- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Mechanical Fractionation of Cultured Neuronal Cells into Cell Body and Neurite Fractions
(*contributed equally to this work) Published: Vol 11, Iss 11, Jun 5, 2021 DOI: 10.21769/BioProtoc.4048 Views: 5754
Reviewed by: Edgar Soria-GomezMaria DermitFanny Ehret

Original research article
The authors used this protocol in:

Jun 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Many cells contain spatially defined subcellular regions that perform specialized tasks enabled by localized proteins. The subcellular distribution of these localized proteins is often facilitated by the subcellular localization of the RNA molecules that encode them. A key question in the study of this process of RNA localization is the characterization of the transcripts present at a given subcellular location. Historically, experiments aimed at answering this question have centered upon microscopy-based techniques that target one or a few transcripts at a time. However, more recently, the advent of high-throughput RNA sequencing has allowed the transcriptome-wide profiling of the RNA content of subcellular fractions. Here, we present a protocol for the isolation of cell body and neurite fractions from neuronal cells using mechanical fractionation and characterization of their RNA content.
Graphic abstract:
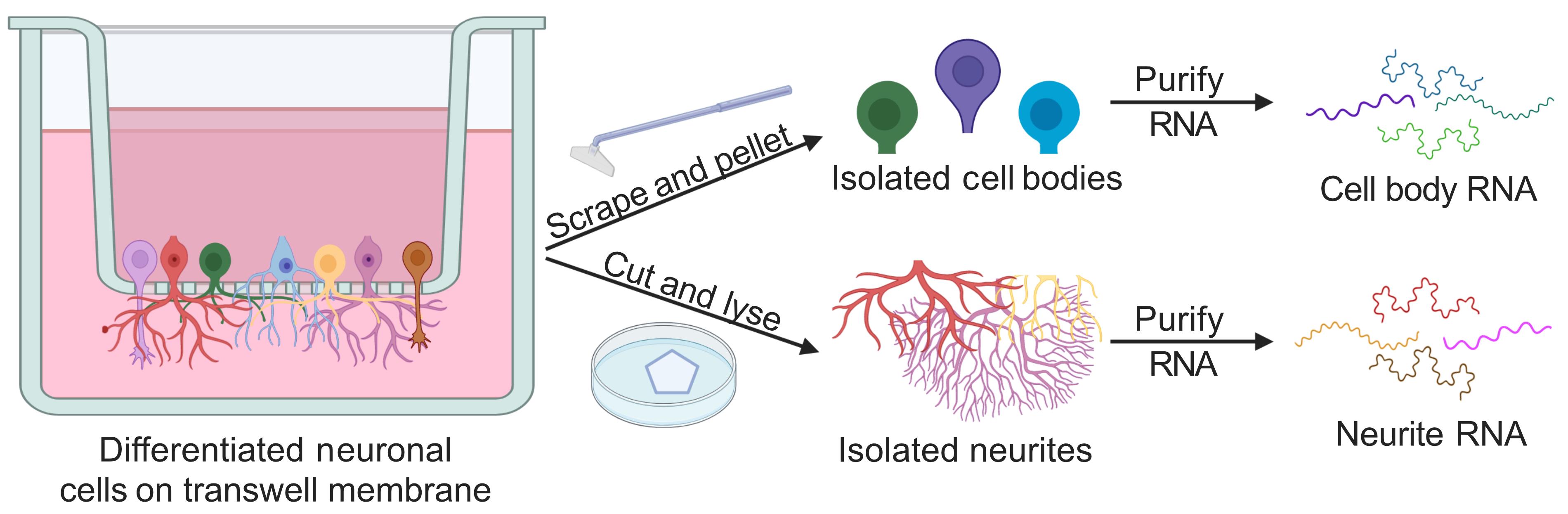
Fractionation of neuronal cells and analysis of subcellular RNA contents
Background
In eukaryotic cells, proteins are asymmetrically distributed to define spatially specialized regions. In cells that have complex morphologies and/or large sizes,like neurons, this asymmetry is often more extreme. These extended morphologies require a regulated and efficient sorting process to ensure that proteins are correctly localized. For many proteins, this process is facilitated through the transport of RNA molecules to the site of protein function (Engel et al., 2020). On-site translation of these RNAs produces a protein that is immediately correctly localized. In neurons, this is a widespread process as up to 1000 different RNA species are enriched in the projections of these cells relative to their cell bodies (Cajigas et al., 2012; Taliaferro et al., 2016).
RNA localization is widely used as a gene expression regulatory strategy and contributes to a diverse set of biological processes, including mating-type switching in yeast (Bertrand et al., 1998), developmental patterning in Drosophila (Ephrussi et al., 1991; Lécuyer et al., 2007), and nutrient response in intestinal epithelial cells (Moor et al., 2017). The misregulation of RNA localization is associated with a range of neurological diseases (Wang et al., 2016), including spinal muscular atrophy (Fallini et al., 2011), amyotrophic lateral sclerosis (Chu et al., 2019; Briese et al., 2020), and Fragile X Syndrome (Dictenberg et al., 2008; Goering et al., 2020).
Despite the increasing recognition of the role of RNA localization in promoting a range of cellular functions, several questions remain unanswered. Among them is perhaps one of the simplest questions that exists regarding RNA localization: what RNAs exist at a given subcellular location and what are their relative abundances? To answer this question, at least in the context of neuronal cells, a variety of techniques have been developed and applied, including laser capture microdissection (Zivraj et al., 2010), compartmentalized culture chambers (Gumy et al., 2011), growth on microfluidic devices (Nijssen et al., 2018), and microdissection of rodent brains (Cajigas et al., 2012).
In this protocol, we describe a similar approach to separate neuronal cells into neurite and cell body fractions. This technique relies on microporous culture membranes. Cells are cultured on top of these membranes, which have pores (usually 1-3 μm in diameter) large enough to allow neurites to pass through them to the underside but small enough to keep the cell bodies on the top of the membrane. After growth on the membranes, the cells can be mechanically fractionated by scraping the top of the membrane with a cell scraper and removing the dislodged cell bodies. Neurites remain attached to the underside of the membrane and can be lysed for RNA extraction. Following RNA isolation, the RNA content of the fractions can be analyzed by reverse transcription-quantitative PCR (RT-qPCR) or by high-throughput RNA sequencing.
Although the focus of this manuscript is the fractionation of neuronal cells, this method can also be applied to cellular protrusions from a variety of cell types, including fibroblasts (Mili et al., 2008) and migrating cancer cells (Mardakheh et al., 2015; Dermit et al., 2020). In the context of neuronal cells, this procedure has been successfully used with neuronal cell lines (Taliaferro et al., 2016; Goering et al., 2020), primary mouse cortical neurons (Taliaferro et al., 2016), and iPS-derived neurons (Goering et al., 2020; Hudish et al., 2020). It is likely compatible with most neuronal cell types.
Materials and Reagents
Deep well 6-well cell culture plates (Corning, catalog number: 353502, store at room temperature)
Microporous transwell membranes (transwell cell culture inserts), 1 μm pore diameter (Corning, catalog number: 353102, store at room temperature)
Matrigel (VWR, catalog number: 47743-706, store at -20°C)
RNase-free pipetting equipment (e.g., aerosol-resistant filter tips)
Cell lifter/scraper (Fisher, catalog number: 07-200-364, store at room temperature)
Quick RNA Microprep kit (Zymo, catalog number: R1051, store at room temperature)
Cell culture media (dependent on the needs of the specific cells being grown)
Mouse anti-beta actin antibody (Sigma, catalog number: A5441)
Mouse anti-histone H3 antibody (Abcam, catalog number: 10799)
iScript reverse transcription supermix (BioRad, catalog number: 1708841)
Taqman probes and master mix (ThermoFisher, catalog number: 4444556)
MOPS running buffer (Invitrogen, catalog number: NP0001)
Protein sample buffer (Invitrogen, catalog number: NP0008, store at room temperature)
Equipment
Cell culture tabletop centrifuge (for example, Eppendorf, model: 5702R)
Benchtop microcentrifuge (for example, Eppendorf, model: 5424R)
SDS page and western blotting materials
Thermocycler
qPCR-enabled thermocycler
Procedure
Prepare transwell membranes
Dilute Matrigel to 0.2% in cell culture media (see Note 1).
Place the transwell membranes upside down in a 15 cm cell culture plate. Add 1 ml of diluted matrigel on the top, coating the bottom (underside) of each transwell membrane.
Incubate at 37°C for 1 h.
Prepare cells
In the meantime, wash cells with 1× PBS and trypsinize them.
Pellet by centrifuging at 500 × g for 5 min.
Resuspend in cell culture media to a concentration of 500,000 cells per ml. Two milliliters of this cell suspension is needed for each transwell membrane (see Note 2).
Plate cells
Remove Matrigel solution from the transwell membranes.
In each well of a deep well 6-well plate, place 4 ml of cell culture media.
Put one transwell filter in each well.
Place 2 ml of cell solution (Step B3) onto each filter.
If a media change following plating is necessary (e.g., a change into differentiation-inducing media), allow cells to attach for 1 h, then replace the media above and below the membrane. Change pipettes in between dealing with the solutions above and below the membrane to avoid introducing cells into the lower chamber.
Allow cells to incubate at 37°C for 48 h.
Fractionate cells
Note: For a description of this procedure, see Video 1.
 Video 1. Mechanical fractionation of neuronal cells for subcellular RNA analysis. This video details the procedures for mechanical fractionation of neuronal cells using microporous membranes and highlights key points of the protocol.
Video 1. Mechanical fractionation of neuronal cells for subcellular RNA analysis. This video details the procedures for mechanical fractionation of neuronal cells using microporous membranes and highlights key points of the protocol.Gently remove media above and below the membrane by aspiration.
Replace the media with PBS, using 2 ml below the membrane and 1 ml above the membrane.
Remove the cell bodies on the top of each membrane.
Gently but thoroughly scrape the top of each membrane with a cell lifter, making sure to get the edges of the membrane where it joins the plastic housing (Figure 1A) (see Note 3).
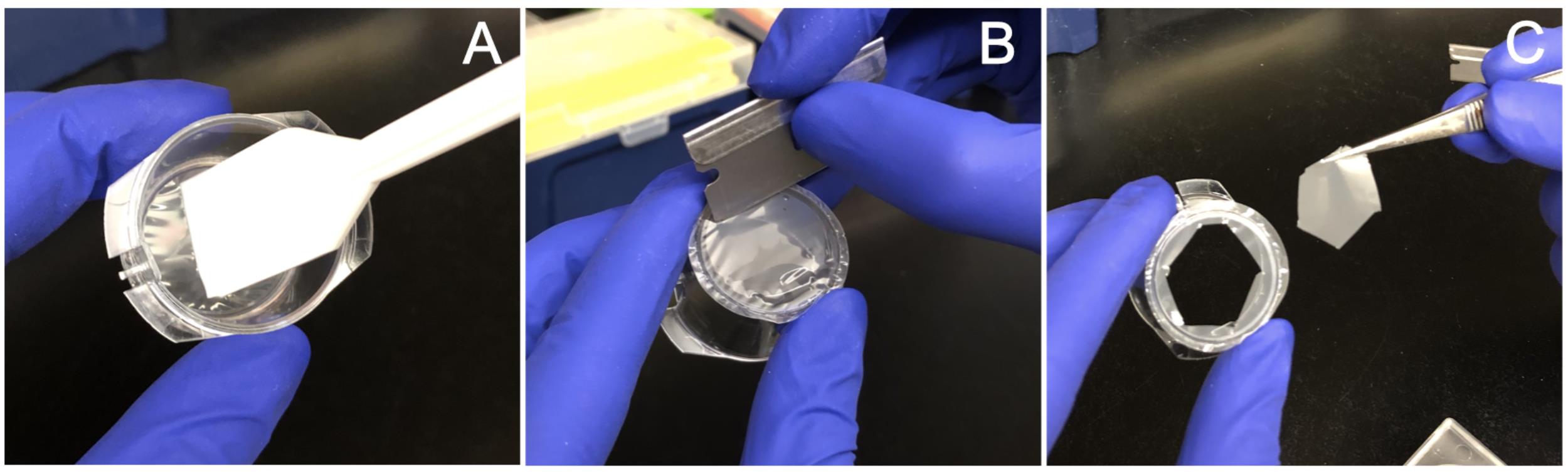
Figure 1. Mechanical fractionation of cells. A. The top of the membrane is scraped to remove cell bodies. B. The membrane is then removed from the plastic housing using a razor blade. C. The cut membrane is placed in a dish of lysis buffer using tweezers.Transfer 700 μl of the cell body suspension into a 15 ml conical tube on ice.
Tilting the membrane, scrape any remaining cell bodies into the remaining 300 μl of PBS.
Thoroughly transfer the PBS into the 15 ml conical tube.
Place the membrane upside down on a clean surface (for example, the lid of the 6-well plate).
Repeat Steps D3a through D3e for the remaining membranes.
After scraping and removing the cell bodies from all six membranes, place 550 μl of RNA lysis buffer from the Zymo RNA Microprep kit into a 6 cm dish.
Remove each membrane from its plastic housing.
Using a fresh razor blade, cut each membrane to remove it from the housing, leaving approximately 3 mm around the edge (Figure 1B) (see Note 4).
Using tweezers, carefully put the released membrane into the RNA lysis buffer with the neurite-containing side facing down (Figure 1C).
Repeat for all six membranes, placing all in the same 6 cm dish.
Incubate the dish at room temperature with rocking for 15 min to lyse neurites.
In the meantime, centrifuge cell bodies at 2,000 × g at 4°C for 7 min. Resuspend in 600 μl of PBS (i.e., 100 μl per membrane).
Reserve samples for western blotting.
Take 10 μl of the 600 μl cell body suspension. Add 90 μl of Protein Sample Buffer.
Take 50 μl of the 550 μl neurite lysate. Add to 50 μl of Protein Sample Buffer. Guanidine in the RNA lysis buffer will precipitate from solution upon addition to Protein Sample Buffer, but that is expected.
Store these samples at -20°C until analysis of fractionation efficiency by western blotting.
Isolate RNA
Take 100 μl of cell body suspension and isolate RNA according to the instructions of the Zymo Quick RNA Microprep kit, beginning with the addition of 350 μl of RNA lysis buffer.
Take the remaining 500 μl of neurite lysate and isolate RNA according to the kit’s instructions, beginning with the addition of 500 μl of 95-100% ethanol.
Elute RNA
Elute RNA in 15 μl RNase free water (see Note 5).
Analysis of fractionation efficiency using western blotting (see Note 6)
Heat the samples that were reserved for western blotting (Step D8) for 5 min at 98°C.
Load 5 μl of cell body sample per lane.
Load 15 μl of neurite sample per lane while it is still hot. This helps any precipitated guanidine to dissolve and facilitates loading.
Run a SDS-PAGE gel and transfer proteins to a nitrocellulose or PVDF membrane.
Blot with primary antibodies.
Use the β-actin antibody at 1:5,000 dilution.
Use the histone H3 antibody at 1:10,000 dilution.
Probe with an appropriate mouse secondary antibody.
Image blot. See Figure 2 for a blot depicting an efficient fractionation.
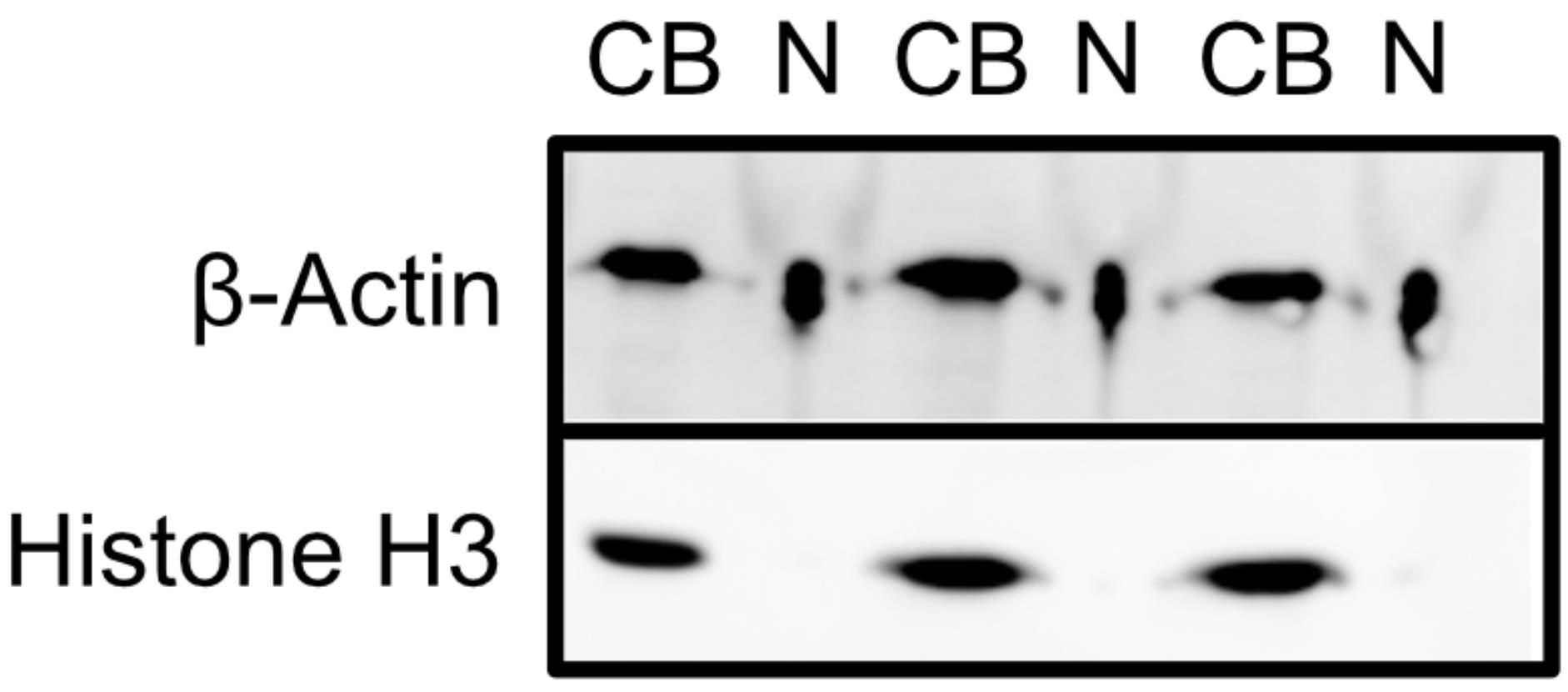
Figure 2. Assessment of fractionation efficiency by western blotting. Cell body samples are indicated by CB, and neurite samples are indicated by N.
Analysis of fractionation efficiency using RT-qPCR (see Note 7)
Reverse transcribe 100 ng of RNA from each sample (see Note 8)
Combine 2 μl of 5× iScript RT master mix, RNA, and water into a 10 μl reaction.
Incubate in a thermocycler for 5 min at 25°C, 20 min at 46°C, and 1 min at 95°C.
Dilute reaction with water to a 20 μl final volume (see Note 9).
Perform qPCR
Using a qPCR master mix of your choice, perform qPCR to quantify the desired marker RNA molecules in each fraction. Use 2 μl of cDNA from Step F1c per reaction. It is usually best to quantify the ratio of two RNA molecules in each sample, with one of the two known to be neurite enriched. The accuracy of the ratio in each sample can be improved using TaqMan qPCR probes to allow simultaneous quantification of the two species in the same reaction.
Assess qPCR results
From the qPCR results, calculate the relative abundance of the neurite-enriched and control RNAs in the cell body and neurite fractions. If the fractionation was successful, the ratio of the neurite-enriched RNA to control RNA will be higher in the neurite fraction than in the cell body fraction. See Figure 3 for qPCR results from a successful fractionation. The increased variation in neurite quantification relative to cell body quantification is typical and likely reflects the technical variation inherent to differences in fractionation efficiency between replicates.
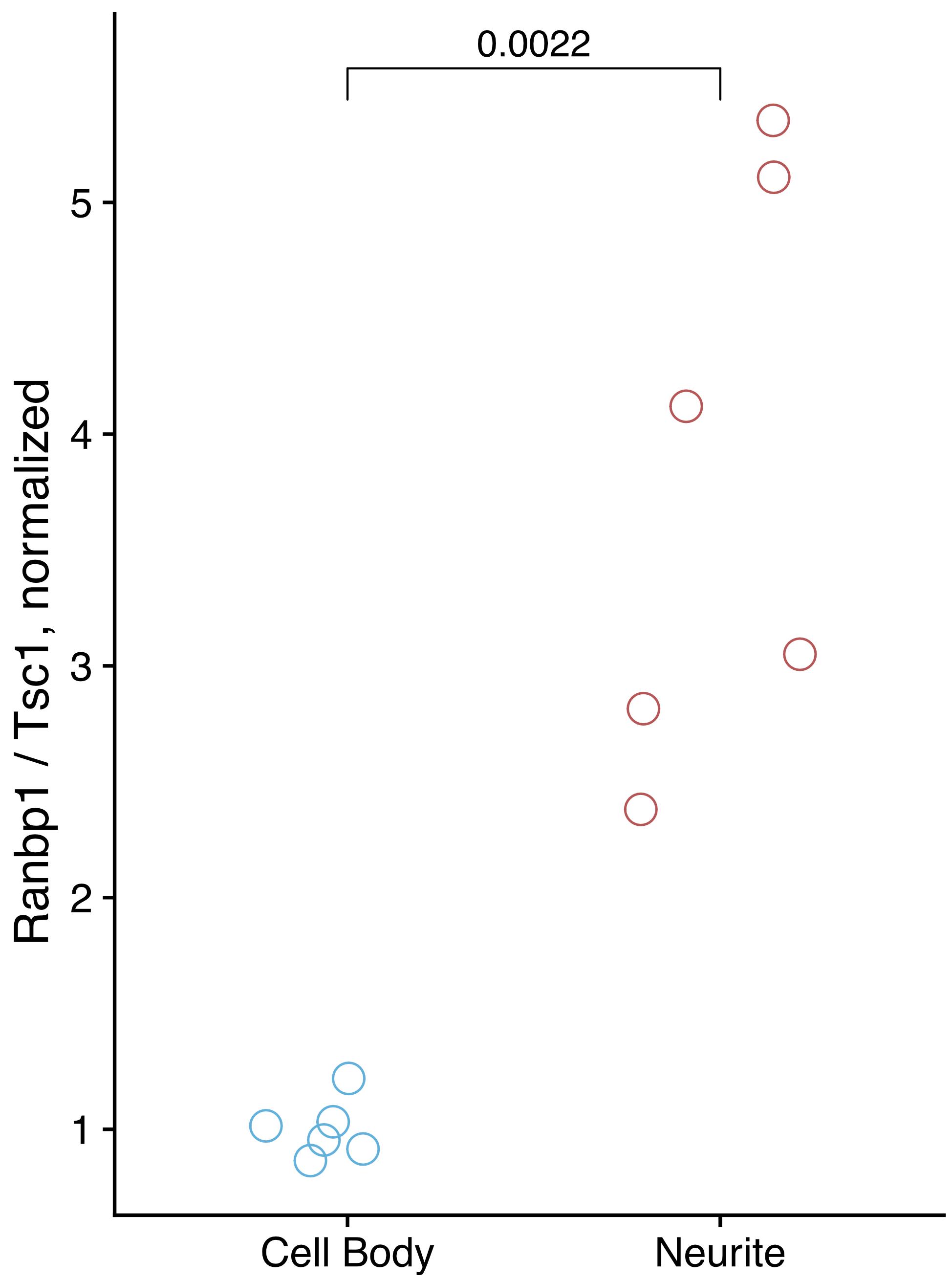
Figure 3. qPCR results from a successful fractionation. Ranbp1 RNA is known from previous experiments to be enriched in neurites, while Tsc1 is known from previous experiments to be enriched in cell bodies. The relative amounts of these RNA species were quantified in cell body and neurite samples using Taqman qPCR.
Construction of high-throughput sequencing libraries
Use the purified RNA to make high-throughput RNA sequencing libraries. Several commercial kits are available for this purpose, although we have had consistently good results using the mRNA Hyperprep kit from KAPA (Kapa KK8580). Importantly, provide the same amount of input RNA (e.g., 100 ng) for all samples in the kit, even though significantly more RNA can be isolated from cell bodies than from neurites.
Data analysis
Analysis of western blot results can be done by visual inspection. Generally, there should be very little to no signal from histone H3 in the neurite samples. Significant histone H3 signal in the neurite samples is indicative of a poor fractionation. The level of β-actin signal may or may not be similar between the cell body and neurite fractions. What is important to consider is the relative ratio of β-actin to histone H3 signal in the two fractions.
Identification of RNAs that are differentially localized between the two fractions can be done using standard differential gene expression techniques with high-throughput sequencing data.
Notes
We recommend thawing the Matrigel stock overnight in a 2°C to 8°C refrigerator. Additionally, dilute matrigel in cold cell culture media as it starts to form a gel at 10°C. As matrigel is very viscous, aspirate and dispense the stock slowly.
This concentration of cells ensures that they are essentially confluent when plated on the membrane. This can be desirable because it can result in neurite outgrowth being forced down through the pores of the membrane rather than laterally across the surface. If this is not desirable, adjust the cell concentration accordingly.
Using too much pressure when scraping can result in the membrane being torn away from the plastic housing. If this happens, discard the torn membrane and move to the next one.
It is often difficult to completely remove cell bodies from the corner formed by the membrane and the plastic housing. For this reason, it is often best to avoid this area when cutting the membrane out of the housing. Do not worry about removing all of the membrane when cutting.
When this procedure is performed with N2A or CAD mouse neuronal cell lines, expect 5-10 μg of RNA from the cell body sample and 500-1,000 ng of RNA from the neurite sample. This is the expected amount when all wells of a 6-well plate are combined.
The fractionation efficiency can be assessed by probing the cell body and neurite fractions for specific proteins. β-Actin should be present in both fractions, whereas histone H3, being nuclear, should be restricted to the cell body fraction. The detection of significant histone H3 signal in the neurite fraction indicates poor fractionation efficiency. The high amount of salt in the neurite fractions may cause them to appear compressed during imaging or while running the gel. This is a purely cosmetic defect, and the ability to detect protein bands within these samples is not hindered.
The efficiency of fractionation can also be assessed using RT-qPCR. This requires knowledge of RNA species enriched in each fraction. We have observed that mRNAs encoding ribosomal proteins are reproducibly neurite-enriched across several neuronal cell types and species. In this example, we use Ranbp1 and Tsc1 RNAs as markers, which we have previously observed to be neurite-enriched and cell body-enriched, respectively.
It should be noted that qPCR requires ~100 ng RNA from both cell body and neurite fractions. Cells from two wells are enough to serve as one replicate for qPCR. Thus, a full 6-well plate can be split into three replicates for qPCR. We highly recommend performing a minus RT control to ensure that there is no genomic DNA contamination in the samples.
The dilution factor for the RT reaction depends on the expression of the genes that need to be tested. For highly expressed genes, the RT reaction can be diluted to 50 μl final volume.
Acknowledgments
This work was funded by the National Institutes of Health (R35-GM133885) (JMT), the Boettcher Foundation (Webb-Waring Early Career Investigator Award AWD-182937), a Predoctoral Training Grant in Molecular Biology (NIH-T32-GM008730) (RG), and the RNA Bioscience Initiative at the University of Colorado Anschutz Medical Campus (RG and JMT). The protocol presented here was derived from previous work published by the authors (Taliaferro et al., 2016; Goering et al., 2020).
Competing interests
The authors declare no financial or non-financial competing interests.
Ethics
No animal or human subjects were used during this study.
References
- Bertrand, E., Chartrand, P., Schaefer, M., Shenoy, S. M., Singer, R. H. and Long, R. M. (1998). Localization of ASH1 mRNA particles in living yeast. Mol Cell 2(4): 437-445.
- Briese, M., Saal-Bauernschubert, L., Luningschror, P., Moradi, M., Dombert, B., Surrey, V., Appenzeller, S., Deng, C., Jablonka, S. and Sendtner, M. (2020). Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol Commun 8(1): 116.
- Cajigas, I. J., Tushev, G., Will, T. J., tom Dieck, S., Fuerst, N. and Schuman, E. M. (2012). The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 74(3): 453-466.
- Chu, J. F., Majumder, P., Chatterjee, B., Huang, S. L. and Shen, C. J. (2019). TDP-43 Regulates Coupled Dendritic mRNA Transport-Translation Processes in Co-operation with FMRP and Staufen1. Cell Rep 29(10): 3118-3133 e3116.
- Dermit, M., Dodel, M., Lee, F.C.Y., Azman, M.S., Schwenzer, H., Jones, J.L., Blagden, S.P., Ule, J., and Mardakheh, F.K. (2020). Subcellular mRNA Localization Regulates Ribosome Biogenesis in Migrating Cells. Dev Cell 55: 298-313.e10.
- Dictenberg, J. B., Swanger, S. A., Antar, L. N., Singer, R. H. and Bassell, G. J. (2008). A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev Cell 14(6): 926-939.
- Engel, K. L., Arora, A., Goering, R., Lo, H. G. and Taliaferro, J. M. (2020). Mechanisms and consequences of subcellular RNA localization across diverse cell types. Traffic 21(6): 404-418.
- Ephrussi, A., Dickinson, L. K. and Lehmann, R. (1991). Oskar organizes the germ plasm and directs localization of the posterior determinant nanos. Cell 66(1): 37-50.
- Fallini, C., Zhang, H., Su, Y., Silani, V., Singer, R. H., Rossoll, W. and Bassell, G. J. (2011). The survival of motor neuron (SMN) protein interacts with the mRNA-binding protein HuD and regulates localization of poly(A) mRNA in primary motor neuron axons. J Neurosci 31(10): 3914-3925.
- Goering, R., Hudish, L. I., Guzman, B. B., Raj, N., Bassell, G. J., Russ, H. A., Dominguez, D. and Taliaferro, J. M. (2020). FMRP promotes RNA localization to neuronal projections through interactions between its RGG domain and G-quadruplex RNA sequences. Elife 9: e52621.
- Gumy, L. F., Yeo, G. S., Tung, Y. C., Zivraj, K. H., Willis, D., Coppola, G., Lam, B. Y., Twiss, J. L., Holt, C. E. and Fawcett, J. W. (2011). Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA 17(1): 85-98.
- Hudish, L.I., Bubak, A., Triolo, T.M., Niemeyer, C.S., Sussel, L., Nagel, M., Taliaferro, J.M., and Russ, H.A. (2020). Modeling Hypoxia-Induced Neuropathies Using a Fast and Scalable Human Motor Neuron Differentiation System. Stem Cell Reports 14 (6): 1033-1043.
- Lécuyer, E., Yoshida, H., Parthasarathy, N., Alm, C., Babak, T., Cerovina, T., Hughes, T. R., Tomancak, P. and Krause, H. M. (2007). Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell 131(1): 174-187.
- Mardakheh, F. K., Paul, A., Kumper, S., Sadok, A., Paterson, H., McCarthy, A., Yuan, Y. and Marshall, C. J. (2015). Global Analysis of mRNA, Translation, and Protein Localization: Local Translation Is a Key Regulator of Cell Protrusions. Dev Cell 35(3): 344-357.
- Mili, S., Moissoglu, K. and Macara, I. G. (2008). Genome-wide screen reveals APC-associated RNAs enriched in cell protrusions. Nature 453(7191): 115-119.
- Moor, A. E., Golan, M., Massasa, E. E., Lemze, D., Weizman, T., Shenhav, R., Baydatch, S., Mizrahi, O., Winkler, R., Golani, O., Stern-Ginossar, N. and Itzkovitz, S. (2017). Global mRNA polarization regulates translation efficiency in the intestinal epithelium. Science 357(6357): 1299-1303.
- Nijssen, J., Aguila, J., Hoogstraaten, R., Kee, N. and Hedlund, E. (2018). Axon-Seq Decodes the Motor Axon Transcriptome and Its Modulation in Response to ALS. Stem Cell Reports 11(6): 1565-1578.
- Taliaferro, J. M., Vidaki, M., Oliveira, R., Olson, S., Zhan, L., Saxena, T., Wang, E. T., Graveley, B. R., Gertler, F. B., Swanson, M. S. and Burge, C. B. (2016). Distal Alternative Last Exons Localize mRNAs to Neural Projections. Mol Cell 61(6): 821-833.
- Wang, E. T., Taliaferro, J. M., Lee, J. A., Sudhakaran, I. P., Rossoll, W., Gross, C., Moss, K. R. and Bassell, G. J. (2016). Dysregulation of mRNA Localization and Translation in Genetic Disease. J Neurosci 36(45): 11418-11426.
- Zivraj, K. H., Tung, Y. C., Piper, M., Gumy, L., Fawcett, J. W., Yeo, G. S. and Holt, C. E. (2010). Subcellular profiling reveals distinct and developmentally regulated repertoire of growth cone mRNAs. J Neurosci 30(46): 15464-15478.
Article Information
Copyright
![]() Arora et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Arora et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Arora, A., Goering, R., Lo, H.-Y. G. and Taliaferro, J. M. (2021). Mechanical Fractionation of Cultured Neuronal Cells into Cell Body and Neurite Fractions. Bio-protocol 11(11): e4048. DOI: 10.21769/BioProtoc.4048.
- Goering, R., Hudish, L. I., Guzman, B. B., Raj, N., Bassell, G. J., Russ, H. A., Dominguez, D. and Taliaferro, J. M. (2020). FMRP promotes RNA localization to neuronal projections through interactions between its RGG domain and G-quadruplex RNA sequences. Elife 9: e52621.
Category
Molecular Biology > RNA > RNA extraction
Neuroscience > Basic technology
Cell Biology > Cell isolation and culture > Cell differentiation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.