- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

ATAC-Seq-based Identification of Extrachromosomal Circular DNA in Mammalian Cells and Its Validation Using Inverse PCR and FISH
(*contributed equally to this work) Published: Vol 11, Iss 9, May 5, 2021 DOI: 10.21769/BioProtoc.4003 Views: 9287
Reviewed by: Manasi K. MayekarAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2020
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Recent studies from multiple labs including ours have demonstrated the importance of extrachromosomal circular DNA (eccDNA) from yeast to humans (Shibata et al., 2012; Dillon et al., 2015; Møller et al., 2016; Kumar et al., 2017; Turner et al., 2017; Kim et al., 2020). More recently, it has been found that cancer cells obtain a selective advantage by amplifying oncogenes on eccDNA, which drives genomic instability (Wu et al., 2019; Kim et al., 2020). Previously, we have purified circular DNA and enriched the population using rolling circle amplification followed by high-throughput sequencing for the identification of eccDNA based on the unique junctional sequence. However, eccDNA identification by rolling circle amplification is biased toward small circles. Here, we report a rolling circle-independent method to detect eccDNA in human cancer cells. We demonstrate a sensitive and robust step-by-step workflow for finding novel eccDNAs using ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) combined with a Circle_finder bioinformatics algorithm to predict the eccDNAs, followed by its validation using two independent methods, inverse PCR and metaphase FISH (Fluorescence in situ Hybridization).
Keywords: Circular DNABackground
Extrachromosomal circular DNAs (eccDNAs) are unique DNA molecules that carry genetic information in addition to the chromosomal DNAs. These eccDNAs have been found in different organisms from yeast to humans (Shibata et al., 2012; Dillon et al., 2015; Møller et al., 2016; Kumar et al., 2017; Turner et al., 2017; Kim et al., 2020). The length of eccDNAs ranges from small (less than 1kb, also called microDNAs) to large (megabase-long). While the small eccDNAs with micro homology ends may promote genetic heterogeneity (Shibata et al., 2012) or produce short RNAs if transcribed (Paulsen et al., 2019), the long eccDNAs may harbor complete genes and regulatory elements such as enhancers (Morton et al., 2019; Wu et al., 2019; Koche et al., 2020). Emerging evidence suggests that eccDNAs could play underappreciated roles in regulating gene expression and genome instability, which ultimately contributes to the selective advantage of cells (Gresham et al. 2010, Koo et al., 2018; Hull et al., 2019). In particular, oncogene-carrying eccDNAs are highly amplified in human cancers and correlate with open chromatin, increased oncogene expression, and chromosome structural rearrangement, in addition to being associated with poor outcomes (Wu et al., 2019; Kim et al., 2020; Koche et al., 2020). Uncovering eccDNAs in the circulation also makes them prospective targets for diagnostic purposes (Kumar et al., 2017; Sin et al., 2020).
The growing research on eccDNAs calls for tool development for eccDNA discovery. Historically, eccDNAs of various sizes were detected by karyotyping, electron microscopy, Southern blotting, and 2-D gel electrophoresis (reviewed in Paulsen et al., 2018). More recently, various high-throughput sequencing (HTS) technologies have been exploited to facilitate the discovery of new eccDNAs (Shibata et al., 2012; Møller et al., 2015; Kim et al., 2020). The basic idea for eccDNA detection via HTS methods is based on their distinctive circular feature – eccDNAs of high confidence can be identified from paired-end reads that (1) could not map as inward pairs on the linear genome, and (2) contain the unique circular junctional sequence that represents the chromosome breakage/ligation point. A unique junctional sequence (shown as “E-A” in Figure 1A) that is not present in the normal reference genome could be formed through ligation of the two ends of a linear DNA, thus creating the circular DNA. However, the majority of eccDNA sequencing pipelines utilize multiple displacement amplification (MDA), an efficient method to amplify small amounts of DNA via rolling circle amplification, which would preferentially amplify short circles. Therefore, we sought to develop an MDA-independent pipeline that incorporates several additional validation assays.
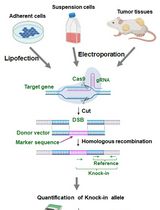
Recently, we demonstrated a robust workflow to detect and validate new eccDNAs from human cancer cell cultures (Kumar et al., 2020). Specifically, ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) and a Circle_finder algorithm was employed for new eccDNA prediction. ATAC-seq, first developed in 2013, utilizes engineered Tn5 transposase to cut open chromatin regions (including eccDNAs that are less chromatinized) and insert transposase-associated adapter DNAs (Buenrostro et al., 2013 and 2015). The Circle-finder algorithm (refer to Software for link access) predicts eccDNAs from paired-end sequencing based on: (1) the presence of split reads (one read maps to two sites in the genome); (2) the two fragments on the split read maps on the same chromosome and same strand; and (3) the continuous read maps between the two fragments on the split read and on the opposite strand to the split read. Predicted eccDNAs can be evaluated by two independent validation assays (Figure 1). Inverse PCR (Figure 1B) will specifically amplify eccDNAs with a primer pair that spans the unique junctional sequence (shown as “E-A”); such a primer pair faces outward on genomic DNA and results in no amplification. Alternatively, eccDNA can be visually confirmed by metaphase FISH (Figure 1C), which can detect both genomic DNA signals overlapping with main chromosomes and signals from eccDNAs that do not overlap with chromosomes.

Figure 1. Overview of eccDNA identification and validation by ATAC-seq, inverse PCR, and metaphase FISH
Materials and Reagents
50 ml Falcon conical tubes (Fisher Scientific, catalog number: 1443222)
15 ml Falcon conical tubes (Fisher Scientific, catalog number: 1495949B)
1.5 ml DNA LoBind tubes (Eppendorf, catalog number: 022431021)
1 ml pipette tip
100-200 μl pipette tip
100 mm Petri dish
0.2 μm filter
Microscope glass slides (Fisher Scientific, catalog number: 4951F-001)
22 mm × 50 mm cover glass (Fisher Scientific, catalog number: 12-545E)
Parafilm (Thermo Fisher Scientific, catalog number: S37440)
Aluminum foil (Thermo Fisher Scientific, catalog number: 14-648-236)
Mammalian cells in culture. In this protocol, we used the ovarian cancer cell line, OVCAR8, and the prostate cancer cell line, C4-2B, cultured in RPMI medium (Corning, catalog number: 10-040-CV) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, catalog number: 26140079) and 1% penicillin-streptomycin (Thermo Fisher Scientific, catalog number: 15-140-122)
SybrGold dye (Invitrogen, catalog number: S11494)
0.5% trypsin-EDTA (Thermo Fisher Scientific, catalog number: 15400054)
UltraPure DNase/RNase-free distilled water (Thermo Fisher Scientific, catalog number: 10977015)
1 M Tris-HCl, pH 7.5 (Thermo Fisher Scientific, catalog number: 15567027, store at 4°C, shelf life: 6 months)
5 M NaCl solution (Thermo Fisher Scientific, catalog number: AM9760G, store at room temperature)
1 M MgCl2 solution (Thermo Fisher Scientific, catalog number: AM9530G, store at room temperature)
Dulbecco’s phosphate-buffered saline or DPBS, no calcium, no magnesium (Thermo Fisher/Gibco, catalog number: 14190144, store at 4°C, shelf life: 36 months)
10% Nonidet P40 substitute (Millipore/Sigma-Aldrich, catalog number: 11332473001, store at 4°C, keep protected from light, shelf life: 24 months)
10% (w/v) Tween-20 (Millipore/Sigma-Aldrich, catalog number: 11332465001, store at 4°C under inert gas and keep protected from light, shelf life: 24 months)
20 mg/ml digitonin in DMSO (Promega, catalog number: G9411, store in -20 °C)
DNA Clean & Concentrator Kit (ZYMO, catalog number: D4033)
Nextera DNA Sample Preparation Kit (Illumina, catalog number: FC-121-1030, store at -20°C)
Note: This kit has been discontinued and can be purchased separately: Tagmentation DNA Enzyme/TDE (Illumina, catalog number: 15027865) and Tagmentation DNA Buffer/TDB (Illumina, catalog number: 15027866).Nextera Index Kit, 24 indexes (Illumina, catalog number: 15055289, store at -20°C)
NEBNext High-Fidelity 2× PCR Master Mix (New England Biolabs, catalog number: M0541, store at -20°C)
Phosphate-buffered saline, pH 7.4 (Thermo Fisher Scientific, catalog number: 10010023)
Qiagen HiSpeed Plasmid Midi Kit (Qiagen, catalog number: 12643)
Isopropanol (Fisher Chemical, catalog number: A516-500)
Ethanol (Thermo Fisher Scientific, catalog number: A4094)
Glycogen (Thermo Fisher Scientific, catalog number: AM9510)
Plasmid-safe ATP-dependent DNase (Lucigen, catalog number: E3101K)
QIAquick PCR Purification Kit (Qiagen, catalog number: 28104)
KOD Hot-Start DNA Polymerase (Millipore/Sigma-Aldrich, catalog number: 71086)
Thymidine (Millipore/Sigma-Aldrich, catalog number: T1895)
10 mg/ml KaryoMax Colcemid solution in PBS (Thermo Fisher Scientific, catalog number: 15212012)
Potassium chloride (Millipore/Sigma-Aldrich, catalog number: P9541)
Formamide (Millipore/Sigma-Aldrich, catalog number: 47670)
Sodium chloride (Thermo Fisher Scientific, catalog number: BP358)
Sodium citrate (Millipore/Sigma-Aldrich, catalog number: W302600)
BAC FISH Probe label with 5-fluorescein (Empire Genomics, catalog number: RP11-732I3 and RP11-765O11)
Rubber cement (Elmer’s Rubber Cement, catalog number: EPIE904)
Nonidet P-40 (Sigma, catalog number: I8896)
Dextran sulfate (Thermo Fisher Scientific, catalog number: BP1585)
VectaShield Mounting Medium with DAPI (Vector Laboratories, catalog number: H-1200-10)
Nail polish (OPI Nail Lacquer)
Nikon immersion oil for the confocal microscope (Thermo Fisher Scientific, catalog number: 12-624-66A)
1% (10 mg/ml) digitonin (see Recipes, store at -20°C as aliquotes, stable for 6 months)
ATAC-Resuspension Buffer (see Recipes)
ATAC-Lysis Buffer (see Recipes)
ATAC-Wash Buffer (see Recipes)
ATAC-Reaction Mastermix (see Recipes)
100 mM thymidine solution (see Recipes)
75 mM KCl Hypotonic Solution (see Recipes)
Carnoy’s Fixative Solution (see Recipes)
20× Saline-Sodium-Citrate (SSC) buffer (see Recipes)
Hybridization Buffer (see Recipes)
FISH Denaturation Buffer (see Recipes)
FISH Wash Buffer 1 (see Recipes)
FISH Wash Buffer 2 (see Recipes)
Equipment
Cell culture incubator
Tissue culture hood
Tabletop microcentrifuge (Eppendorf, model: 5424)
Thermomixer (Thermo Scientific, catalog number: 13687711)
PCR machine with heated lid (Eppendorf, model: Mastercycler Pro)
Tabletop centrifuge (Eppendorf, model: 5804)
Water bath (Thermo Fisher Scientific, Isotemp)
Coplin jar (Local Company)
Hybridization chamber (Thermo Fisher Scientific, Isotemp)
Chemical fume hood (Bellco Glass Inc.)
Brightfield microscope (Olympus)
Confocal microscope (Nikon, model: Ti-E eclipse series)
Computer with enough data storage capacity up to TB
Software
Circle_finder (github, https://github.com/pk7zuva/Circle_finder/blob/master/circle_finder-pipeline-bwa-mem-samblaster.sh); pre-requisite installation to run Circle_finder: bedtools (https://github.com/arq5x/bedtools2), samtools (http://samtools.sourceforge.net), parallel (https://www.gnu.org/software/parallel/), bwa (https://github.com/lh3/bwa), samblaster (https://github.com/GregoryFaust/samblaster)
Cutadapt (https://cutadapt.readthedocs.io/en/stable/)
AR Elements Software (Nikon, Japan)
ImageJ (NIH, USA)
Procedure
ATAC-seq from cultured mammalian cells
Nuclei isolation from cultured mammalian cells
Pellet 50,000 mammalian cells in culture into a 1.5 ml DNA LoBind tube.
Note: Check cell viability prior to the experiment by Trypan Blue staining and ensure cell viability is at least 95%. Please refer to the original ATAC-seq protocol (Corces, 2017) for treatment of cells with DNase to remove extracellular DNAs or to separate cells via ficoll gradient if viability is lower than 95%.
Wash cells in ice-cold DPBS twice at 500 × g.
Add 50 μl ice-cold ATAC-LB to each tube, pipette up and down 3 times with a 100-200 μl pipette tip. Incubate on ice for 3 min.
Note: We have used a 3-min lysis time with several cell lines, including HCT116, OVCAR8, and C4-2B. The lysis time may need to be increased for specific tough-to-lyse cell lines. The efficiency of cell lysis can be checked by Trypan Blue staining under the microscope (blue staining suggests successful lysis).
Immediately dilute the 50 μl lysate by adding 1 ml ice-cold ATAC-WB into the tube. Invert tube 3 times to mix. Spin at 500 × g for 10 min at 4°C.
Carefully remove the supernatant (containing the cytoplasmic fraction*), first by a 1-ml pipette tip followed by a 100-200-μl pipette tip. The pellet now contains the nuclei, which should be used for the tagmentation reaction immediately.
*Note: The cytoplasmic fraction can be saved for RNA extraction later.
Transposition/tagmentation reaction and clean-up
Resuspend the nuclei pellet in 50 μl ATAC-RM by pipetting up and down 6 times. Incubate in a thermomixer at 37°C for 30 min with 1,000 RPM constant mixing.
Add 250 μl DNA Binding Buffer (from Zymo DNA Clean and Concentrator Kit) to each 50 μl ATAC reaction, mix well by pipetting up and down 3 times. Transfer 300 μl mixture to each column sitting on a collection tube (both from Zymo DNA Clean and Concentrator Kit), spin down at 15,000 × g for 30 s and discard liquid in the collection tube.
Wash column with 200 μl DNA Wash Buffer (from Zymo DNA Clean and Concentrator Kit), spin down at 15,000 × g for 30 s.
Note: Make sure ethanol is added to the Wash Buffer according to the manufacturer’s guidelines.
Repeat the wash with 200 μl DNA Wash Buffer. Spin down at 15,000 × g (or maximum speed) for 2 min to ensure efficient removal of all buffers.
Discard the previous collection tube. Carefully displace the column into a new 1.5-ml tube. Add 21 μl ddH2O to the center of the column and spin down at 15,000 × g (or maximum speed) for 30 s to collect eluted DNA. Tagmented DNA can be stored at -20°C (in low DNA-binding tube) at this point if not proceeding to the next step immediately.
DNA library PCR amplification and clean-up
Set up PCR reaction in 0.2-ml PCR tubes on ice:
20 μl eluted DNA
2.5 μl 25 μM Nextera i5 primer (from Nextera index kit)
2.5 μl 25 μM Nextera i7 primer (from Nextera index kit)
25 μl 2X NEBNext master mix
Note: If multiple samples will be pooled for sequencing, different combinations of i5 and i7 indexed primers need to be used for each sample. Please refer to specific NGS sequencing platform for recommendation about choice of index combinations. For Illumina sequencing with Nextera index, please refer to Illumina Index Adapters Pooling Guide.
PCR amplification for 10 cycles with the following PCR cycling program (lid set to 105°C):
Step 1. 72°C, 5 min
Step 2. 98°C, 30 s
Step 3. 98°C, 10 s
Step 4. 63°C, 30 s
Step 5. 72°C, 1 min
(Repeat steps 3-5 for a total of 5-10 cycles)
Step 6. 4°C on hold
Note: The optimal number of PCR cycle needs to be optimized for each specific cell line to ensure enough final products while avoiding over-amplification. The cycle number can be optimized by testing a range of different cycle numbers (lowest from 5 cycles to ensure sequencing adaptors attached to DNA fragments) using a small portion of the tagmented DNA. The amplification can be monitored by qPCR or run on an agarose gel (Figure 2).
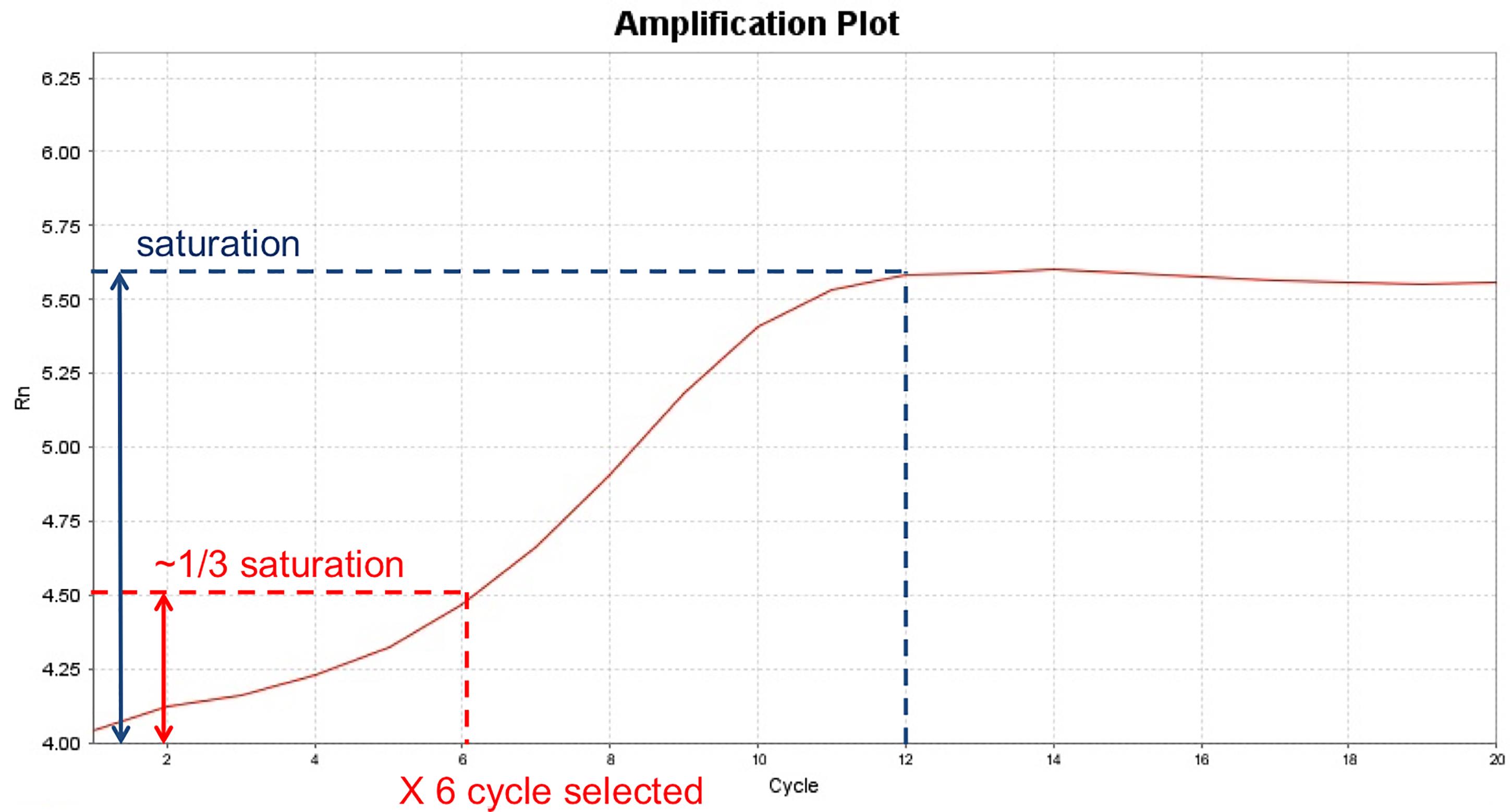
Figure 2. Example of optimizing ATAC-seq PCR cycle number by a qPCR method. Shown here is an example of using qPCR to determine the optimal number of PCR cycles for ATAC-seq library preparation in C4-2 cells. A separate ATAC reaction with the same number of starting materials was set up in parallel to the actual ATAC-seq reaction, and finished Step A2. The qPCR reaction was set up similar to Step A3a, with the addition of 1:400 dilution of SybrGold Dye (Invitrogen S11494), and set up in a qPCR-compatible plate. The qPCR program was set up the same as Step A3b with real-time fluorescence reading for a total of 20 cycles. Upon finishing qPCR, the Rn (normalized reporter) value is plotted against the cycle number and the saturation point (indicated by the dashed blue line) is determined from the plot. From the Rn-Cycle plot, the cycle number is selected when it reaches 1/3 of the saturation signal (indicated by the red dashed line). In this particular example, a PCR cycle number of 6 was selected. If a separate ATAC reaction is not intended, a small portion (for example, 1/16) of the reaction after Step A2 can be used as input for the qPCR reaction; and the remaining reaction can be used for the final PCR amplification.Clean up the 50 μl PCR reaction using a ZYMO DNA Clean and Concentrator Kit (similar to above). Elute in 15 μl H2O. Confirm the size distribution of the amplified library using a Bioanalyzer HS DNA Chip (Figure 3).
Submit ATAC-seq libraries for paired-end Illumina sequencing.
Identify potential eccDNA genomic coordinates using the circle_finder algorithm
Remove the adaptor sequence using the cutadapt program (Martin, 2011) with the following parameters: cutadapt -a ADAPT1 -A ADAPT2 -o out1.fastq -p out2.fastq in1.fastq in2.fastq.
Note: ADAPT1 = CTGTCTCTTATACACATCTCCGAGCCCACGAGAC, ADAPT2 = CTGTCTCTTATACACATCTGACGCTGCCGACGA. in1.fastq and in2.fastq are input fastq files before adaptor removal. out1.fastq and out2.fastq are paired-end fastq files after adaptor removal.
The Circle_finder pipeline first maps the paired-end reads (read length should be >75 bases long) onto the genome (in this case hg38 genome build) using bwa aligner (bwa-mem) (Li, 2013). While mapping paired-end reads to the genome, Circle_finder collects those paired-end reads where one read is mapped in a contiguous manner and the partner read is mapped in a non-contiguous (split-read) manner, supposing one end maps on the body of the circular DNA and the other on the circular DNA ligation junction. Returning to the list of paired-end IDs that mapped uniquely to three sites (one contiguously mapped reads and two-position for reads mapped in a split-read manner) in the genome, the pipeline identifies paired-end IDs where the contiguously mapped read is between the two split reads and on the opposite strand (Figure 1A). The start of the first split read and the end of the second read is annotated as the start and end of the eccDNA. The pipeline (a single script – see next step) to identify eccDNA from paired-end sequencing data of read length >than 75 bases long coming from a specific locus (nonchimeric eccDNA) of any length is available through our GitHub page (refer to software section).
Generate the genome index file: this only needs to be generated once for a specific genome. For example, command to generate an index file for human genome hg38: bwa index hg38.fa.
Note: hg38.fa is the fasta file for the genome of interest (hg38 in this case). This will generate an index under the same name as the genome fasta file name.
Use the bash script with the following logic: #Usage: bash Script_name “Number of processors” “/path-of-whole-genome-file/hg38.fa” “paired-end fastq file 1” “paired-end fastq file 2” “minNonOverlap between two split reads” “Sample name” “genome build”. An example script is shown here (code as a single line):
#bash /path-of-script/directory/microDNA-pipeline-bwa-mem-samblaster.sh 16 /path-of-script-directory/hg38.fa S1_R1.fastq S1_R2.fastq 10 S1 hg38
Note 1: The pipeline takes seven arguments as below.
Argument 1 = “Number of processors”;
Argument 2 = “Genome fasta file” for example “hg38.fa”;
Argument 3 = “paired-end fastq file 1”;
Argument 4 = “paired-end fastq file 2”;
Argument 5 = “minNonOverlap between two split reads”, for example 10;
Argument 6 = “Sample name”, user may choose any name for their sample;
Argument 7 = “genome build”, user may choose their genome build, such as hg38.
Note 2: The chance of identifying eccDNA depends on sufficient sequencing depth and read length to cover the eccDNA-specific junctional sequence. Here, we used around 100 million read pairs with a 150-bp read length on the Illumina HiSeq platform.
The output file from “circle_finder” will be a file named “microDNA-JT.txt,” which contains four columns including chromosome number, start position of eccDNA coordinate, end position of eccDNA coordinate, and number of junctional tags.
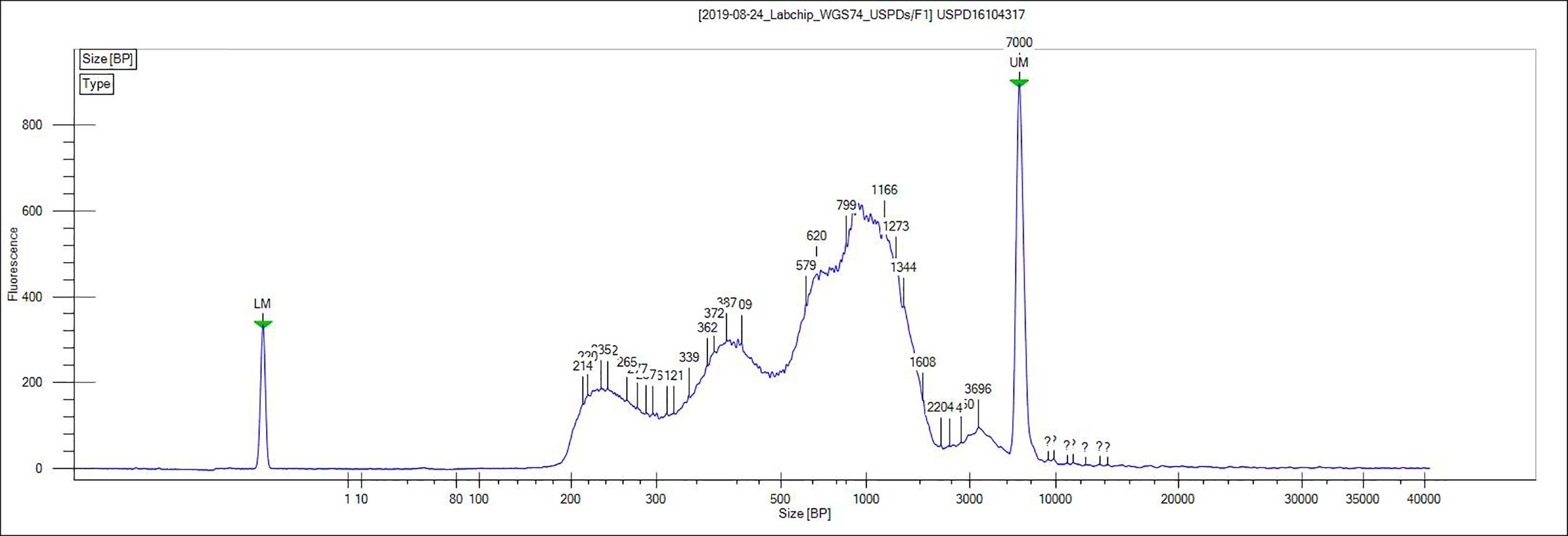
Figure 3. Representative ATAC-seq library size distribution. ATAC-seq was performed on OVCAR8 nuclei. A characteristic ladder distribution is detected by the bioanalyzer due to the nucleosome arrangement on chromatin.
Validation of selective eccDNA by inverse PCR
Note: EccDNA should be treated delicately. EccDNA are prone to shearing and degradation when frozen, vortexed, or kept for long-term storage at a low concentration of DNA.
Culturing and harvesting cells
Harvest 107-108 human cancer cells by trypisinization into a 15-ml tube.
Centrifuge at 300 × g, 4°C for 5 min, remove media by aspiration.
Resuspend cells with 10 ml ice-cold phosphate-buffered saline (PBS).
Centrifuge at 300 × g for 5 min.
Remove PBS by careful aspiration.
Repeat washing steps for a total of two washes, immediately proceed to next step.
EccDNA isolation by plasmid columns
Resuspend cells in 6 ml Buffer P1 from the Qiagen HiSpeed Plasmid Midi Kit.
Note: Add RNase A solution to Buffer P1 prior to experiment and store at 4°C.
Add 6 ml Buffer P2 from the Qiagen HiSpeed Plasmid Midi Kit and mix by gently inverting 4-6 times.
Incubate on the bench at room temperature for 5 min.
Add 6 ml pre-chilled Buffer P3 from the Qiagen HiSpeed Plasmid Midi Kit and mix by gently inverting 4-6 times.
Set up QIAfilter Cartridge from the Qiagen HiSpeed Plasmid Midi Kit by removing the outlet nozzle cap and sitting the cartridge on top of a waste container (such as a 50-ml conical flask). Add the cell lysate (total of 18 ml) into the QIAfilter Cartridge with the cap attached. Incubate at room temperature for 10 min.
While waiting, set up HiSpeed Midi Tip from the Qiagen Plasmid Midi Kit on top of a waste container (such as a 50-ml conical flask) and equilibrate with 4 ml Buffer QBT.
Remove cap from the QIAfilter Cartridge and filter the cell lysate into the HiSpeed Tip by gently inserting the plunger into the cartridge.
After the cell lysate has passed through the HiSpeed Tip, add 10 ml Buffer QC.
After Buffer QC has passed through the HiSpeed Tip, move it to a clean 50-ml conical tube.
Add 5 ml Buffer QF to elute DNA from the HiSpeed Tip.
Add 3.5 ml isopropanol and mix gently by inversion, incubate at room temperature for 5 min.
Attach the QIAprecipitator Module from the Qiagen HiSpeed Plasmid Midi Kit onto a 20-ml syringe after removing the plunger from the syringe.
Add the DNA-isopropanol solution from Step B2k to the syringe and gently push the solution through the QIAprecipitator Module into a waste container.
Remove the 20-ml syringe from the QIAprecipitator Module, pull out the plunger, re-attach the 20-ml syringe to the QIAprecipitator Mocule, add 20 ml 70% ethanol to the syringe and gently push the plunger to wash the QIAprecipitator Module.
Dry the QIAprecipitator Module by pushing air through the module several times until no more liquid can be pushed out. Dry the QIAprecipitator Module outlet with absorbent paper (such as kimwipes).
Detach the Module from the 20-ml syringe. Attach a new 5-ml syringe (without plunger) to the QIAprecipitator Module.
Add 1 ml Buffer TE from the Qiagen HiSpeed Plasmid Midi Kit to the syringe and push through the QIAprecipitator Module with the plunger into a 1.5-ml Eppendorf tube.
Perform ethanol precipitation: Split the 1000 μl DNA solution to 500 μl between two 1.5-ml Eppendorf tubes. Add 1,000 μl 100% ethanol and 1 μg glycogen to each of the tubes. Mix by pipetting up and down gently. Centrifuge in a tabletop centrifuge at 13,000 rpm for 30 min.
Remove supernatant as much as possible with 1,000 μl and 100 μl pipette tips without dislodging the DNA pellet. The pellet should be visible at the bottom of the tube. Air dry for 5 min.
Resuspend the DNA in each tube in 20 μl Buffer TE from the Qiagen HiSpeed Plasmid Midi Kit and combine to one tube (40 μl volume).
Further enrichment of eccDNAs by DNase digestion to remove linear DNA
To 40 μl DNA, add 128 μl ddH2O, 8 μl 25 mM ATP, 20 μl 10× Reaction Buffer (from the Plasmid-safe ATP-dependent DNase Kit), and 4 μl Plasmid-safe ATP-dependent DNase. Incubate at 37°C overnight (10-12 h).
Purify the DNA using the QIAquick PCR Purification Kit by adding 5 volumes of Buffer PB (1,000 μl) to 1 volume of the digested DNA solution (200 μl) and mix gently by inversion. Add the mixture to a QIAquick Spin Column, centrifuge for 1 min at 17,900 × g.
Discard the flowthrough and wash the QIAquick Spin Column with 750 μl Buffer PE (containing ethanol) by centrifugation for 1 min at 17,900 × g. Repeat the wash step for a total of two washes.
Elute the DNA with 50 μl Buffer EB from the QIAquick PCR Purification Kit.
Repeat digestion and purification steps until the DNA concentration no longer decreases. The concentration begins high (>500 ng/μl) and should decrease to a low level (<20 ng/μl).
Note: For this protocol, we perform a total of 2 rounds of digestion and purification. To optimize the number of digestion/purification rounds needed for each specific cell line, DNA concentration should be measured by Qubit before and after each round of digestion/purification. The concentration will stop decreasing after a certain number of digestion/purification rounds, indicating that the contaminating linear DNA has been successfully removed.
Inverse PCR to detect specific eccDNA using an outward-directed primer set
Perform PCR with primers that target the junction sequence of the eccDNA using the KOD Hot-Start PCR Kit.
Add 25 μl Xtreme buffer, 10 μl dNTPs (2 μM each), 1.5 μl each primer (10 pmol/μl), 1 μl KOD Xtreme Hot-Start Polymerase, 200 ng purified DNA, and enough ddH2O for the final solution to be 50 μl.
PCR amplification using a PCR machine with heated lid (lid set to 105°C):
Step 1. 94°C, 2 min
Step 2. 98°C, 10 s
Step 3. 68°C, 1 min/kb
(Repeat Step 2-3 for a total of 30 cycles)
The PCR product can then be visualized on an agarose gel and sequenced by Sanger sequencing with the primers used for PCR amplification (Figure 4).
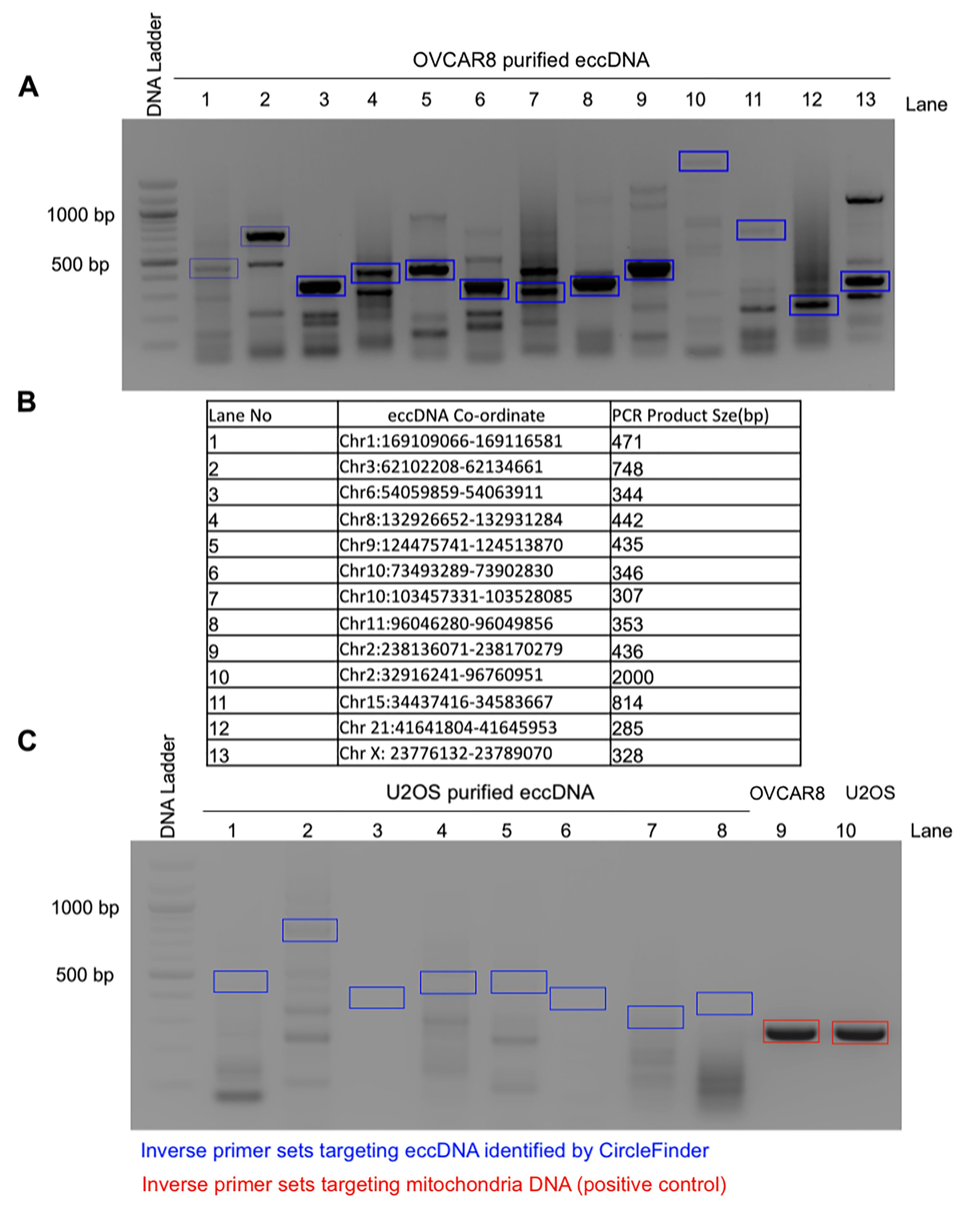
Figure 4. Representative eccDNA identified from the ATAC-seq library and validated by inverse PCR in OVCAR8 cells. A. EccDNAs were amplified using inverse PCR primers (shown in blue boxes), gel purified, and validated for the presence of junctional sequences by Sanger sequencing. Example Sanger sequencing results can be found in Figure 3D of the original manuscript (Kumar et al., 2020). B. The table represents the distribution of eccDNA on different chromosomes with coordinates and their expected PCR product size; the numbers represent the different lanes on the gel. C. As a negative control, the same inverse PCR primers were used on purified eccDNAs from U2OS cells (lanes 1-8). As a positive control, inverse PCR primers against mitochondrial DNA were used on eccDNAs (lanes 9-10).
Metaphase spread and FISH detection of specific circular DNAs
Prepare the cells for metaphase spread
Seed 5 × 105 cells in each of five 100 mm Petri dishes 24 h before thymidine block.
Note: Starting cell number is important to get enough mitotic cells for eccDNA detection.
Add 100 mM thymidine solution to a final concentration of 2 mM to each of the 100 mm plates for 16 h. Release cells for 9 h by replacing regular medium without thymidine solution. Repeat another thymidine block (2 mM final concentration, 16 h) to arrest the cells at the G1-S boundary and release cells for 3 h in regular medium.
Add 0.1 μg/ml final concentration Colcemid for 9 h to collect the mitotic cells.
Note: Check cells under a microscope to confirm the round shaped mitotic cells.
Gently shake off the floating mitotic cells from the culture dish and collect into a 15-ml Falcon tube by centrifuging at 300 × g for 5 min, and wash the pellet twice with cold PBS.
Note: Mitotic cells are fragile, so it is very important to use a low speed during centrifugation.
Gently resuspend the pellet in 5 ml 75 mM KCl Hypotonic Solution and incubate at 37°C in a water bath for 30 min. Invert the tube gently every 10 min to ensure the cells are in suspension. Add 1 ml Carnoy’s Fixative Solution to the cell suspension dropwise using a 1,000 μl pipette tip and mix by inverting the tube slowly to keep the metaphase chromosome intact.
Centrifuge cells at 300 × g for 5 min and carefully aspirate most of the KCl solution, leaving about 300 μl. Resuspend cell pellet by gently tapping on the tube.
Fix the resuspended cells by adding 5 ml ice-cold Carnoy’s Fixative Solution dropwise using a 1,000 μl pipette tip and invert the tube slowly to mix the cells.
Note: It is important to invert the tube slowly to avoid fragmentation of the mitotic chromosomes.
Note: At this stage, fixed cells can be stored at 4°C for several months.
Centrifuge the cells at 300 × g for 5 min and resuspend the pellet gently in 1-2 ml Carnoy’s Fixative Solution.
Humidify the glass slides, putting on a box at 55°C by slanting at a 45-degree angle, and add several drops of fixative cell suspension from 15-20 cm above the slides.
Note: It is very important to humidify the glass slides for proper disruption of the nuclear membrane and also to drop the fixative cell suspension from the above-mentioned height for proper spreading of the mitotic chromosomes.
Dry the slides at room temperature away from light and stain with VectaShield Mounting Medium containing DAPI to view the mitotic chromosome spread under a microscope. The dry slides containing the mitotic spreads can be stored at 4°C for several months.
Denaturation of slides containing metaphase DNA
Pre-warm 100 ml FISH Denaturation Buffer at 73°C for 5 min.
Immerse the glass slides containing metaphase spread in a Coplin jar containing pre-warmed FISH Denaturation Buffer for 5 min.
Immerse the slides in a Coplin jar containing 1× PBS, pH 7.4 for 5 min.
Dehydrate the slides serially by immerging the slides in 70%, 85%, and 100% ethanol for 2 min each.
Air dry the slides until all the ethanol has evaporated.
Probe denaturation and hybridization
Note: It is important to keep the FISH probe protected from light. Aluminium foil is used to cover the slides or hybridization chamber.
Denature the FISH probe in Hybridization Buffer (19 μl Hybridization Buffer + 1 μl labeled probe) at 73°C for 5 min and immediately chill on ice.
Apply the probe mixture onto the previously prepared, air-dried metaphase spread slide and cover with a coverslip. Seal the coverslip with rubber cement and place the slides in the humidified box and hybridize at 37°C in the hybridization chamber overnight. Parafilm is used to ensure sealing of the humidified box.
After hybridization, immerse the slides in 1× PBS and remove the rubber cement and coverslip gently.
Wash the slides in a Coplin jar with pre-warmed FISH Wash Buffer 1 at 73°C for 5 min.
Wash the slides with FISH Wash Buffer 2 at room temperature for 5 min.
Air dry the slides in the dark at room temperature, mount with VectorShield DAPI medium, and seal with nail polish.
Image and data analysis
Capture the images under a confocal microscope with a 63× oil-immersion objective. Set the laser power to output 40% and acquire the image with the full region of interest (512 × 512) at 300 ms exposure times. For the detection of eccDNA, OVCAR8 (experimental cell line) and C4-2 (negative control cell line where we do not see any extrachromosomal signal) were probed against the eccDNA locus Chr2:238136071-238170279 or Chr10:103457331-103528085 (Figure 5). Potential eccDNA signals (indicated by the red arrows) are located off the main chromosomes, while the chromosomal signals overlap with the main chromosomes and usually appear as doublet signals (indicated by the yellow arrows).
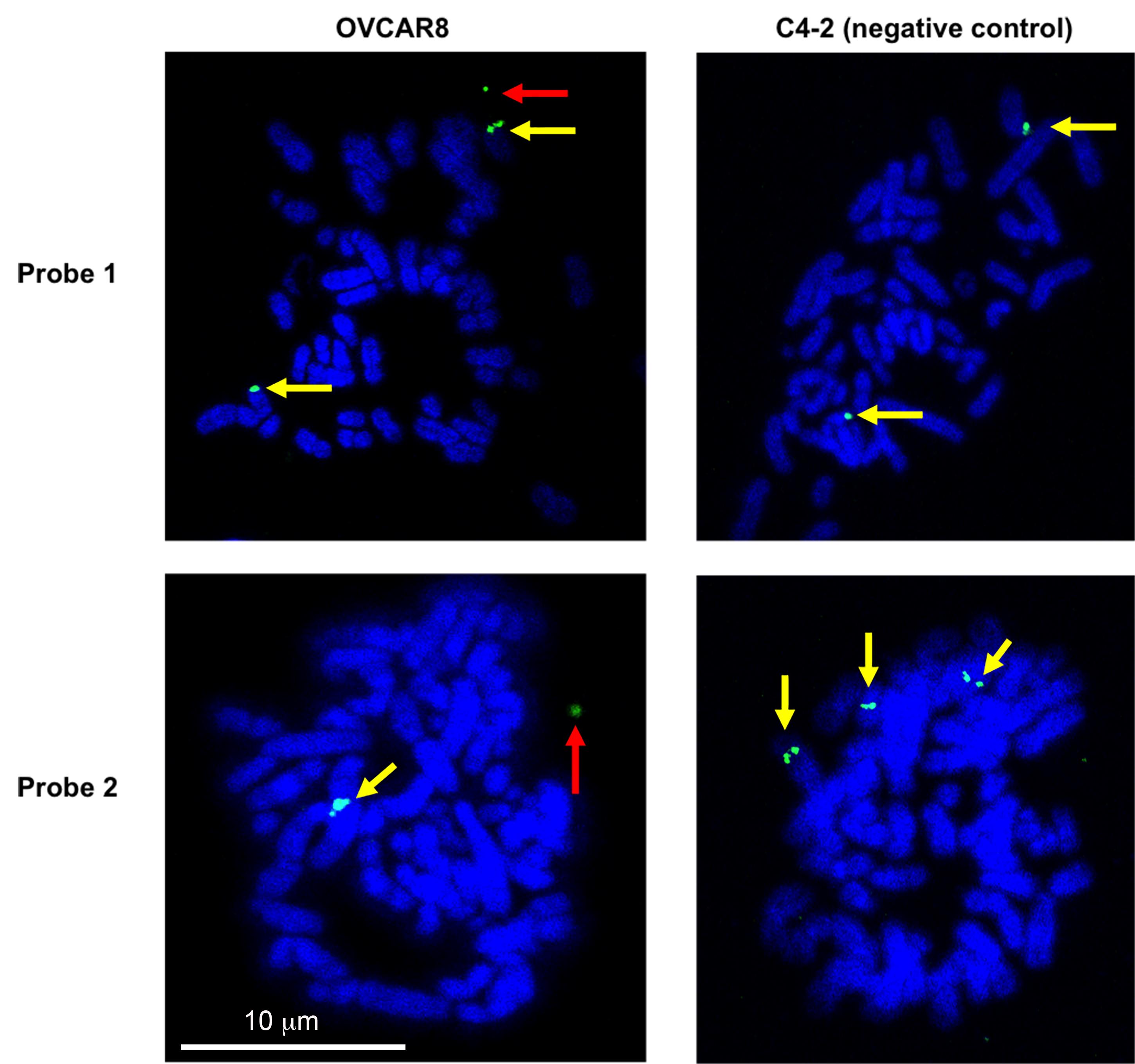
Figure 5. Validation of eccDNA in OVCAR8 cells by metaphase FISH. Metaphase spread of the chromosomes was carried out and the eccDNAs were identified by FISH. A representative eccDNA locus Chr2:238136071-238170279 (top row – probe 1) or Chr10:103457331-103528085 (bottom row – probe 2) was identified from OVCAR8 ATAC-seq and used for specific BAC probe design. The metaphase spreads from C4-2B cells on the left show no extrachromosomal circular DNA (negative control), while the spreads from OVCAR8 cells on the right confirm the presence of an extrachromosomal eccDNA signal (green: BAC probe, blue: DAPI). The red arrow indicates the eccDNA signals (which can be a singlet or a doublet due to replication of the eccDNA). The yellow arrows mark chromosomal DNA signals (which is usually a doublet but can be a singlet because the signal is seen from only a single chromatid).
Data analysis
The OVCAR8 and C4-2B ATAC-seq data has been deposited in Gene Expression Omnibus (accession: GSE145409). Following the Circle-finder algorithm, the identified eccDNA coordinates output can be found in the Supplementary File under the same GEO accession. Hundreds of potential eccDNAs were identified from this dataset, including small circles of less than 1 kb and large circles of 400 kb encompassing several genes [refer to Figure 3 in the original manuscript (Kumar et al., 2020)].
To validate the potential eccDNAs, PCR-based validation was used for 11 eccDNAs from OVCAR8 and 6 eccDNAs from C4-2B. The primer is designed based on the junctional sequence identified from ATAC-seq and will specifically amplify eccDNA but not the genomic DNA (unless tandem duplication). In total, 13 out of 17 eccDNAs were validated by this method [refer to Figure 3 in the original manuscript (Kumar et al., 2020)].
FISH on metaphase spreads can be used to visually validate the presence of selective eccDNAs, but preferentially with large circles. Here, we were able to detect FISH signals off chromosomes corresponding to 34-kb and 71-kb eccDNAs in OVCAR8 cells (Figure 5). To understand the general distribution of eccDNA signals in a cell population, we counted eccDNA FISH signals in more than 20 cells and found a distribution between 0 and 4 eccDNA FISH signals in each cell examined [refer to Figure 4 in the original manuscript (Kumar et al., 2020)].
Recipes
1% (10 mg/ml) Digitonin
40 μl 20 mg/ml digitonin stock solution in DMSO
40 μl ddH2O
Store at -20°C as aliquots, stable for 6 months
ATAC-Resuspension Buffer (ATAC-RSB) (50 ml)
500 μl 1 M Tris-HCl, pH 7.4 (10 mM final concentration)
100 μl 5 M NaCl (10 mM final concentration)
150 μl 1 M MgCl2 (3 mM final concentration)
49.25 ml ddH2O
ATAC-Lysis Buffer (ATAC-LB) (500 μl for 8 reactions)
5 μl 10% Nonidet P40 substitute (0.1% final concentration)
5 μl 10% Tween-20 (0.1% final concentration)
5 μl 1% digitonin (0.01% final concentration)
485 μl ATAC-RSB
Prepare fresh, keep on ice
ATAC-Wash Buffer (ATAC-WB) (10 ml for 8 reactions)
100 μl 10% Tween-20 (0.1% final concentration)
9.9 ml ATAC-RSB
Prepare fresh, keep on ice
ATAC-Reaction Mastermix (ATAC-RM) (450 μl for 8 reactions)
225 μl 2× TDB buffer (from Nextera kit, 25 μl per reaction)
22.5 μl TDE transposase (from Nextera kit, 2.5 μl per reaction)
148.5 μl DPBS
4.5 μl 1% Digitonin (0.01% final concentration)
4.5 μl 10% Tween-20 (0.1% final concentration)
45 μl H2O
Prepare fresh, keep on ice
100 mM Thymidine Solution
Dissolve 242 mg thymidine powder in 10 ml cell culture grade water and filter through a 0.2-μm filter
75 mM KCl Hypotonic Solution
Dissolve 559 mg KCl in 100 ml cell culture grade water and filter through a 0.2-μm filter
Carnoy’s Fixative Solution
Add 75 ml methanol to 25 ml glacial acetic acid (v/v) to make 100 ml fixative solution
Prepare under a chemical fume hood
20× SSC Buffer
Mix 87.5 g NaCl and 44.1 g sodium citrate in 400 ml water and adjust the pH with a few drops of 12 N hydrochloric acid to pH 7.0
Adjust the volume with water to 500 ml and filter through a 0.2-μm filter
Hybridization Buffer
10 ml 20× SSC buffer pH 7.0
50 ml 100% formamide solution
10 g dextran sulfate
Adjust volume to 100 ml with water
Prepare under chemical fume hood
FISH Denaturation Buffer
70 ml 100% formamide solution
10 ml 20× SSC buffer
20 ml sterile water
Prepare under chemical fume hood
FISH Wash Buffer 1
2 ml 20× SSC buffer
3 ml 10% NP-40
95 ml water
FISH Wash Buffer 2
10 ml 20× SSC buffer
1 ml 10% NP-40
89 ml water
Acknowledgments
This work was supported by R01 CA060499 and P30 CA044579 to AD and Cancer Training Grant support from T32 CA009109 (PI: Amy Bouton) to TP. We thank all members of the Dutta Lab for many helpful discussions.
Competing interests
The authors declare no competing interests.
References
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. and Greenleaf, W. J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10(12): 1213-1218.
- Buenrostro, J. D., Wu, B., Chang, H. Y. and Greenleaf, W. J. (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109: 21 29 21-21 29 29.
- Corces, M. R., Mumbach, M. R., Greenleaf, W. J., Montine, T. J., Khavari, P. A., Kundaje, A., Risca, V. I., Orloff, L. A., Kasowski, M., Carter, A. C., Cho, S. W., Trevino, A. E., Kathiria, A., Wu, B., Montine, K. S., Rubin, A. J., Satpathy, A. T., Vesuna, S., Sinnott-Armstrong, N. A., Greenside, P. G., Hamilton, E. G. and Chang, H. Y. (2017). Omni-ATAC-seq: Improved ATAC-seq protocol. Protocol Exchange10.1038/protex.2017.096.
- Dillon, L. W., Kumar, P., Shibata, Y., Wang, Y. H., Willcox, S., Griffith, J. D., Pommier, Y., Takeda, S. and Dutta, A. (2015). Production of Extrachromosomal MicroDNAs Is Linked to Mismatch Repair Pathways and Transcriptional Activity. Cell Rep 11(11): 1749-1759.
- Gresham, D., Usaite, R., Germann, S. M., Lisby, M., Botstein, D. and Regenberg, B. (2010). Adaptation to diverse nitrogen-limited environments by deletion or extrachromosomal element formation of the GAP1 locus. Proc Natl Acad Sci U S A 107(43): 18551-18556.
- Hull, R. M., King, M., Pizza, G., Krueger, F., Vergara, X. and Houseley, J. (2019). Transcription-induced formation of extrachromosomal DNA during yeast ageing. PLoS Biol 17(12): e3000471.
- Kim, H., Nguyen, N. P., Turner, K., Wu, S., Gujar, A. D., Luebeck, J., Liu, J., Deshpande, V., Rajkumar, U., Namburi, S., Amin, S. B., Yi, E., Menghi, F., Schulte, J. H., Henssen, A. G., Chang, H. Y., Beck, C. R., Mischel, P. S., Bafna, V. and Verhaak, R. G. W. (2020). Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet 52(9):891-897.
- Koche, R. P., Rodriguez-Fos, E., Helmsauer, K., Burkert, M., MacArthur, I. C., Maag, J., Chamorro, R., Munoz-Perez, N., Puiggros, M., Dorado Garcia, H., Bei, Y., Roefzaad, C., Bardinet, V., Szymansky, A., Winkler, A., Thole, T., Timme, N., Kasack, K., Fuchs, S., Klironomos, F., Thiessen, N., Blanc, E., Schmelz, K., Kunkele, A., Hundsdorfer, P., Rosswog, C., Theissen, J., Beule, D., Deubzer, H., Sauer, S., Toedling, J., Fischer, M., Hertwig, F., Schwarz, R. F., Eggert, A., Torrents, D., Schulte, J. H. and Henssen, A. G. (2020). Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat Genet 52(1): 29-34.
- Koo, D. H., Molin, W. T., Saski, C. A., Jiang, J., Putta, K., Jugulam, M., Friebe, B. and Gill, B. S. (2018). Extrachromosomal circular DNA-based amplification and transmission of herbicide resistance in crop weed Amaranthus palmeri. Proc Natl Acad Sci U S A 115(13): 3332-3337.
- Kumar, P., Dillon, L. W., Shibata, Y., Jazaeri, A. A., Jones, D. R. and Dutta, A. (2017). Normal and Cancerous Tissues Release Extrachromosomal Circular DNA (eccDNA) into the Circulation. Mol Cancer Res 15(9): 1197-1205.
- Kumar, P., Kiran, S., Saha, S., Su, Z., Paulsen, T., Chatrath, A., Shibata, Y., Shibata, E. and Dutta, A. (2020). ATAC-seq identifies thousands of extrachromosomal circular DNA in cancer and cell lines. Sci Adv 6(20): eaba2489.
- Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997.
- Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads.EMBnet J 17: 10-12.
- Møller, H. D., Larsen, C. E., Parsons, L., Hansen, A. J., Regenberg, B. and Mourier, T. (2016). Formation of extrachromosomal circular DNA from long terminal repeats of retrotransposons in Saccharomyces cerevisiae. G3 (Bethesda) 6(2): 453-462.
- Møller, H. D., Parsons, L., Jorgensen, T. S., Botstein, D. and Regenberg, B. (2015). Extrachromosomal circular DNA is common in yeast. Proc Natl Acad Sci U S A 112(24): E3114-E3122.
- Morton, A. R., Dogan-Artun, N., Faber, Z. J., MacLeod, G., Bartels, C. F., Piazza, M. S., Allan, K. C., Mack, S. C., Wang, X., Gimple, R. C., Wu, Q., Rubin, B. P., Shetty, S., Angers, S., Dirks, P. B., Sallari, R. C., Lupien, M., Rich, J. N. and Scacheri, P. C. (2019). Functional enhancers shape extrachromosomal oncogene amplifications. Cell 179(6): 1330-1341 e13.
- Paulsen, T., Kumar, P., Koseoglu, M. M. and Dutta, A. (2018). Discoveries of extrachromosomal circles of DNA in mormal and tumor cells. Trends Genet 34(4): 270-278.
- Paulsen, T., Shibata, Y., Kumar, P., Dillon, L. and Dutta, A. (2019). Small extrachromosomal circular DNAs, microDNA, produce short regulatory RNAs that suppress gene expression independent of canonical promoters. Nucleic Acids Res 47(9): 4586-4596.
- Shibata, Y., P. Kumar, R. Layer, S. Willcox, J. R. Gagan, J. D. Griffith and A. Dutta. (2012). extrachromosomal microDNAs and chromosomal microdeletions in normal tissues. Science 336(6077): 82-86.
- Sin, S. T. K., Jiang, P., Deng, J., Ji, L., Cheng, S. H., Dutta, A., Leung, T. Y., Chan, K. C. A., Chiu, R. W. K. and Lo, Y. M. D. (2020). Identification and characterization of extrachromosomal circular DNA in maternal plasma. Proc Natl Acad Sci U S A 117(3): 1658-1665.
- Turner, K. M., Deshpande, V., Beyter, D., Koga, T., Rusert, J., Lee, C., Li, B., Arden, K., Ren, B., Nathanson, D. A., Kornblum, H. I., Taylor, M. D., Kaushal, S., Cavenee, W. K., Wechsler-Reya, R., Furnari, F. B., Vandenberg, S. R., Rao, P. N., Wahl, G. M., Bafna, V. and Mischel, P. S. (2017). Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543(7643): 122-125.
- Wu, S., Turner, K. M., Nguyen, N., Raviram, R., Erb, M., Santini, J., Luebeck, J., Rajkumar, U., Diao, Y., Li, B., Zhang, W., Jameson, N., Corces, M. R., Granja, J. M., Chen, X., Coruh, C., Abnousi, A., Houston, J., Ye, Z., Hu, R., Yu, M., Kim, H., Law, J. A., Verhaak, R. G. W., Hu, M., Furnari, F. B., Chang, H. Y., Ren, B., Bafna, V. and Mischel, P. S. (2019). Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 575(7784): 699-703.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Su, Z., Saha, S., Paulsen, T., Kumar, P. and Dutta, A. (2021). ATAC-Seq-based Identification of Extrachromosomal Circular DNA in Mammalian Cells and Its Validation Using Inverse PCR and FISH. Bio-protocol 11(9): e4003. DOI: 10.21769/BioProtoc.4003.
- Kumar, P., Kiran, S., Saha, S., Su, Z., Paulsen, T., Chatrath, A., Shibata, Y., Shibata, E. and Dutta, A. (2020). ATAC-seq identifies thousands of extrachromosomal circular DNA in cancer and cell lines. Sci Adv 6(20): eaba2489.
Category
Cancer Biology > General technique > Genetics > Genome-wide analysis
Cancer Biology > Genome instability & mutation > Cell biology assays > DNA structure and alterations
Molecular Biology > DNA > Extrachromosomal DNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.