in ≥50% of disease or control samples are included in the differential analysis.
Perform differential analysis of N-glycopeptide abundance in Microsoft Excel using an unpaired Student’s t-test to compare the normalized N-glycopeptide abundance values for each 18O-labeled N-glycosite-containing peptide in disease samples with those values in control samples.
Identify N-glycopeptides with significantly altered, normalized N-glycopeptide abundance in the disease state using the thresholds of ±1.3-fold change over the control group (i.e., disease/control ratio >1.3 or <0.77) and P < 0.05.
Generate a volcano plot using GraphPad Prism to visualize the results of the differential analysis of N-glycopeptide abundance (e.g., Figure 2A in Zhang et al., 2020).
Integrative N-glycoproteomics and proteomics analysis of N-glycosylation site occupancy
Perform integrative analysis by comparing the N-glycoproteome data with the proteome data from the same samples measured using the same instrument for LC-MS/MS analysis.
Analyze the proteome data by performing differential expression analysis as described in Zhang et al. (2018) to identify proteins with significantly altered normalized protein abundance in the disease state using the thresholds of ±1.3-fold change over the control group and P < 0.05.
Determine the fold change in N-glycosylation site occupancy for each N-glycosite in disease versus control as the fold change in the normalized N-glycopeptide abundance of the N-glycosite-containing peptide divided by the fold change in the normalized protein abundance of the corresponding glycoprotein.
Identify N-glycosites with altered N-glycosylation site occupancy in the disease state using the thresholds of ±1.3-fold change in N-glycosylation site occupancy in disease versus control.
Perform qualitative assessment of whether an N-glycosite is exclusively occupied in either disease or control samples to identify N-glycosites with a complete loss or gain of N-glycosylation in the disease state.
Identification of differentially N-glycosylated proteins and dysregulated N-glycosylation-affected processes
Identify differentially N-glycosylated proteins in a disease as the N-glycoproteins containing in vivo N-glycosites with altered N-glycosylation site occupancy and/or a complete loss or gain of N-glycosylation in the disease state.
Define hyperglycosylated proteins in a disease as the N-glycoproteins containing N-glycosites with increased N-glycosylation site occupancy and/or a gain of N-glycosylation in the disease state.
Define hypoglycosylated proteins in a disease as the N-glycoproteins containing N-glycosites with decreased N-glycosylation site occupancy and/or a complete loss of N-glycosylation in the disease state.
Define aberrantly N-glycosylated proteins as the N-glycoproteins containing both hyper-glycosylated and hypoglycosylated N-glycosites.
Perform Gene Ontology (GO) enrichment analysis of the generated datasets of differentially N-glycosylated proteins using the MetaCore bioinformatics software as described in Zhang et al. (2020) to reveal dysregulated N-glycosylation-affected biological processes in the disease state.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) Grants RF1AG057965 (to L.L.) and R56 AG059714 (to L.S.C.). This protocol was adapted from our recently published research paper (DOI: 10.1126/sciadv.abc5802). The Emory Center for Neurodegenerative Disease Brain Bank was supported in part by NIH Grants P50 AG025688 and P30 NS055077.
Competing interests
The authors declare that they have no competing interests.
Ethics
Research related to this work was performed in accordance with the NIH guidelines for research involving human tissues and with the ethical standards and principles of the Declaration of Helsinki. Human postmortem brain tissues were obtained from the Emory Center for Neurodegenerative Disease Brain Bank, and brain tissues were acquired with Institutional Review Board (IRB) approval and informed consent from the subject or their family.
References
- Cherepanova, N., Shrimal, S. and Gilmore, R. (2016). N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr Opin Cell Biol 41: 57-65.
- Freeze, H.H., Eklund, E.A., Ng, B.G. and Patterson, M.C. (2015). Neurological aspects of human glycosylation disorders. Annu Rev Neurosci 38: 105-125.
- Karpievitch, Y.V., Dabney, A.R. and Smith, R.D. (2012). Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinformatics 13 Suppl 16: S5.
- Kuster, B. and Mann, M. (1999). 18O-labeling of N-glycosylation sites to improve the identification of gel-separated glycoproteins using peptide mass mapping and database searching. Anal Chem 71(7): 1431-1440.
- Ma, C., Qu, J., Meisner, J., Zhao, X., Li, X., Wu, Z., Zhu, H., Yu, Z., Li, L., Guo, Y., Song, J. and Wang, P.G. (2015). Convenient and Precise Strategy for Mapping N-Glycosylation Sites Using Microwave-Assisted Acid Hydrolysis and Characteristic Ions Recognition. Anal Chem 87(15): 7833-7839.
- Moremen, K.W., Tiemeyer, M. and Nairn, A.V. (2012). Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol 13(7): 448-462.
- Pinho, S.S. and Reis, C.A. (2015). Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 15(9): 540-555.
- Reily, C., Stewart, T.J., Renfrow, M.B. and Novak, J. (2019). Glycosylation in health and disease. Nat Rev Nephrol 15(6): 346-366.
- Schedin-Weiss, S., Winblad, B. and Tjernberg, L.O. (2014). The role of protein glycosylation in Alzheimer disease. FEBS J 281(1): 46-62.
- Wisniewski, J.R. (2017). Filter-Aided Sample Preparation: The Versatile and Efficient Method for Proteomic Analysis. Methods Enzymol 585: 15-27.
- Wisniewski, J.R., Zougman, A., Nagaraj, N. and Mann, M. (2009). Universal sample preparation method for proteome analysis. Nat Methods 6(5): 359-362.
- Zhang, Q., Ma, C., Chin, L.S. and Li, L. (2020). Integrative glycoproteomics reveals protein N-glycosylation aberrations and glycoproteomic network alterations in Alzheimer's disease. Sci Adv 6(40).
- Zhang, Q., Ma, C., Gearing, M., Wang, P.G., Chin, L.S. and Li, L. (2018). Integrated proteomics and network analysis identifies protein hubs and network alterations in Alzheimer's disease. Acta Neuropathol Commun 6(1): 19.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Zhang, Q., Ma, C., Li, L. and Chin, L. (2021). Differential Analysis of N-glycopeptide Abundance and N-glycosylation Site Occupancy for Studying Protein N-glycosylation Dysregulation in Human Disease . Bio-protocol 11(12): e4059. DOI: 10.21769/BioProtoc.4059.
- Zhang, Q., Ma, C., Chin, L.S. and Li, L. (2020). Integrative glycoproteomics reveals protein N-glycosylation aberrations and glycoproteomic network alterations in Alzheimer's disease. Sci Adv 6(40).
Category
Systems Biology > Proteomics
Neuroscience > Cellular mechanisms > Intracellular signalling
Molecular Biology > Protein > Detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.