- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Click Chemistry for Imaging in-situ Protein Palmitoylation during the Asexual Stages of Plasmodium falciparum
Published: Vol 11, Iss 9, May 5, 2021 DOI: 10.21769/BioProtoc.4002 Views: 5714
Reviewed by: Vasudevan AchuthanRakesh ChatrikhiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Palmitoylation refers to the modification of the cysteine thiols in proteins by fatty acids, most commonly palmitic acid, through ‘thioester bond’ formation. In vivo, palmitoylation of proteins is catalyzed by palmitoyl acyltransferases (PATs or DHHC-PATs). Palmitoylation has recently emerged as a crucial post-translational modification in malarial parasites. The expression and activity of palmitoyl transferases vary across different developmental stages of the malarial parasite’s life cycle. The abundance of palmitoylated proteins at a given stage is a measure of overall PAT activity. The PAT activity can also change in response to external signals or inhibitors. Here, we describe a protocol to ‘image’ palmitoyl-transferase activity during the asexual stages using Click Chemistry and fluorescence microscopy. This method is based on metabolic labeling of a clickable analog of palmitic acid by parasitic cells, followed by CuAAC (Copper-catalyzed Alkyne-Azide Cycloaddition reaction) Click Chemistry to render palmitoylated proteins fluorescent. Fluorescence allows the quantitation of intracellular palmitoylation in parasite cells across various development stages. Using this method, we observed that intracellular palmitoylation increases as the parasite transitions from ring to schizont stages and appears to be most abundant during the schizont stages in Plasmodium falciparum.
Keywords: Protein palmitoylationBackground
Quantitation of the relative abundance of a known palmitoylated protein allows quantitation of intracellular palmitoyl-transferase activity. Palmitoylated proteins can be captured and quantitated using acyl biotinyl exchange chemistry followed by western blotting or mass spectroscopy. Quantitation can also be achieved by in-gel visualization of palmitoylated proteins rendered fluorescent after metabolic labeling and Click Chemistry (Martin, 2013). Previously, radiolabeled fatty acids were used to monitor the in-vivo palmitoylation of proteins; however, such methods do not allow visualization of palmitoylated proteins or imaging of global PAT activity in-situ. The method described here is a modification of procedures that use Click Chemistry with proximity ligation to study palmitoylation of proteins in cells (Hannoush and Arenas-Ramirez, 2009; Gao and Hannoush, 2014). This method has high sensitivity and reproducibility in our hands and can be reliably used to measure in-situ protein palmitoylation or palmitoyl-transferase activity at the ‘single-cell’ level. We have used it to study palmitoylation during different stages of malarial parasites. This method can be applied to the high-throughput screening of inhibitors of palmitoyl-transferases (also called DHHC-PATs; DHHC refers to Aspartate (D), Histidine (H), Histidine (H), Cysteine (C), a motif required for palmitoyl-transferase activity) by adapting the protocol to a multi-well format and using fluorescence as a readout.
Materials and Reagents
Consumables
Costar® 6-well clear TC-treated multiple-well plates (Individually Wrapped, Sterile, catalog number: 3516)
Cell culture dish, 150 mm (BD, catalog number: 353025)
2 ml microcentrifuge tubes (Tarsons, catalog number: 500020)
1.5 ml microfuge tubes (Tarsons, catalog number: 500010)
Glass slides (GEM Microscopy slides, catalog number: 051)
Coverslips (Blue star)
KimwipesTM (Sigma-Aldrich, catalog number: Z671584-1EA)
10 ml serological pipette (Falcon®, catalog number: 357551)
Powder-free gloves
Nail polish
3D7 strain of Plasmodium falciparum (MR4)
Human blood O+ (packed red blood cells) (Rotary Blood Bank)
AlbuMAXTM II (Thermo, catalog number: 11021037)
RPMI Media (Thermo, catalog number: 23400-021)
Chemicals and Reagents
Gentamycin (Sigma, catalog number: G1272-10ML)
DAPI ProLongTM Gold Antifade Mountant (Thermo, catalog number: P36966)
Sorbitol (Sigma, catalog number: S3889-500G)
Giemsa stain (Sigma, catalog number:48900-1L)
DMSO HybrimaxTM (Sigma, catalog number: D2650)
Hypoxanthine (Sigma, catalog number: H9377)
Triton X-100 (Sigma-Aldrich, catalog number: 93443)
Methanol (Sigma, catalog number: 34860)
DPBS or Dulbecco’s Modified PBS (Thermo, catalog number: 14190250)
Bovine Serum Albumin (Sigma, catalog number: A7638)
Glucose HybimaxTM (Sigma-Aldrich, catalog number: G5146)
Sodium bicarbonate/NaHCO3 (Sigma-Aldrich, catalog number: S5761)
Click Chemistry Reagents
17-Octadecynoic acid (Sigma, catalog number: O8382)
Oregon Green 488 Azide (Thermo, catalog number: O10180)
Copper Sulfate/CuSO4 (Sigma-Aldrich, catalog number: 451657)
Tris(2-carboxyethyl) phosphine hydrochloride/TCEP (Sigma-Aldrich, catalog number: C4706)
Stock Solutions
RPMI complete medium (1 L) (see Recipes)
5% Sorbitol solution (0.5 L) (see Recipes)
17-ODA stock solution (see Recipes)
Oregon Green 488 azide solution (see Recipes)
TCEP solution (see Recipes)
CuSO4 solution (see Recipes)
Permeabilization buffer (see Recipes)
Blocking buffer (see Recipes)
Triton X-100 10% solution (see Recipes)
Equipment
-80°C Freezer (Thermo)
Culture hood (Esco Class-II type Bio-safety cabinet)
37°C incubator (Automatic CO2 incubator)
Confocal microscope (Nikon A1)
Orbital shaker
Tabletop centrifuge (Eppendorf, model: 5180R)
Swing-bucket rotor (Eppendorf, catalog number: 5810718007)
Stainless-steel tweezers (Sigma, catalog number: T5915-1E, any good tweezers can be used)
Coplin jars (Eisco Labs, catalog number: BI0108PK3)
Procedure
Principle
The copper-catalyzed alkyne azide cycloaddition (CuAAC) reaction, also known as azide-alkyne Huisgen cycloaddition, is a ‘Click Chemistry’ reaction (Wang et al., 2003). Click Chemistry is a set of simple chemical reactions driven by a high thermodynamic driving force, which proceed to completion at room temperature, usually without the formation of side products. The CuAAC reaction combines an alkyne-tagged molecule with an azide-tagged molecule in the presence of copper (I) to give rise to a 1-4 cycloaddition product via a [3+2] cycloaddition reaction. CuAAC is a bio-orthogonal reaction, which implies no effect of other chemical reactions or metabolites within cells. 17-ODA acts as a mimic of endogenous palmitic acid, but it is ‘clickable’ unlike natural fatty acids (capable of undergoing a click reaction) due to the presence of a terminal alkyne moiety.
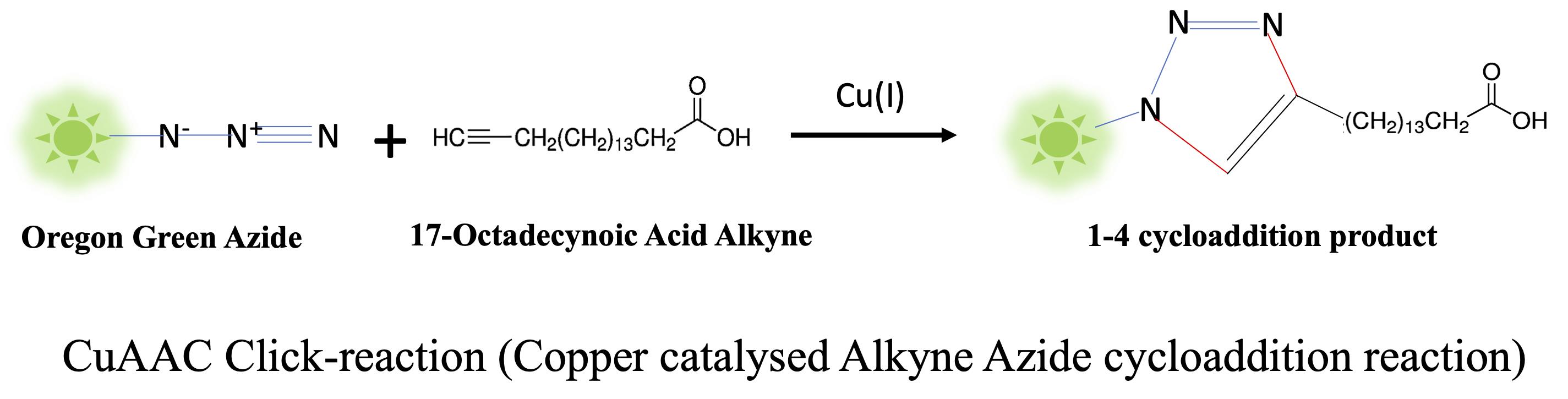
Figure 1. CuAAC click reaction between the fluorescent reporter Oregon Green 488 Azide and 17-Octadecynoic Acid Alkyne (17-ODA). The reaction occurs between the azide and alkyne moieties to give a 1-4 cycloaddition reaction.
DHHC-PATs cannot discriminate between alkyne fatty acids and natural endogenous fatty acids. Hence, they label the intracellular palmitoyl proteins when mimics such as 17-ODA are supplied exogenously in the growth medium (Hannoush and Arenas-Ramirez, 2009; Gao and Hannoush, 2014; Siddiqui et al., 2020).
Parasite culture and metabolic labeling
The 3D7 strain of Plasmodium falciparum is cultured as per the modified method of Trager and Jensen’s method (Trager and Jensen, 1976; Radfar et al., 2009). The culture is started with a synchronized early ring-stage frozen glycerol stock with at least a 10% parasitemia (Parasitemia refers to the number of infected/parasitized red blood cells per 100 red blood cells). The 3D7 strain of Plasmodium falciparum is cultured on O+ human red blood cells (Packed red blood cells obtained from healthy blood donors) in complete RPMI medium (see Recipes), since the malarial parasites grow inside red blood cells. The culture is maintained below 5% parasitemia and 1-2% hematocrit (Hematocrit value is the percentage of red blood cells (v/v) in the culture medium) by splitting the culture and reducing parasitemia when required. The hematocrit is maintained at 1-2% by adding fresh red blood cells to the culture. The parasite is cultured under 5% carbon dioxide or a gas mixture composed of 96% nitrogen, 3% carbon dioxide, and 1% oxygen. The amount of culture required for this experiment is small, and a 150-mm cell culture dish has enough material to obtain parasites for metabolic labeling and Click Chemistry.
The parasite culture is synchronized by sorbitol treatment, which brings about osmotic lysis of the trophozoite and schizont stages of the parasite but spares the early ring stages. The culture suspension is centrifuged at 4,000 rpm for 10 min in an Eppendorf 5180R centrifuge using a swing-bucket rotor. The pellet is treated with a 5% solution of sorbitol in water, usually in 5–10-fold excess of the culture pellet volume, and gently resuspended in sorbitol solution and allowed to incubate for 10-15 min at room temperature. This suspension is centrifuged at 4,000 rpm for 10 min and the supernatant is discarded until a clear solution is obtained.
It is good practice to label distinct homogenous stages of intra-erythrocytic development of the parasite with a clickable fatty acid probe (17-ODA) using sorbitol-synchronized parasite cultures. Sorbitol treatment of a mixed culture eliminates the late trophozoite and schizont stages but spares the ring stages, ensuring a homogenous population of parasites (see Figure 2a-2c).
Note: Sorbitol treatment is most effective when the culture contains a mixed population of rings, trophozoites, and schizonts. See Recipes to make the sorbitol solution.
Giemsa staining of parasite smears allows judgment of the state of synchronization for the parasite culture before and after sorbitol treatment. For labeling, parasites are maintained at a parasitemia of 5% and a hematocrit value of 1%. Parasites at the ring, trophozoite, or schizont stage (42-44 h post-invasion or h.p.i) are incubated in a 6-well plate in complete RPMI medium supplemented with 17-ODA, a clickable surrogate of palmitic acid. The optimal 17-ODA concentration requires optimization. We found 100 µM to be the optimal concentration of 17-ODA that yielded the best labeling of intra-erythrocytic malarial parasites.
Note: Usually, a concentration of 25-50 µM is sufficient for other cell types, but the asexual stages of malarial parasites develop inside red blood cells and may therefore require a higher concentration of 17-ODA in the culture medium for efficient labeling.
The ring/trophozoite/schizont cultures are allowed to incubate at 37°C for 8-10 h in complete RMPI medium supplemented with 17-ODA, which allows metabolic labeling of cellular palmitoyl-proteins with 17-ODA. After incubation, the culture medium and parasitized red blood cells are aspirated from 6-well plate wells, transferred to 2-ml microcentrifuge tubes, and centrifuged at 4,000 rpm in an Eppendorf tabletop centrifuge for 10 min (see Figure 2e).
Note: It is advisable to reserve a well for parasite cells incubated in culture medium lacking 17-ODA. The smears prepared from this well can serve as a control for the specificity of the click reaction.
Smear preparation and sample fixation
The infected and uninfected red blood cells are visible at the bottom of the microcentrifuge tube as a loose pellet (at 1% hematocrit, the total pellet volume is 1 ml packed red blood cells containing 5% infected cells if the parasitemia is 5% in 100 ml culture; 20 µl in 2 ml). The supernatant is aspirated using a pipette and discarded, and the pellet is washed twice with DPBS (Dulbecco’s Modified PBS). The culture pellet is gently resuspended in RPMI (1:1 v/v; pellet: RPMI); 10-20 µl suspension is used to make smears (see Figure 2e).
Note: Smears should also be made from uninfected red blood cells incubated in 17-ODA-supplemented medium to observe the background signal due to fluorescence.
Preparing thin smears ensures an even distribution of cells. The slides are marked accordingly to indicate the sample, and allowed to air dry inside a laminar flow biosafety hood for 20-30 min. Over-drying of smears is not recommended and once dried, smears are fixed using chilled methanol in a Coplin jar for 10-15 min. The slides are removed after 15 min and air-dried again.
The smears are placed into small Petri dishes and washed three times with 1× PBS while shaking gently. The smears are washed again with 0.1% Triton X-100 in PBS for permeabilization. The permeabilized, fixed smears are now ready for Click Chemistry to be performed.
In-situ Click Chemistry in malarial parasite cells
The Click reaction with Oregon Green 488 Azide proceeds in the dark at room temperature. Metabolic labeling with 17-ODA labels the parasite proteins with 17-ODA due to DHHC-PAT activity (Siddiqui et al., 2020). The CuAAC click reaction causes chemo-selective ligation between chemically complementary azide and alkyne functions, since the fatty acid probe (17-ODA) is thio-esterified to palmitoyl proteins (Figure 2f, reaction is shown in the box) and alkyne function is available to undergo a click reaction. A click reaction combines the fluorescent reporter Oregon Green 488 Azide with the alkyne function of 17-ODA and renders 17-ODA-labeled palmitoyl proteins fluorescent (Figure 2f).
The click reaction cocktail is prepared as follows. For 100 μl reaction cocktail, add the following components sequentially to 94 μl filtered PBS: 2 μl Oregon Green 488 Azide (Invitrogen, final concentration of 0.1 mM), 2 μl 0.1 mM Tris (2-carboxyethyl) phosphine (TCEP) solution, and 2 μl 0.1 mM CuSO4 solution. The click reaction cocktail should cover the entire surface of the smear.

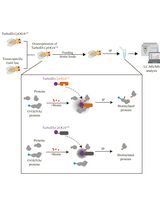
Figure 2. Layout of the protocol. (a). Asexual stages of the parasite are allowed to grow on human blood in complete RPMI medium. (b). Parasite cultures are subjected to centrifugation. (c). Parasite culture pellets are treated with sorbitol to synchronize cultures. (d). Synchronized cultures are allowed to grow in complete RPMI medium supplemented with 17-ODA. (e). Cultures are centrifuged gently to obtain a loose pellet, which is then resuspended to make smears. (f). Smears are processed and Click Chemistry is performed (the box in blue shows the click reaction between 17-ODA-labeled parasite proteins and Oregon Green 488 Azide). (g). Confocal imaging of smears for each parasite stage is performed and raw data are acquired. (h). The raw data are processed and analyzed to quantitate the fluorescence in different asexual stages of the parasite.Notes:
This image was created using BioRender.
The sequence in which reagents for the click reaction are added is crucial and should be adhered to during the reaction. Always prepare fresh TCEP and CuSO4 for each experiment and use immediately.
Each slide is placed in a small Petri dish for incubation, with two small pieces of tissue paper or cotton soaked in water placed alongside to retain moisture and prevent drying of the smears during the incubation period.
Oregon Green 488 Azide is fluorescent and should be protected from light; therefore, the reaction is allowed to proceed for 40 min in the dark at room temperature.
After incubation, each slide is washed with 10-15 ml filtered PBS in a Petri dish with gentle rotation. This is performed two or three times to remove any ingredients of the click reaction that are non-specifically bound to the smear/slide surface. In particular, non-specific binding of Oregon Green 488 Azide to the smear or slide surface may lead to a high background fluorescence and must be removed by gentle and repeated washing.
The excess liquid on the slide surface should be aspirated using a pipette, and the remaining moisture should be wiped with KimwipesTM without touching or abrading the smear.
Slide mounting and fixation
Wear powder-free gloves when touching the coverslips or slides from this stage onward. The slides are mounted with DAPI ProLongTM Gold Antifade Mountant; 20-30 µl is poured on the smear and a coverslip gently placed on top, which should be pressed gently on the smear to avoid the formation of air bubbles.
Note: It is advisable to use tweezers to hold the coverslips. Coverslips should be immersed in 70% ethanol overnight to render them sterile and dirt-free. Coverslips tend to stick together; make sure only one coverslip is picked up.
The excess mountant is gently wiped off the slide using KimwipesTM, and the edges of the coverslips are sealed to the slide surface using nail polish. The nail polish is allowed to dry and slides are then stored in the dark at 4°C until image acquisition.
Note: It is recommended that the image acquisition be performed within a day or two to obtain the best results.
Data acquisition and analysis
Any confocal microscope can be used for capture and acquisition of fluorescence.
The raw data are processed using the NIS image analysis software. Since the fluorescence due to Oregon Green 488 Azide corresponds to palmitoylated proteins, fluorescence is a direct measure of palmitoyl-transferase activity at a given stage.
It is recommended to score a minimum of 10-20 parasite cells corresponding to each stage (rings, trophozoite, and schizont) for each experimental replicate. Data from three experimental replicates are required to obtain a mean fluorescence value with standard deviation and statistical significance.
Fluorescence intensity for each cell is determined by drawing an ROI (Region of Interest) around each fluorescent cell using the NIS image analysis software toolbox and measuring the fluorescence intensity. Such an analysis is also possible using the open source software, ImageJ or Fiji.
Note: It is essential to use the same ROI to measure the fluorescence intensity for each cell from a replicate.
Fluorescence intensities for cells from a given stage are required to calculate the mean fluorescence intensity and standard deviation. However, it is important to note that each cell’s fluorescence intensity should be corrected by subtracting the average background fluorescence intensity.
For this purpose, an average background fluorescence intensity is calculated by measuring the fluorescence intensities of uninfected red blood cells (labeled with 17-ODA).
Corrected fluorescence intensity is determined by subtracting the average background from the fluorescence intensity of each cell.
Note: The average background fluorescence intensity may vary among experiments and replicates; therefore, it is recommended to use the background fluorescence value for a given sample/slide to calculate the corrected fluorescence values for cells on the same slide/smear.
We calculated the average fluorescence intensity across the various asexual stages of the parasite and found that palmitoylation increases as the parasite transitions from the ring to the schizont stage (see Figure 3). The highest fluorescence is observed for the schizont stages of Plasmodium falciparum, which suggests that PAT activity is highest during this stage (Figure 3A).
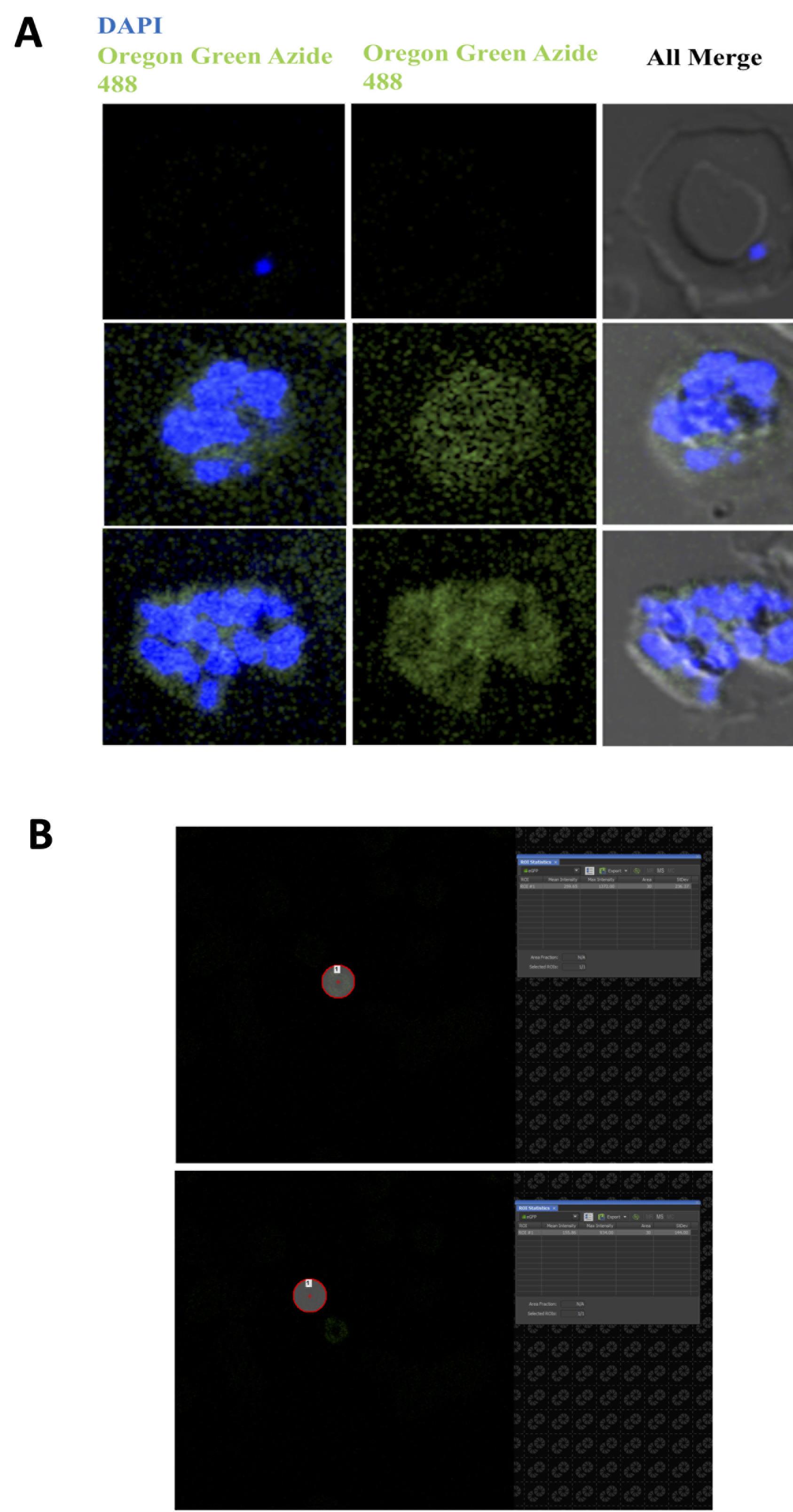
Figure 3. Images of intra-erythrocytic parasites rendered fluorescent after Click Chemistry. A. Metabolic labeling of the ring, trophozoite, and schizont stages with 17-ODA, followed by CuAAC Click Chemistry to render palmitoylated proteins fluorescent using Oregon Green 488 Azide. B. Representative images showing an ROI drawn around the parasite to measure the fluorescence intensity in uninfected and infected red blood cells.
Recipes
RPMI complete medium (1 L)
Measure the following components and mix:
15.87 g RPMI-1640
2 g NaHCO3
2 g glucose
5 g Albumax II
0.0250 g hypoxanthine
1 ml (10 mg/ml) gentamycin
Add autoclaved or double-distilled water to 1 L
Filter sterilize (0.22 μm) and store at 4°C
Before use, warm to 37°C
5% (w/v) Sorbitol solution (0.5 L)
Weigh 25 g sorbitol
Add double-distilled water to a final volume of 0.5 L
Stir and filter sterilize (0.22 μm)
Before use, warm to 37°C
17-ODA stock solution
Dissolve 5 mg lyophilized 17-ODA powder in 178.57 µl DMSO to a concentration of 100 mM in the original vial containing 17-ODA
Transfer to a sterile 1.5-ml microfuge tube. Sonicate for 2-3 min in a bath-sonicator to effectively dissolve the powder. Do not over sonicate
Aliquots of 10 µl can be stored in a -80°C freezer for up to 6 months
Notes:
Avoid freeze-thaw cycles of the fatty acid probe solution, as this may adversely affect the quality of this reagent. Once thawed, an aliquot should not be reused or refrozen. It is always good to prepare fresh 17-ODA for labeling cells.
Add the appropriate volume of 17-ODA stock to complete RPMI medium prewarmed to 37°C. Allow 1-2 h for precomplexing with serum albumin.
Oregon Green 488 azide solution
Dissolve 0.5 mg Oregon Green 488 azide in 156.8 μl DMSO (to a final concentration of 5 mM) in a 1.5-ml microcentrifuge tube
Sonicate for 5 min, divide the solution into aliquots (10 μl per tube) and store at -80°C
Oregon Green 488 azide is light-sensitive and should be stored protected from light. Avoid freeze-thaw of its stock
TCEP solution
TCEP is always prepared fresh and used immediately
Dissolve 7.2 mg TCEP in 0.5 ml Milli-Q purified water (to a final concentration of 50 mM) in a 1.5-ml microfuge tube
Vortex the mixture to dissolve TCEP
CuSO4 solution
CuSO4 is always prepared fresh and used immediately
Dissolve 79.8 mg CuSO4 in 10 ml Milli-Q purified water (to a final concentration of 50 mM) in a 15-ml conical centrifuge tube
Vortex the mixture to dissolve CuSO4
Permeabilization buffer
Add 10 ml 10% (v/v) Triton X-100 (to a final concentration of 0.1% (v/v)) to 990 ml PBS
Stir the solution with a stir bar to dissolve Triton X-100
Use a pH meter to verify the pH (7.2 ± 0.1)
The buffer should be filtered through a 0.2-µm filter and can be stored at room temperature for up to 2 months
Blocking buffer
Add 25 g BSA (to a final concentration of 5% (w/v)) and 15 ml 10% (v/v) Triton X-100 (to a final concentration of 0.3% (v/v)) to 485 ml PBS
Stir the solution with a stir bar to dissolve BSA and Triton X-100. Avoid frothing
The pH should be 7.2. Prepare fresh each time
Triton X-100 10% solution
Add 10 ml Triton X-100 to 90 ml PBS (1×) and stir gently until a homogenous solution is obtained. Use a wide-bore tip to aspirate Triton X-100 from the bottle
Acknowledgments
The author wishes to acknowledge the fellowship support from CSIR, Government of India. Thanks also to the Rotary Blood bank for providing the O+ blood used for culturing malarial parasites and to the confocal facility at ICGEB. This protocol is modified for labeling malarial parasites from the original protocol described in Gao, X. and Hannoush, R. N. (2014). Single-cell in situ imaging of palmitoylation in fatty-acylated proteins.Nat Protoc 9(11): 2607-2623.
Competing interests
The author declares no competing interests.
References
- Martin, B. R. (2013). Nonradioactive analysis of dynamic protein palmitoylation. Curr Protoc Protein Sci 73: 14 15 11-14 15 19.
- Hannoush, R. N. and Arenas-Ramirez, N. (2009). Imaging the lipidome: omega-alkynyl fatty acids for detection and cellular visualization of lipid-modified proteins. ACS Chem Biol 4(7): 581-587.
- Gao, X. and Hannoush, R. N. (2014). Single-cell in situ imaging of palmitoylation in fatty-acylated proteins. Nat Protoc 9(11): 2607-2623.
- Wang, Q., Chan, T. R., Hilgraf, R., Fokin, V. V., Sharpless, K. B. and Finn, M. G. (2003). Bioconjugation by copper(I)-catalyzed azide-alkyne [3+2] cycloaddition. J Am Chem Soc 125(11): 3192-3193.
- Siddiqui, M. A., Singh, S., Malhotra, P. and Chitnis, C. E. (2020). Protein S-palmitoylation is responsive to external signals and plays a regulatory role in microneme secretion in Plasmodium falciparum merozoites. ACS Infect Dis 6(3): 379-392.
- Trager, W. and Jensen, J. B. (1976). Human malaria parasites in continuous culture. Science 193(4254): 673-675.
- Radfar, A., Meńdez, D., Moneriz, C., Linares, M., Marín-García, P., Puyet, A., Diez, A. and Bautista, J. M. (2009). Synchronous culture of Plasmodium falciparum at high parasitemia levels. Nat Protoc 4(12): 1899-1915.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Siddiqui, M. A. (2021). Click Chemistry for Imaging in-situ Protein Palmitoylation during the Asexual Stages of Plasmodium falciparum. Bio-protocol 11(9): e4002. DOI: 10.21769/BioProtoc.4002.
Category
Biochemistry > Protein > Posttranslational modification
Microbiology > Microbial biochemistry > Lipid
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.