- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Generation of Mouse Pluripotent Stem Cell-derived Trunk-like Structures: An in vitro Model of Post-implantation Embryogenesis
Published: Vol 11, Iss 11, Jun 5, 2021 DOI: 10.21769/BioProtoc.4042 Views: 6016
Reviewed by: Giusy TornilloDavid TurnerStefano Vianello
Original research article
The authors used this protocol in:

Dec 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Post-implantation mammalian embryogenesis involves profound molecular, cellular, and morphogenetic changes. The study of these highly dynamic processes is complicated by the limited accessibility of in utero development. In recent years, several complementary in vitro systems comprising self-organized assemblies of mouse embryonic stem cells, such as gastruloids, have been reported. We recently demonstrated that the morphogenetic potential of gastruloids can be further unlocked by the addition of a low percentage of Matrigel as an extracellular matrix surrogate. This resulted in the formation of highly organized trunk-like structures (TLSs) with a neural tube that is frequently flanked by bilateral somites. Notably, development at the molecular and morphogenetic levels is highly reminiscent of the natural embryo. To facilitate access to this powerful model, here we provide a detailed step-by-step protocol that should allow any lab with access to standard cell culture techniques to implement the culture system. This will provide the user with a means to investigate early mid-gestational mouse embryogenesis at an unprecedented spatiotemporal resolution.
Keywords: Trunk-like structuresBackground
Gastrulation and early organogenesis represent developmental events that are crucial for the successful generation of a functional body plan. In mammals, these processes start just after the embryo implants in utero and within few days, a variety of morphologically and functionally diverse tissues emerge. It is currently difficult to study these highly dynamic changes in vivo, and ex vivo culture of post-implantation mouse embryos is laborious, costly, and requires rigorous training, which often renders it impractical for most laboratories. These impediments have led to extensive efforts to model post-implantation and early mid-gestational development in vitro using embryonic stem cells (reviewed in Shahbazi and Zernicka-Goetz 2018; Shahbazi et al., 2019; Baillie-Benson et al., 2020; Veenvliet and Herrmann, 2021). In particular, post-implantation development can be modeled with gastruloids, mouse or human embryonic stem cell (mESC/hESC) aggregates that self-organize (van den Brink et al., 2014 and 2020; Moris et al., 2020). The original mouse gastruloid culture protocol resulted in elongated structures with embryo-like expression domains similar to the post-occipital mouse embryo and with correct positioning of the three body axes, but limited morphogenesis (van den Brink et al., 2014; Baillie-Johnson et al., 2015; Beccari et al., 2018a and 2018b; Turner et al., 2017). More recent efforts have managed to introduce embryo-like morphological features by changing the cellular environment, such as the formation of somite-like structures or a heart tube (van den Brink et al., 2020; Rossi et al., 2021). Further advances have demonstrated that the addition of an extracellular matrix (ECM) surrogate to gastruloids can trigger a more embryo-like architecture with a gut tube as well as somites flanking a neural tube (Veenvliet et al., 2020). We dubbed these embryonic organoids trunk-like structures (TLSs), since they resemble the core part of the trunk of an early mid-gestational embryo (~embryonic stage (E) 8.5-9). Importantly, during the timeframe of TLS induction (96-120 h post-aggregation), the gene regulatory programs are highly similar to the developing embryo. Moreover, the segmentation clock, an oscillator driving the rhythmic deposition of somites in vivo, is active at an embryo-like pace in the TLS (Pourquié, 2003; Veenvliet et al., 2020).
The TLS model is easy to access, track, manipulate, and scale, which makes it a powerful tool to study post-implantation and early mid-gestational mammalian development in a dish. Here, we provide a comprehensive step-by-step procedure to facilitate the generation of trunk-like structures. We also describe how to process TLSs for downstream analysis, including whole-mount immunofluorescent staining and (single cell) RNA sequencing.
Materials and Reagents
Pipet tips, variable volumes (Biozym, SafeSeal SurPhob VT)
1.5 ml tubes (Sarstedt, catalog number: 72.706)
15 ml Falcon tubes (Sarstedt, catalog number: 62.554.502)
50 ml Falcon tubes (Sarstedt, catalog number: 62.547.254)
6 cm cell culture plates (Sarstedt, catalog number: 83.3901.300)
Ultra-low attachment 96-well plates (Corning, Costar, catalog number: CLS7007)
6-well cell culture plates (Corning, catalog number: 3516)
10 cm cell culture plates (Corning, catalog number: 430167)
Luna cell counting slides (Logos Biosystems, catalog number: L12001)
µ-Slide 8-well glass bottom (Ibidi, catalog number: 80827)
Flowmi cell strainers 40 µm (Merck, catalog number: BAH136800040)
Bottle top vacuum filter unit (Corning, catalog number: CLS431096)
KnockOut DMEM (Gibco, catalog number: 10829018)
100× Penicillin (5000 U/ml)-Streptomycin (5,000 µg/ml) (Lonza, catalog number: DE17-603E)
100× Glutamine, 200 mM (Lonza, catalog number: BE17-605E)
100× Nucleosides (Sigma, catalog number: ES-008D)
Gibco 2-Mercaptoethanol, 55 mM solution in DPBS (Gibco, catalog number: 21985023)
Fetal Calf Serum (FCS), both regular (Pan Biotech, catalog number: P30-3306) and qualified and embryonic stem cell culture tested (Pan Biotech, catalog number: P30-2602)
TrypLE (Gibco, catalog number: 12604013) OR 0.05% Trypsin-EDTA (1x) (Gibco, catalog number: 25300-054)
NDiff 227 medium (Takara, catalog number: Y40002)
CHIR99021 InSolution (Sigma, catalog number: 361571) OR 10 mM in dimethyl sulfoxide (DMSO) (Tocris Biosciences, catalog number: 4423)
LDN193189 (Reprocell, catalog number: 04-0074-10)
DMSO (Sigma, catalog number: D2650)
Matrigel Growth Factor Reduced (GFR), Phenol Red-free (Corning, catalog number: 356231) – multiple lots/batches have been tested yielding similar results in terms of trunk-like-structure generation efficiency
Gelatin 2% solution (Sigma, catalog number: G1393)
DPBS, w/o MgCl2/CaCl2 (Gibco, catalog number: 14190144)
PBS with MgCl2/CaCl2 (Sigma, catalog number: D8662)
Murine Leukemia Inhibitory Factor (LIF) ESGROTM (107U/ml) (Millipore, catalog number: ESG1107)
Trypan Blue (Bio-Rad, catalog number: 1450021)
UltraPure Dnase/Rnase-Free Distilled Water (Invitrogen, catalog number: 10977049)
Reagent Reservoirs 60 ml (Merck, catalog number: BR703411)
Bovine Serum Albumin powder (BSA) (Sigma, catalog number: A2153)
Dulbecco’s Modified Eagle's Medium (DMEM) 4,500 mg/ml glucose, without sodium pyruvate (Lonza, catalog number: BE12-733F)
Cell culture grade water (Lonza, catalog number: BE17-724Q)
0.1% Gelatin solution (see Recipes)
Mouse embryonic fibroblast (MEF) medium (see Recipes)
Mouse embryonic stem cell (mESC) medium (see Recipes)
PBS/0.5% BSA solution (see Recipes)
Equipment
Biological safety cabinet (Thermo Fisher Scientific, model: Herasafe KS12)
Clean horizontal laminar flow hood (Thermo Fisher Scientific, model: HeraGuard ECO)
Cell culture incubator (Thermo Fisher Scientific, model: Heracell Vios 160i)
Cell culture centrifuge (Eppendorf, model: Centrifuge 5804R)
Variable volume pipets and multichannel pipets (Eppendorf, model: Research® plus pipette)
Horizontal light source, Light ring (Nikon, P-DF LED Darkfield Unit) or other stereomicroscope stand
Automated cell counter (Logos biosystems, Luna automated cell counter, L10001)
Cell culture water bath (LAUDA Aqualine, catalog number: AL18)
Tissue culture vacuum pump (Vacuubrand, catalog number: 20727200)
Microcentrifuge (Eppendorf, model: 5424R)
Equipment set up:
Cell culture incubators are set to 37°C, 5% CO2.
NOTE: We have also successfully generated TLSs at 7.5% CO2, but routinely use 5%.
Cell culture water bath (set to 37°C).
All centrifugation steps are performed at room temperature, unless otherwise indicated.
Procedure
Seeding mouse embryonic fibroblasts (MEFs)
NOTES:- Seed MEFs at least one day prior to seeding the mESCs.
- Pre-warm MEF medium in the water bath for at least 20 min before starting.
- MEF plates should be used within one week of seeding.
Coat a 6 cm cell culture plate with 3 ml 0.1% gelatin solution.
NOTE: Gelatin-coated culture plates have to be prepared fresh on the day of seeding MEFs and cannot be stored.
Leave the plate at room temperature for 15 min.
Next, thaw a vial of mitotically inactive MEFs at 37°C in the water bath.
NOTES:
Inactive MEFs are mitotically inactivated in-house using mitomycin C treatment (3 h at 37°C).
You need 1.0 × 106 MEFs to coat a 6 cm cell culture plate. Thaw the appropriate number depending on the number of 6 cm cell culture plates needed.
Add the MEFs to a 15 ml Falcon tube containing 5 ml pre-warmed MEF medium.
Centrifuge the cells at 200 × g for 5 min.
While centrifuging, aspirate gelatin from each 6 cm plate and add 2 ml MEF medium.
Aspirate the supernatant from the 15 ml tube containing MEFs and resuspend the cell pellet at a concentration of 1.0 × 106 cells per ml.
NOTE: Viable MEFs are counted at the time of freezing and there is no need to count them again after thawing.
Add 1 ml cell suspension to each prepared 6 cm plate.
Place the plate in the incubator and swirl the plate to ensure even distribution of cells.
Seeding mouse embryonic stem cells (mESCs)
NOTES:Pre-warm mESC medium in the water bath for at least 20 min before starting.
We routinely use mESCs with an F1G4 genetic background for TLS protocol generation (George et al., 2007).
Thaw a vial of mESCs at 37°C in the water bath immediately before plating.
NOTE: You need 3.5 × 105 mESCs for a 6 cm MEF-coated plate. Thaw the appropriate number depending on the number of 6 cm cell culture plates needed.
Add the mESCs to a 15 ml Falcon tube containing 5 ml pre-warmed mESC medium.
Centrifuge the cells at 200 × g for 5 min.
While centrifuging, aspirate MEF medium from each 6 cm plate containing MEFs and add 2 ml mESC medium.
Aspirate the supernatant from the 15 ml tube containing mESCs and resuspend the cell pellet at a concentration of 3.5 × 105 cells per ml.
NOTE: Viable mESCs are counted at the time of freezing and there is no need to count them again after thawing.
Add 1 ml cell suspension to each MEF-coated 6 cm plate.
Place the plate in the incubator and swirl the plate to ensure even distribution of cells.
Replace the medium daily with 3 ml fresh mESC medium.
Passaging mESCs
NOTES:Passage mESCs every 48 h at a splitting ratio of 1:8-1:10. Colony density and morphology should look similar to that shown in Figure 1A. Do not let your culture overgrow (Figure 1B).
The splitting time and ratios detailed here are optimized for the mESC lines used in Veenvliet et al. (2020). Based on the proliferation rate of the mESC line used, splitting times and ratios may need to be adjusted. This may be especially true if transgenic lines and/or mESCs with different genetic backgrounds are used.
Pre-warm mESC medium and TrypLE in the water bath for at least 20 min before starting.
Prepare MEF-coated plates one day prior to passaging mESCs.
Instead of TrypLE, 0.05% Trypsin-EDTA can be used.
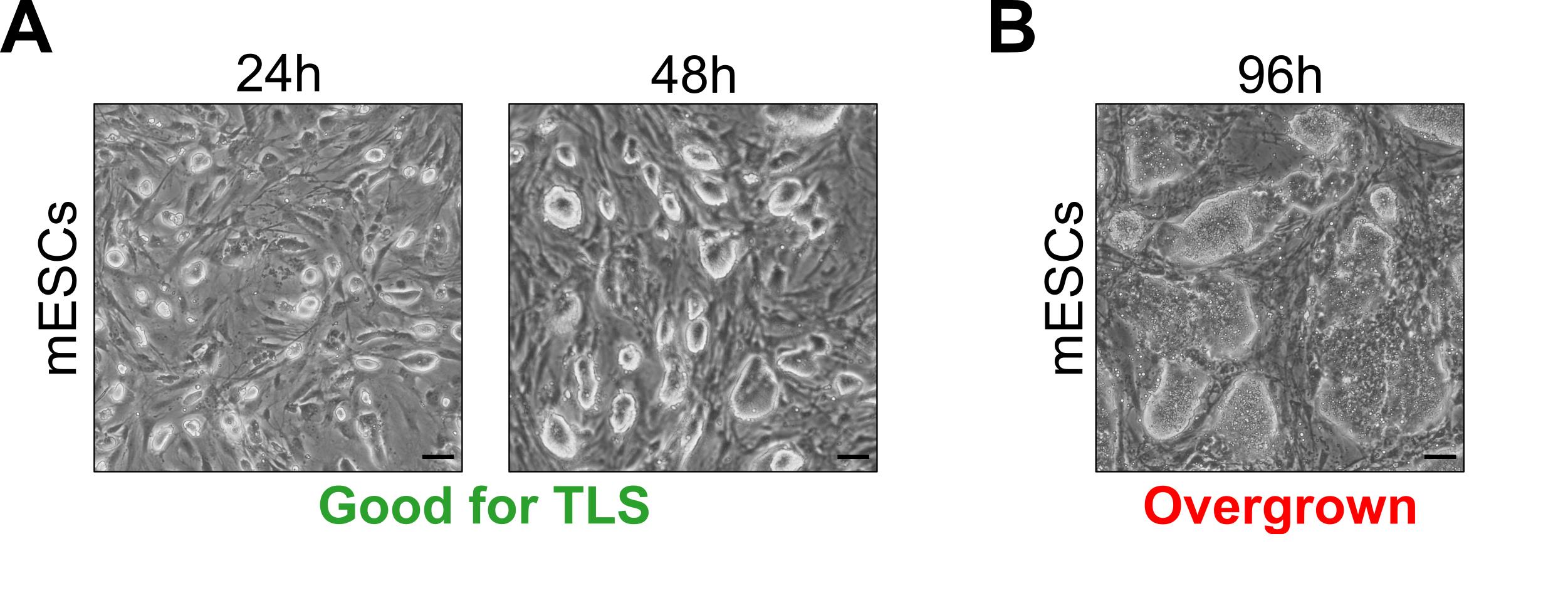
Figure 1. Optimal embryonic stem cell culture densities for successful TLS generation. A. mESC culture densities suitable for TLS generation (24 h and 48 h after seeding). B. mESC culture density unsuitable for TLS generation (96 h after seeding). Scale bars for all panels, 50 μm.Aspirate the medium from the mESC plate and wash with 3 ml DPBS.
Aspirate the DPBS and add 1 ml TrypLE.
Ensure that the plate surface is evenly covered with TrypLE and place it in the incubator at 37°C for 5 min.
After 5 min, dislodge the colonies with a P1000 pipet set to 800 μl by pipetting up and down in the plate 20 times.
Inactivate the TrypLE by adding 1 ml mESC medium and pipet further to obtain a single cell suspension.
Transfer the cell suspension to a 15 ml Falcon tube and wash the plate with an additional 3 ml mESC medium to recover all cells. Transfer these cells to the same 15 ml tube.
Centrifuge the cells at 200 × g for 5 min.
While centrifuging, aspirate the MEF medium from the previously prepared 6 cm plate containing MEFs and add 2 ml mESC medium.
Aspirate the supernatant from the 15 ml tube containing mESCs and resuspend the cell pellet in 2 ml mESCs medium.
Add the appropriate amount of cell suspension to each MEF-coated 6 cm plate (ratio 1:8-1:10 → 200-250 μl). Adjust the final volume to 3ml.
Place the plate in the incubator and swirl the plate to ensure even distribution of cells.
Replace the medium daily with 3 ml fresh pre-warmed mESC medium.
Generation of trunk-like structures (TLSs)
NOTES:The input cell number for each well detailed here is optimized for the mESC lines used in Veenvliet et al. (2020). Based on the proliferation rate of the mESC line (especially for transgenic lines and/or mESCs with a different genetic background), the cell amount may need to be adjusted to reach the same efficiency reported in Veenvliet et al. (2020).
We recommend first optimizing the standard gastruloid protocol for new cell lines, using gastruloid elongation efficiency as a fast experimental readout (Cermola et al., 2021). In our experience, good gastruloid elongation efficiency (>95%) is essential to achieve a similar TLS efficiency to that reported in Veenvliet et al. (2020). A routine optimization procedure involves the seeding of 100-600 mESCs per well, with a stepwise increase of 50 cells.
mESCs must be in culture for at least one passage before starting.
Pre-warm mESC medium and TrypLE in the water bath for at least 20 min before starting.
Here, we use commercially available, quality controlled NDiff 227 medium (N2B27). We and others have successfully generated gastruloids with home-made N2B27 (Beccari et al., 2018b); however, in our hands, more robust results of the gastruloid and TLS protocols are obtained with the NDiff 227 medium.
Instead of TrypLE, 0.05% Trypsin-EDTA can be used.
A schematic overview of the TLS generation protocol indicating critical timepoints is provided in Figure 2.
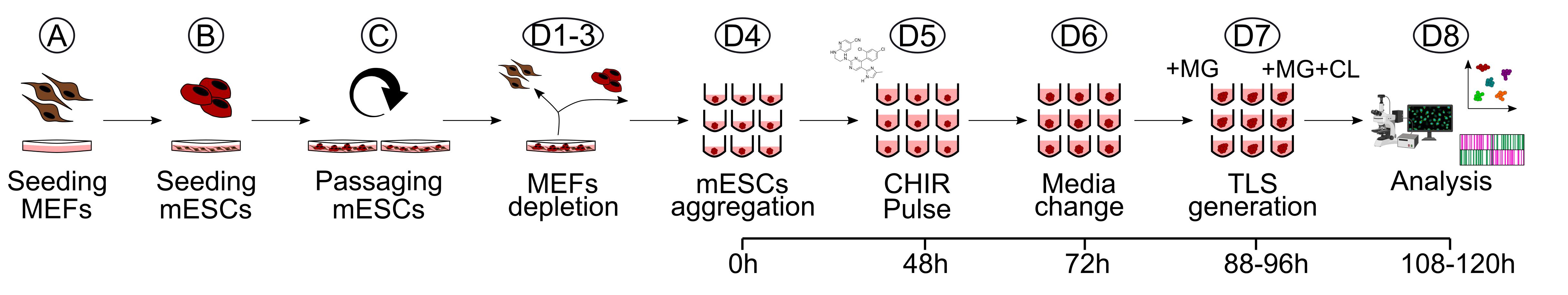
Figure 2. Schematic overview of the TLS generation protocol. Workflow for the generation of trunk-like-structures (TLS) from seeding of MEFs to downstream analysis. MG, Matrigel; CL, CHIR+LDN; MEFs, mouse embryonic fibroblasts; mESCs, mouse embryonic stem cells.
Coat three wells of a 6-well plate with 2 ml 0.1% gelatin solution for each 6 cm plate that will be used for TLS generation.
Incubate the 6-well plate at room temperature for 15 min.
Aspirate gelatin solution and add 1 ml mESC medium to each well.
Store plate in the incubator until use.
Aspirate the medium from the mESC plate and wash with 3 ml DPBS.
Aspirate the DPBS and add 1 ml TrypLE.
Ensure that the plate surface is evenly covered with TrypLE and place it in the incubator at 37°C for 5 min.
After 5 min, dislodge the colonies with a P1000 pipet set to 800 μl by pipetting up and down in the plate 20 times.
Inactivate the TrypLE by adding 1 ml mESC medium and pipet-mix.
Transfer the cell suspension to a 15 ml Falcon tube and wash the plate with an additional 3 ml mESC medium to recover all cells. Transfer these cells to the same 15 ml tube.
Centrifuge the cells at 200 × g for 5 min.
Resuspend the cell pellet in 1 ml mESC medium and pipet up and down 50 times.
NOTE: Here, it is critical to achieve a proper single cell suspension to avoid losing mESCs (or retaining MEFs) during MEF depletion and to ensure the best protocol performance. We recommend checking for a proper single cell suspension under a microscope.
D3. MEF depletion
NOTE: An example how the wells with cells attached to the bottom should look like after each step of MEF depletion is provided in Figure 3.
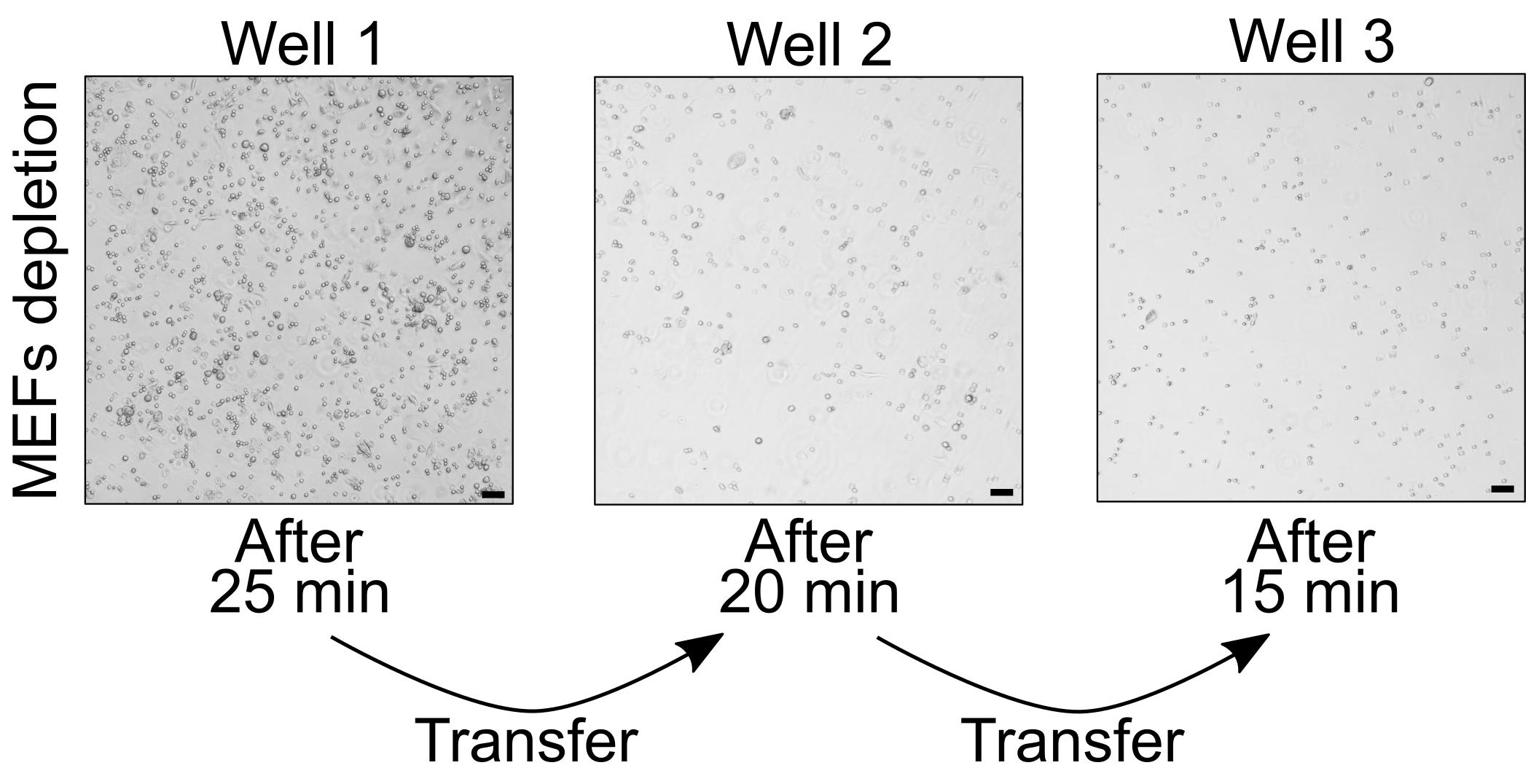
Figure 3. MEF depletion prior to mESC aggregation. MEFs attach to the 0.1% gelatin-coated wells. Scale bars, 50 μm.
NOTE: With consecutive transfers, the amount of attached cells decreases. After the third incubation, MEF depletion is completed and mESCs are ready to be used for aggregation.Transfer the obtained cell suspension to a well of the prepared 6-well plate (see D1).
NOTE: Transfer the amount of mESCs present in one 6 cm plate into one well of the prepared 6-well plate. The presence of too many cells could result in decreased depletion efficiency.
Pipet-mix 10 times.
Place the plate in the incubator and swirl the plate to ensure even distribution of cells. Leave untouched for 25 min.
Next use a P1000 pipet set to 1 ml to carefully transfer all the cells in suspension to another well.
NOTE: It is critical not to dislodge the MEFs, which are attached to the bottom of the wells.
Pipet up and down 10 times in the new well to ensure a single cell suspension.
NOTE: Cells may clump during the incubation; therefore, it is critical to pipet once the cells are transferred to the new well. We recommend confirming under the microscope that you have obtained a proper single cell suspension.
Place the plate in the incubator and swirl the plate to ensure even distribution of cells. Leave untouched for 20 min.
Next, use a P1000 pipet set to 1 ml to carefully transfer all the cells in suspension to another well.
NOTE: It is critical not to dislodge the MEFs, which are attached to the bottom of the wells.
Pipet up and down 10 times in the new well to ensure a single cell suspension.
NOTE: Cells may clump during the incubation; therefore, it is critical to pipet once the cells are transferred in the new well. We recommend confirming under the microscope that you have obtained a proper single cell suspension.
Place the plate in the incubator and swirl the plate to ensure even distribution of cells. Leave untouched for 15 min.
During this last 15-min step, equilibrate the required amount of NDiff 227 in a 10 cm dish in the incubator for at least 20 min. Longer incubation is also possible (e.g., NDiff 227 can be placed in the incubator after step 6. See Table 1 for the volume needed as a function of the number of 96-well plates to seed).
NOTE: NDiff 227 is light-sensitive and should be protected from (direct) light as much as possible.
Carefully transfer all MEF-depleted mESCs to a 15 ml Falcon tube with a P1000 pipet.
NOTE: It is critical not to dislodge the MEFs, which are attached to the bottom of the wells.
D4. mESC aggregation (0 h)
NOTE: The first 96 h of the TLS protocol are similar to the gastruloids protocol (Baillie-Johnson et al., 2015; van den Brink et al., 2014; Beccari et al., 2018b; Anlas et al., 2021). Detailed protocols for gastruloid formation, including troubleshooting, are provided elsewhere (Baillie-Johnson et al., 2015; Beccari et al., 2018b; Anlas et al., 2021).
Centrifuge the cells at 200 × g for 5 min.
Resuspend the cell pellet in 5 ml PBS with MgCl2/CaCl2 and pipet up and down 20 times (Wash 1).
Centrifuge the cells at 200 × g for 5 min.
NOTE: In case of low starting cell numbers, steps 2 and 3 can be omitted. This may however slightly compromise protocol efficiency.
Resuspend the cell pellet in 5 ml pre-equilibrated NDiff 227 and pipet up and down 20 times (Wash 2).
Centrifuge the cells at 200 × g for 5 min.
Resuspend the cell pellet in 500 μl pre-equilibrated NDiff 227 and pipet up and down 30 times. NOTE: It is critical to obtain a single cell suspension prior to counting and plating.
For counting, prepare a 1:2 dilution of the cell suspension by adding 10 μl cell suspension to 10 μl Trypan Blue.
Count using the Luna automated cell counter with the following set up: Dilution factor → 2; Noise reduction → 5; Live detection sensitivity → 5; Roundness → 85%; Min cell size → 10 μm; Max cell size → 20 μm; Declustering level → High.
Transfer the amount of cells needed for the experiment to a new Falcon tube (see Table 1 for the number of cells needed as a function of the number of 96-well plates to seed).
Add the pre-incubated NDiff 227 volume required to bring the cell suspension to a concentration of 5.7 × 103 cells/ml (see Table 1 for the volume to add as a function of the number of 96-well plates to seed).
NOTE: This cell concentration is optimized for an input of 200 cells/well, which has been shown to give high TLS generation efficiency for all cell lines tested (Veenvliet et al., 2020). Similar results were obtained for inputs ranging from 200 to 250 cells/well.
Mix the new cell suspension vigorously and transfer it to a reservoir.
Use a multichannel pipet to transfer 35 μl to each well of an ultra-low attachment 96-well plate. Pipet gently up and down in the reservoir between each transfer.
Gently tap the plate 5 times on a clean bench, transfer to the incubator, and allow undisturbed aggregation for 48 h.
NOTE: Keeping NDiff 227 outside the incubator for longer periods of time (more than 5 min) will lead to disequilibration of the medium. Therefore, try to avoid keeping NDiff 227 or plates with freshly seeded cells in NDiff 227 out the incubator for too long. In the case of handling multiple plates, we recommend putting each plate into the incubator directly after pipetting.
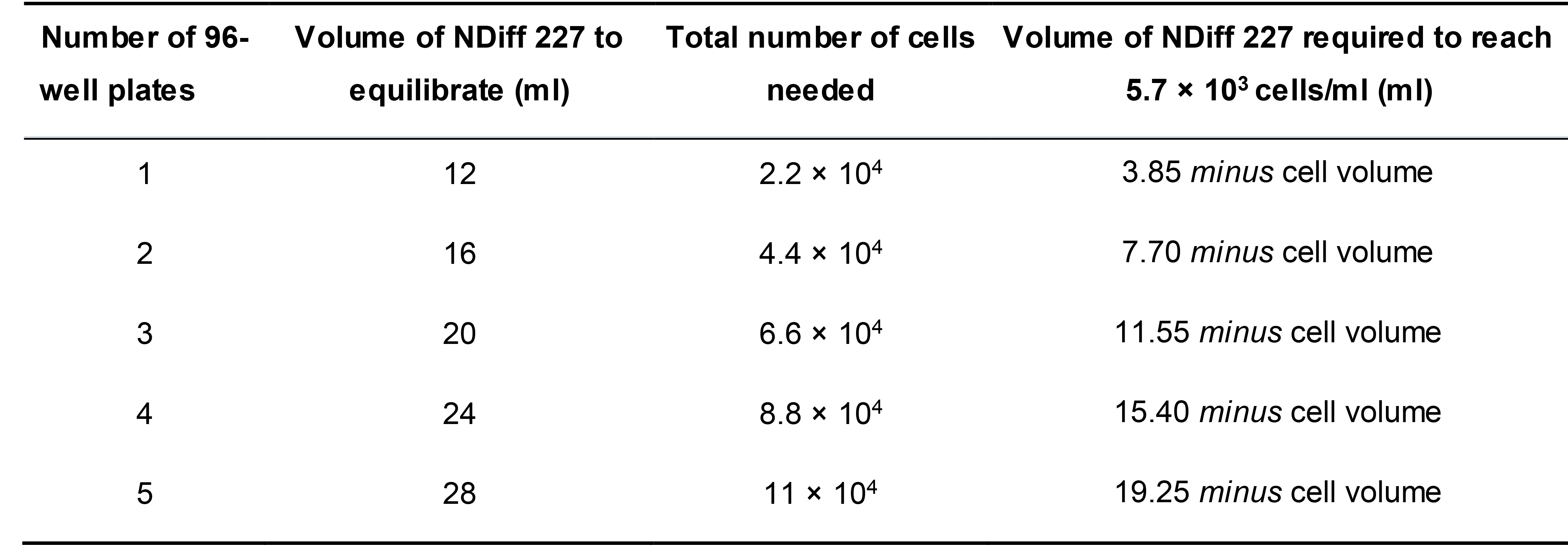
Table 1. Cell numbers and volumes required for mESC aggregation. The amounts are calculated for an input of 200 cells/well in 35 μl. Volumes and cell numbers in column 4 are calculated for 100 samples (instead of 96) per plate plus a 10% excess dead volume. In column 2, the volume of NDiff 227 to equilibrate is calculated based on the amount needed for washing and counting the cells (5.5 ml per experiment, independent of the number of plates, see step D4.4 and D4.6), plus the amount indicated in column 4, plus an extra volume to account for dead space in the dish and evaporation during medium equilibration.
D5. CHIR pulse (48 h)
NOTES:
Start this procedure at least one hour before the end of the 48 h.
Cells at 48 h should have formed one single round aggregate with a diameter measuring 214 ± 13 μm (seeFigure 4A).
CHIR99021 is light-sensitive and should be protected from (direct) light as much as possible.
CHIR99021 should be aliquoted in single-use aliquots in brown (light-protected) sterile tubes upon arrival and not subjected to repeated freeze-thaw cycles.
Equilibrate the needed amount of NDiff 227 in a 10-cm dish in the incubator for at least 20 min. (see Table 2 for the volume needed as a function of the number of 96-well plates used in the experiment).
Transfer the required amount of NDiff 227 to a 50-ml Falcon tube (see Table 2 for the volume needed as a function of the number of 96-well plates used in the experiment).
Table 2. Volumes of NDiff 227, CHIR99021, and Matrigel required during the last three steps (protocol steps D5, D6, D7) of the TLS generation protocol. In column 2, the volume of NDiff 227 to equilibrate is calculated including an excess volume (2 ml for each dish that is used for medium equilibration) to account for dead space in the dish(es) and medium evaporation during equilibration.
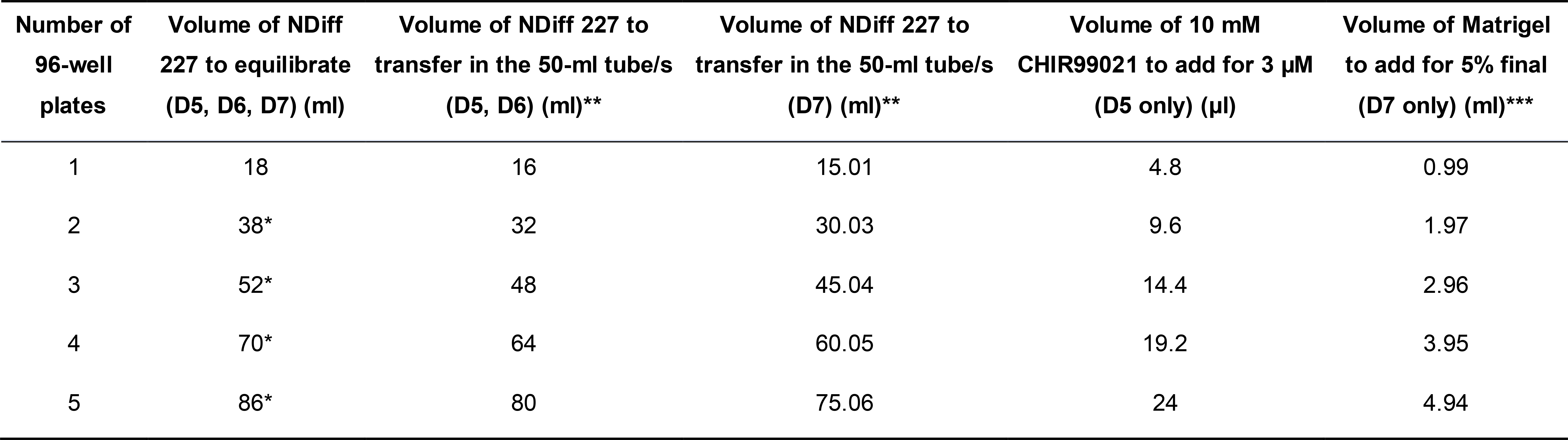
*For volumes higher than 30 ml, use more than one 10-cm plate to equilibrate NDiff 227 in the incubator.
**Use two 50-ml tubes if the volume exceeds 50 ml.
***Note that we calculate 5% v/v Matrigel as a function of the final volume in each well (35 μl + 150 μl = 185 μl) (as opposed to CHIR99021). This means that in the NDiff 227, the concentration of Matrigel is slightly higher than 5% (6.17%). For instance, for 1 plate of TLSs, the volume of Matrigel added is calculated as (0.05*16)*(185/150) = 0.987 ml.
Add CHIR99021 to the NDiff 227 medium to obtain a final concentration of 3 μM.
Mix the medium vigorously and transfer it to a reservoir.
Use a multichannel pipet to add 150 μl CHIR99021-supplemented medium to each well of the plates containing the aggregates.
Gently tap the plate 10 times on a clean bench, transfer to the incubator, and allow further undisturbed development for 24 h.
NOTES:
The tapping is critical to prevent cell aggregates from attaching to the culture plates. Ensure that the aggregates are freely moving immediately after tapping (this can be checked under the microscope).
Take caution to avoid splashing medium on the lid while tapping the plates.
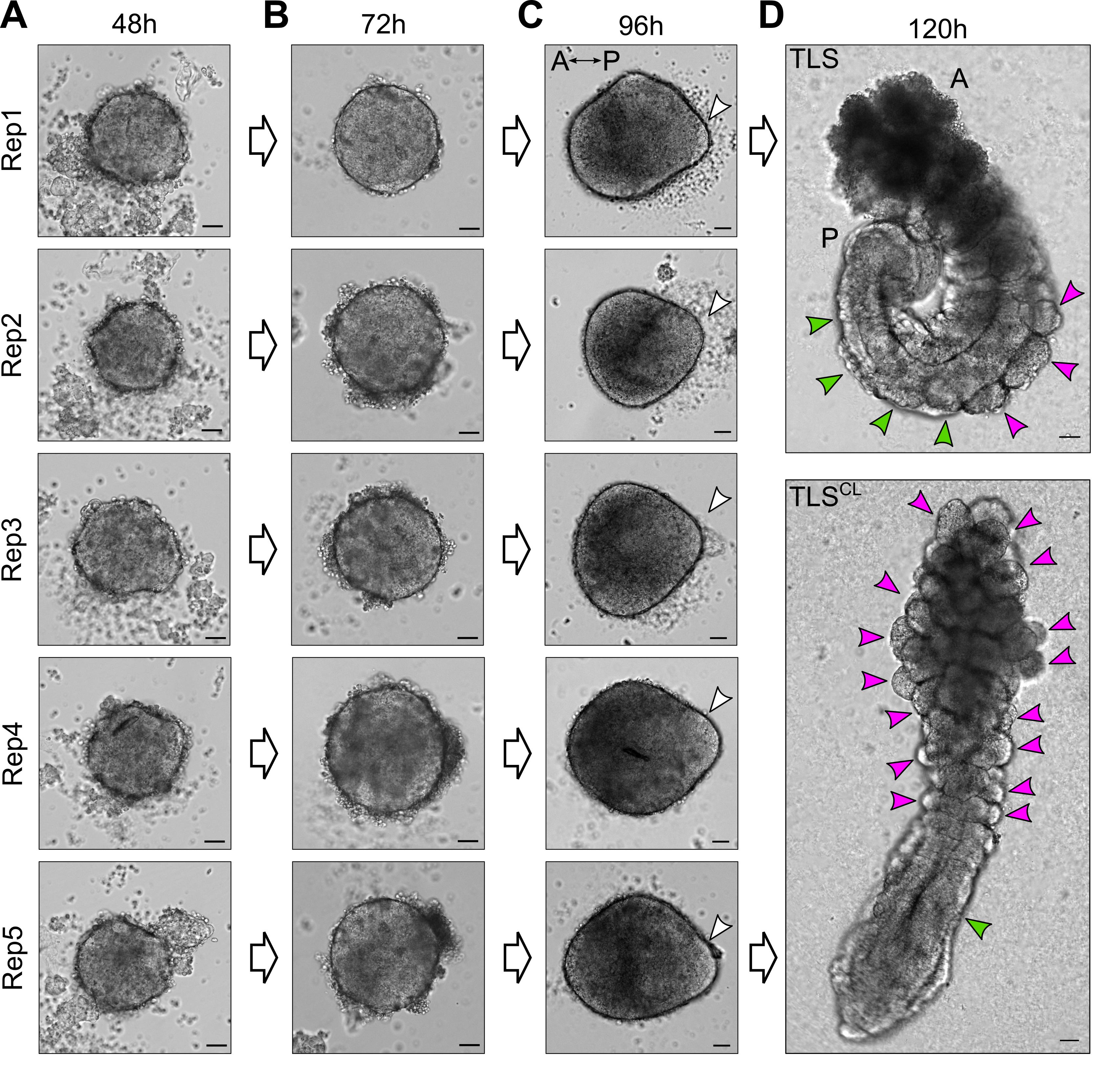
Figure 4. Examples of expected morphology of mESC-derived aggregates at several timepoints during TLS generation. A, B. mESC-derived aggregates at 48 h and 72 h after aggregation are round without clear signs of symmetry breaking. C. At 96 h after aggregation, the structures have clearly broken symmetry and are teardrop shaped. The white arrowheads indicate the posterior pole, where localized expression of Brachyury is expected. Note that, depending on the cell line used, aggregates may establish the teardrop-like morphology prior to 96 h. In that case, structures should be embedded in Matrigel earlier (as soon as the teardrop-like morphology is observed) to achieve optimal TLS efficiency (see main text for details). D. Upon addition of 5% Matrigel, the aggregates will establish an architecture reminiscent of the embryonic trunk, with somites (magenta arrowheads) flanking a neural tube (green arrowheads). Chemical modulation with a WNT agonist (5 μM CHIR99021) and BMP inhibitor (600 nM LDN193189) results in compromised neural tube development and formation of excess somites arranged like a “bunch-of-grapes” (TLSCL). Scale bars for all panels, 50 μm. A, Anterior; P, Posterior.
D6. Media change (72 h)
NOTES:
Start this procedure at least one hour before the end of the 72 h.
Aggregates at 72 h should look like the example given in Figure 4B and measure 244 ± 15 μm in diameter.
If available, perform this step on a clean bench containing a stereoscope or a light ring to help visualize the structures and avoid loss of structures while pipetting off the old medium.
- Equilibrate the required amount of NDiff 227 in a 10-cm dish in the incubator for at least 20 min (see Table 2 for the volume needed as a function of the number of 96-well plates used in the experiment).
- Transfer the required amount of NDiff 227 to a 50-ml tube (see Table 2 for the volume needed as a function of the number of 96-well plates used in the experiment).
- Use a multichannel pipet to remove 150 μl from each well without disturbing the structure.
NOTE: CRITICAL STEP. Avoid losing structures while pipetting off the medium. This is best achieved by keeping the plate under a 30-40° angle and putting the pipet tips against the side opposite to that where the aggregate should be located. As a visual aid, a stereoscope or light ring could be used as stated above (see Figure 5 for a schematic of how to position the plate and tips).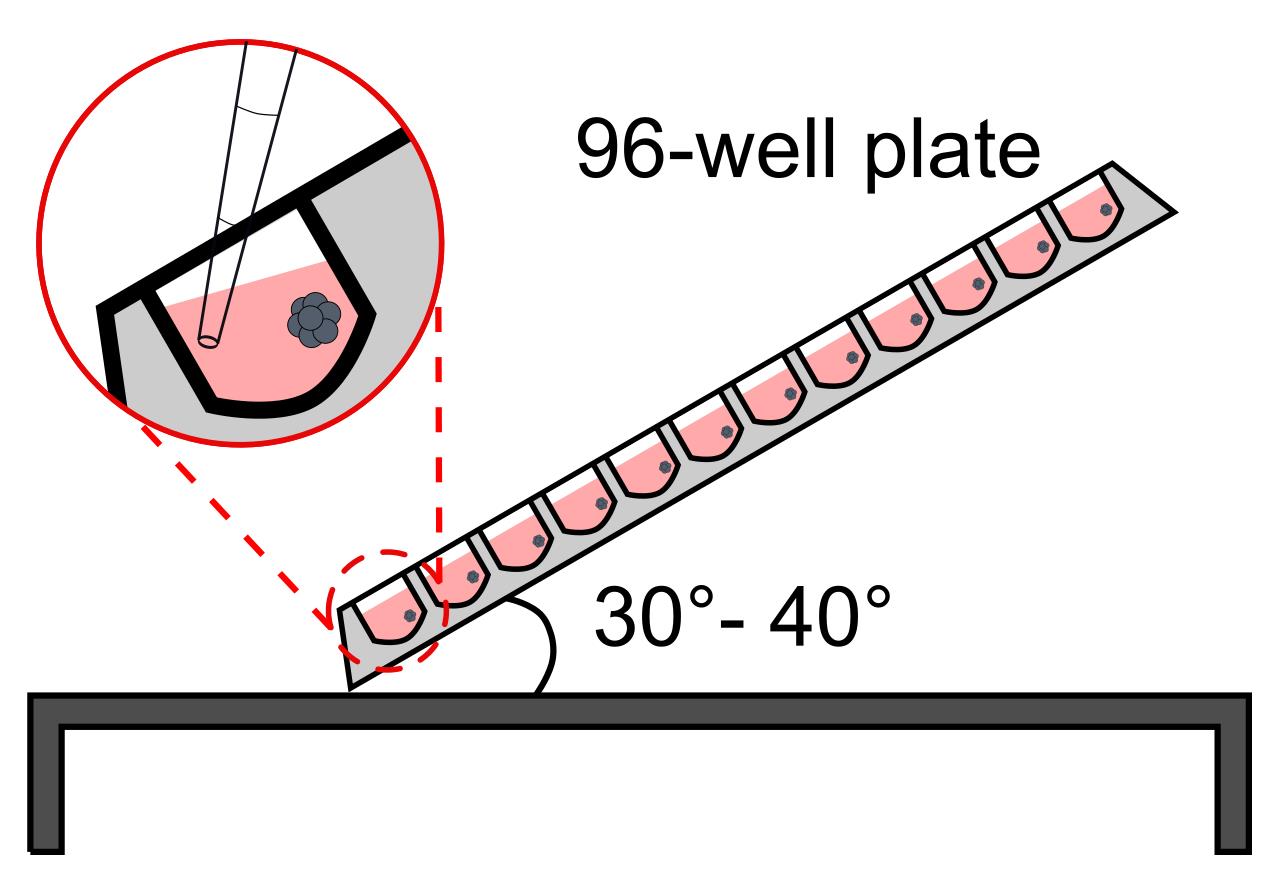
Figure 5. Schematic representation of plate positioning for media changes during the TLS generation procedure. The plate is tilted at a 30-40° angle on a clean bench and the media is carefully aspirated with a multichannel pipet, avoiding disturbance of the aggregates. Pour the pre-equilibrated medium in a reservoir.
Use a multichannel pipet to add 150 μl medium to each well of the plates containing the aggregates.
Gently tap the plate 10 times on a clean bench, transfer to the incubator, and allow further undisturbed development for 16-24 h.
NOTES:
The tapping is critical to prevent cell aggregates from attaching to the culture plates. Ensure that the aggregates are freely moving immediately after tapping (this can be checked under the microscope).
Take caution to avoid splashing medium on the lid while tapping the plates.
Thaw overnight at 4 °C on ice the amount of Matrigel needed the following day (see Table 2 for the volume needed as a function of the number of 96-well plates used in the experiment).
We have used multiple Matrigel batches with comparable results.
D7. TLS generation (88-96 h)
NOTES:
Start monitoring the TLSs around 88 h after aggregation for the appearance of a “teardrop-like” shape (see Figure 4C). Structures should present a longer axis (421 ± 33 μm) and a shorter axis (337 ± 30 μm), with an axis ratio of 0.8 ± 0.07.
Start this procedure immediately when a “teardrop-like” shape is observed in the majority of the TLSs (or latest 96 h after aggregation) to achieve optimal TLS formation efficiency.
If available, perform this step on a clean bench containing a stereoscope or a light ring to help visualize the structures and avoid losing structures while pipetting off the old medium.
If performing chemical modulation at this step, follow the “Variant protocol: chemical modulation during TLS generation.”
Equilibrate the required amount of NDiff 227 in a 10-cm dish in the incubator for at least 20 min (see Table 2 for the volume needed as a function of the number of 96-well plates used in the experiment).
Transfer the required amount of NDiff 227 to a 50-ml Falcon tube and place it on ice (see Table 2 for the volume needed as a function of the number of 96-well plates used in the experiment).
Once the medium has cooled down, supplement it with the correct volume of Matrigel on ice and mix vigorously.
NOTE: It is critical, while handling Matrigel, that every step is performed on ice to avoid clumping. It is also recommended to pre-cool the pipet tips used for the handling of 100% Matrigel by placing the box in the fridge until use; see Table 2 for the volume needed as a function of the number of 96-well plates used in the experiment.
Move the Falcon tube with Matrigel-supplemented medium to room temperature.
Use a multichannel pipet to remove 150 μl from each well without disturbing the structure.
NOTES:
CRITICAL STEP. Avoid losing structures while pipetting out the medium. This is best achieved by keeping the plate under a 30-40° angle and putting the pipet tips against the side opposite to that where the aggregate should be located. As a visual aid, a stereoscope or light ring could be used as stated above (see Figure 5 for a schematic of how to position the plate and tips).
This step should not take more than 5 min after Matrigel-supplemented medium has been placed at room temperature. If there are multiple plates, it is advisable to remove the medium from all plates before equilibrating the Matrigel-supplemented medium at room temperature and keep the structures in the incubator.
Pour the Matrigel-supplemented medium into a reservoir.
Use a multichannel pipet to add 150 μl Matrigel-supplemented medium in each well of the plates containing the aggregates.
Gently tap the plate 10 times on a clean bench and transfer to the incubator.
NOTES:
The tapping is critical to prevent cell aggregates from attaching to the culture plates. Ensure that aggregates are freely moving immediately after tapping (this can be checked under the microscope).
Take caution to avoid splashing medium on the lid while tapping the plates.
If performing Live Cell Imaging after embedding, allow the TLSs to settle for 1 h in the incubator before starting imaging.
Variant protocol: chemical modulation during TLS embedding
Follow “D7. TLS generation (88-96 h)” for all passages except for point 3.
3’. Once the medium has cooled down, supplement it with the correct volume of Matrigel and appropriate chemical compounds for your modulations on ice and mix vigorously.
NOTES:
It is critical, while handling Matrigel, that every step is performed on ice to avoid clumping; see Table 2 for the volume needed as a function of the number of 96-well plates used in the experiment.
Add an equal volume of diluent to the control sample TLSs when performing chemical modulations.
In Veenvliet et al. (2020), we induced excess somite production and compromised neural tube development by supplementing the medium with 5 μM CHIR99021, alone (TLSC) or in combination with 600 nM LDN193189 (TLSCL).
EXPECTED OUTCOME:
TLSs at 120 h should look elongated with a clear anterior and posterior domain (Figure 4D upper panel; Figure 6A). Moreover, they should show clear segmentation (somites) on one or both sides of a tubular structure (neural tube). TLSs subjected to chemical modulation (TLSCL) should display compromised neural tube development and formation of excess somites arranged like a “bunch-of-grapes” (Figure 4D lower panel; Figure 6B).
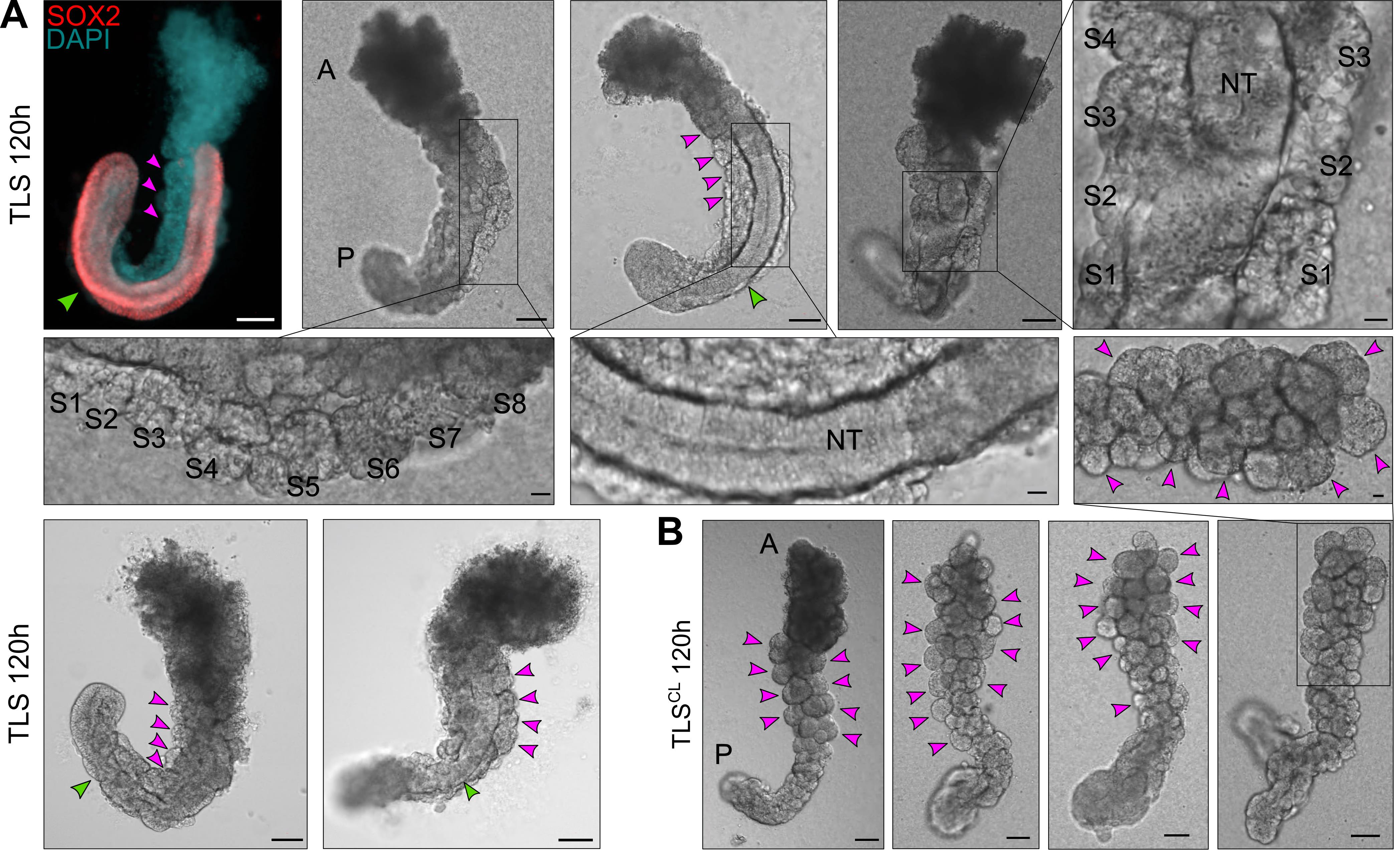
Figure 6. Expected outcome of the TLS protocol. A. Examples of trunk-like structures (TLSs) 24 h after addition of 5% Matrigel (total culture time 120 h). The left upper structure is immunostained with a SOX2 antibody and counterstained with DAPI, labeling the neural tube and nuclei, respectively. Somites are indicated with magenta arrowheads or with S1, S2, etc. (in magnifications); S1-S8, Somite 1-Somite 8. Neural tube (NT) is indicated with a green arrowhead. A, Anterior; P, Posterior. B. Expected outcome of TLSs subjected to chemical modulation (TLSCL) is compromised neural tube development and formation of excess somites arranged like a “bunch-of-grapes.” Somites are indicated with magenta arrowheads. Scale bars, 100 μm (whole structure) or 20 μm (magnifications). A, Anterior; P, Posterior.
D8. TLS analysis (108-120 h)
NOTES:
Depending on the specific biological question, the exact time of analysis may vary (see Veenvliet et al., 2020 for time-resolved expression dynamics).
If available, perform this step on a clean bench containing a stereoscope or a light ring to help visualize the structures and avoid losing structures while processing them.
The following protocol variants are performed (D8’, D8’’, D8’’’) depending on the downstream applications.
Prepare a P200 tip box with the tip cut-off at the 50-μl mark for TLS picking (if using unmarked tips, cut approximately 9 mm off the tip).
Use a P200 pipet set to 50 μl and the cut-off tips to manually pick each individual TLS that needs to be analyzed in a well of an Ibidi 8-chamber plate.
Add 200 μl cold PBS/0.5% BSA solution to each well containing a TLS.
Remove 200 μl with a pipet and perform the same 200 μl cold PBS/0.5% BSA solution wash three times.
Use the fixative of interest to fix TLSs in the Ibidi plate for the downstream protocol.
Perform the rest of the staining protocol in the Ibidi plate and image the structure with the desired microscope and settings.
NOTE: We have so far used 4% PFA fixation for whole-mount immunofluorescence (WIFC) as well as whole-mount in situ hybridization (WISH). A detailed description of the protocols used for WIFC and WISH, including method-specific fixation times and downstream processing, is provided in the Supplemental Information of Veenvliet et al. (2020).
D8’’. RNA extraction
Use a P200 pipet set to 50 μl and the cut-off tips to manually pick and transfer each individual TLS that needs to be analyzed to a 1.5-ml tube containing 1 ml cold PBS/0.5% BSA solution.
NOTE: The number of TLSs that are pooled in one tube depends on the downstream application and experiment.
Centrifuge the TLSs at 200 × g for 1 min at 4°C.
Remove the supernatant with a P1000 pipet while being careful not to disturb the TLS pellet.
Wash the structures with 1 ml cold PBS/0.5% BSA solution.
NOTE: Ensure loosening of the pellet without aspirating into the pipet tip.
Centrifuge the TLSs at 200 × g for 1 min at 4°C.
Remove the supernatant with a P1000 pipette while being careful not to disturb the TLS pellet.
Add the indicated amount of RNA lysis buffer or TRIzol depending on the desired RNA extraction strategy.
D8’’’. 10× Genomics single-cell RNA sequencing
NOTES:
This section explains how to process TLSs to generate a single cell suspension suitable for efficient Gel Bead-in-Emulsion (GEMs) generation. Follow the manufacturer’s instructions for every step after the single cell suspension has been generated and counted.
Pre-warm TrypLE in the water bath for at least 20 min before starting.
Use a P200 pipette set to 20 μl and the cut-off tips to manually pick each individual TLS that needs to be analyzed in a well of an ultra-low attachment 96-well plate containing 200 μl cold PBS.
Transfer each TLS serially five times to new wells containing 200 μl cold PBS.
NOTES:
It is CRITICAL to carry over as little volume as possible from the culture to minimize the amount of Matrigel transferred. Carrying over excess amounts of Matrigel can lead to microfluidics clogging during GEM generation.
Since washing of TLSs is performed in PBS without BSA, the structures may become sticky and get stuck to the tip wall. Avoid this by pipetting a very low volume to maintain the structure at the liquid/air interphase in the tip.
After the five washes, transfer all the structures into a single drop of 200 μl pre-warmed TrypLE in the center of a 6-cm plate.
Transfer the plate to the incubator and allow cell dissociation for 25 min with pipetting every 5 min to ensure that a single cell suspension is achieved.
NOTE: Perform the pipetting steps under a stereoscope to monitor the degree of cell dissociation and ensure no loss of material.
At the end of the 25 min, and after verifying correct achievement of a single cell suspension, transfer the cell suspension to a 1.5-ml tube on ice.
To ensure maximum cell recovery and to quench the trypsinization reaction, wash the part of the plate where the drop was located four times with 200 μl PBS/0.5% BSA solution. Add every wash to the same tube (from step 5) containing the cell suspension.
Filter the cell suspension using a P1000 set to 1 ml through a 40-µm Flowmi Cell Strainer in a new 1.5-ml tube on ice.
Centrifuge the cell suspension at 300 × g for 5 min at 4°C.
Remove 800 μl supernatant with a P1000 while being careful not to disturb the cell pellet.
NOTE: The cell pellet may be very small and barely visible, so be extremely careful during these steps.Wash with 1 ml PBS/0.5% BSA solution.
Centrifuge the cell suspension at 300 × g for 5 min at 4°C.
Remove 800 μl supernatant with a P1000 while being careful not to disturb the cell pellet.
NOTE: The cell pellet may be very small and barely visible, so be extremely careful during these steps.Resuspend the pellet in the remaining 200 μl left in the tube.
Centrifuge the cell suspension at 300 × g for 5 min at 4°C.
Remove the supernatant with a P200 pipet, leaving ~42 μl in the tube.
NOTE: Use another tube containing exactly 42 μl PBS/0.5% BSA solution as a guide to evaluate the approximate volume to leave in the tube.
Resuspend the cell pellet in the ~42 μl left and determine the cell suspension concentration using a manual hemocytometer (analyze a 1:5 cell suspension dilution by adding 2 μl cell suspension to 8 μl Trypan Blue).
Proceed with the desired amount of cells for GEM generation following the manufacturer’s instructions.
Data analysis
All data and analysis needed for the development and characterization of this protocol are available in the main text or Supplemental Information of Veenvliet et al. (2020).
Recipes
0.1% Gelatin solution
Dilute sterile 2% Gelatin to 0.1% in cell culture grade water. Store at 4°C
Mouse embryonic fibroblast (MEF) medium
NOTE: Heat inactivate the FCS for 30 min at 56°C before use.
500 ml Dulbecco’s Modified Eagle's Medium (DMEM)
55 ml regular FCS (Pan Biotech; catalog number: P30-3306)
5.5 ml 100× Glutamine
5.5 ml 100× Penicillin/Streptomycin
Sterile filter
Store at 4°C
Mouse embryonic stem cell (mESC) medium
NOTE: Heat inactivate the FCS for 30 min at 56°C before use.
400 ml Knockout Dulbecco’s Modified Eagle’s Medium (KO-DMEM)
75 ml mESC tested FCS (Pan Biotech; catalog number: P30-2602)
5 ml 100× Glutamine
5 ml 100× Penicillin/Streptomycin
5 ml 100× Nucleosides
1 ml Gibco 2-Mercaptoethanol
Sterile filter
Aliquot mESC medium without LIF in 40-ml portions and freeze at -20°C
Thaw before use, then store at 4°C
Add 1:10,000 LIF immediately before use
Store at 4°C
NOTE: Homemade LIF has also been successfully used; however, the right concentration has to be tested based on the purification protocol and batch concentrations.
PBS/0.5% BSA solution
PBS with MgCl2/CaCl2
0.5% BSA powder
Prepare fresh, sterile filter, keep on ice for the procedure
Store at 4°C
Acknowledgments
This work is based on and adapted from the method published in Veenvliet et al. (2020). We are grateful for the support and feedback received to develop and characterize our in vitro system from present and past members of the Herrmann & Meissner laboratories, in particular Manuela Scholze-Wittler, Dennis Schifferl, Frederic Koch, Abhishek Sampath Kumar, Milena Pustet, Fabian Tobor, Simon Heimann, Lars Wittler, Stefanie Grosswendt, Zachary Smith and Atsuhiro Taguchi. The work was supported by an Alexander von Humboldt Fellowship (J.V.V.), NIH grant HG006193 (A.M.), and the Max Planck Society.
Competing interests
The authors declare no competing interests.
References
- Anlas, K., Baillie-Benson, P., Arato, K., Turner, D. A. and Trivedi, V. (2021). Gastruloids: Embryonic Organoids from Mouse Embryonic Stem Cells to Study Patterning and Development in Early Mammalian Embryos. Methods Mol Biol 2258: 131-147.
- Baillie-Benson, P., Moris, N. and Martinez Arias, A. (2020). Pluripotent stem cell models of early mammalian development. Curr Opin Cell Biol 66: 89-96.
- Baillie-Johnson, P., van den Brink, S. C., Balayo, T., Turner, D. A. and Martinez Arias, A. (2015). Generation of Aggregates of Mouse Embryonic Stem Cells that Show Symmetry Breaking, Polarization and Emergent Collective Behaviour In Vitro. J Vis Exp(105).
- Beccari, L., Moris, N., Girgin, M., Turner, D. A., Baillie-Johnson, P., Cossy, A. C., Lutolf, M. P., Duboule, D. and Arias, A. M. (2018a). Multi-axial self-organization properties of mouse embryonic stem cells into gastruloids. Nature 562(7726): 272-276.
- Beccari, L., Girgin, M., Turner, D., Baillie-Johnson, P., Cossy, A.-C., Moris, N., Lutolf, M., Duboule, D. and Martinez Arias, A. (2018b). Generating Gastruloids from Mouse Embryonic Stem Cells. Protocol Exchange. DOI: 10.1038/protex.2018.094.
- Cermola, F., D’Aniello, C., Tatè, R., De Cesare, D., Martinez-Arias, A., Minchiotti, G. and Patriarca, E. J. J. B. (2021). Gastruloid development competence discriminates different states of pluripotency between naïve and primed.Stem Cell Reports 16(2): 354-369.
- George, S. H., Gertsenstein, M., Vintersten, K., Korets-Smith, E., Murphy, J., Stevens, M. E., Haigh, J. J. and Nagy, A. (2007). Developmental and adult phenotyping directly from mutant embryonic stem cells. Proc Natl Acad Sci U S A 104(11): 4455-4460.
- Moris, N., Anlas, K., van den Brink, S. C., Alemany, A., Schroder, J., Ghimire, S., Balayo, T., van Oudenaarden, A. and Martinez Arias, A. (2020). An in vitro model of early anteroposterior organization during human development. Nature 582(7812): 410-415.
- Pourquié, O. (2003). The segmentation clock: converting embryonic time into spatial pattern. Science 301(5631): 328-330.
- Rossi, G., Broguiere, N., Miyamoto, M., Boni, A., Guiet, R., Girgin, M., Kelly, R. G., Kwon, C. and Lutolf, M. P. (2021). Capturing Cardiogenesis in Gastruloids. Cell Stem Cell 28(2):230-240.e6.
- Shahbazi, M. N., Siggia, E. D. and Zernicka-Goetz, M. (2019). Self-organization of stem cells into embryos: A window on early mammalian development. Science 364(6444): 948-951.
- Shahbazi, M. N. and Zernicka-Goetz, M. (2018). Deconstructing and reconstructing the mouse and human early embryo. Nat Cell Biol 20(8): 878-887.
- Turner, D. A., Girgin, M., Alonso-Crisostomo, L., Trivedi, V., Baillie-Johnson, P., Glodowski, C. R., Hayward, P. C., Collignon, J., Gustavsen, C., Serup, P., Steventon, B., M, P. L. and Arias, A. M. (2017). Anteroposterior polarity and elongation in the absence of extra-embryonic tissues and of spatially localised signalling in gastruloids: mammalian embryonic organoids. Development 144(21): 3894-3906.
- van den Brink, S. C., Alemany, A., van Batenburg, V., Moris, N., Blotenburg, M., Vivie, J., Baillie-Johnson, P., Nichols, J., Sonnen, K. F., Martinez Arias, A. and van Oudenaarden, A. (2020). Single-cell and spatial transcriptomics reveal somitogenesis in gastruloids. Nature 582(7812): 405-409.
- van den Brink, S. C., Baillie-Johnson, P., Balayo, T., Hadjantonakis, A. K., Nowotschin, S., Turner, D. A. and Martinez Arias, A. (2014). Symmetry breaking, germ layer specification and axial organisation in aggregates of mouse embryonic stem cells. Development 141(22): 4231-4242.
- Veenvliet, J. V., Bolondi, A., Kretzmer, H., Haut, L., Scholze-Wittler, M., Schifferl, D., Koch, F., Guignard, L., Kumar, A. S., Pustet, M., Heimann, S., Buschow, R., Wittler, L., Timmermann, B., Meissner, A. and Herrmann, B. G. (2020). Mouse embryonic stem cells self-organize into trunk-like structures with neural tube and somites. Science 370(6522).
- Veenvliet, J. V. and Herrmann, B. G. (2021). Modeling mammalian trunk development in a dish. Dev Biol. 2021 474:5-15.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Bolondi, A., Haut, L., Gassaloglu, S. I., Burton, P., Kretzmer, H., Buschow, R., Meissner, A., Herrmann, B. G. and Veenvliet, J. V. (2021). Generation of Mouse Pluripotent Stem Cell-derived Trunk-like Structures: An in vitro Model of Post-implantation Embryogenesis. Bio-protocol 11(11): e4042. DOI: 10.21769/BioProtoc.4042.
- Veenvliet, J. V., Bolondi, A., Kretzmer, H., Haut, L., Scholze-Wittler, M., Schifferl, D., Koch, F., Guignard, L., Kumar, A. S., Pustet, M., Heimann, S., Buschow, R., Wittler, L., Timmermann, B., Meissner, A. and Herrmann, B. G. (2020). Mouse embryonic stem cells self-organize into trunk-like structures with neural tube and somites. Science 370(6522).
Category
Stem Cell > Embryonic stem cell > Cell differentiation
Developmental Biology > Morphogenesis > Organogenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.