- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Transfection and Activation of CofActor, a Light and Stress Gated Optogenetic Tool, in Primary Hippocampal Neuron Cultures
Published: Vol 11, Iss 8, Apr 20, 2021 DOI: 10.21769/BioProtoc.3990 Views: 3554
Reviewed by: Naushaba HasinAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2020
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Proteins involved in neurodegeneration can be coupled with optogenetic reagents to create rapid and sensitive reporters to provide insight into the biochemical processes that mediate the progression of neurodegenerative disorders, including Alzheimer’s Disease (AD). We have recently developed a novel optically-responsive tool (the ‘CofActor’ system) that couples cofilin and actin (key players in early stage cytoskeletal abnormalities associated with neurodegenerative disorders) with light-gated optogenetic proteins to provide spatial and temporal resolution of oxidative and energetic stress-dependent biochemical events. In contrast to currently available small-molecule based biosensors for monitoring changes in the redox environment of the cell, CofActor is a light-activated, genetically encoded redox sensor that can be activated with precise spatial and temporal control. Here we describe a protocol for the expression and activation of the CofActor system in dissociated hippocampal neuron cultures prepared from newborn mice. Cultures were transfected with Lipofectamine on the fifth day in vitro (DIV5), then exposed to cellular stress inducing stimuli, leading to the formation of actin-cofilin rods that can be observed using live cell imaging techniques. The protocol described here allows for studies of stress-related cytoskeletal dysregulation in live neurons exposed to neurodegenerative stimuli, such as toxic Aβ42 oligomers. Moreover, expression of the sensor in neurons isolated from transgenic mouse models of AD and/or mice KO for proteins involved in AD can advance our understanding of the molecular basis of early cytoskeletal dysfunctions associated with neurodegeneration.
Keywords: Cofilin-actin rodsBackground
The biochemical hallmarks of neurodegenerative disorders (neural fibrils, clumps and tangles, heightened reactive oxygen species (ROS)), have presented numerous obstacles to clinicians as potential diagnostic tools (Kanazawa, 2001; Lee et al., 2001; Polidori et al., 2007; McKhann et al., 2011; López-Otín et al., 2013; Laske et al., 2015; Sweeney et al., 2017). These markers, which include Aβ fibrils and tau tangles in Alzheimer’s Disease (AD), are only accessible via an invasive cerebrospinal fluid assay for peptide mass fingerprinting or post mortem diagnosis (Blennow and Zetterberg, 2009; Anoop et al., 2010; Johansson et al., 2011; Kitamura et al., 2017), ROS can be fleeting and thus challenging to monitor in vivo (Halliwell and Whiteman, 2004; Owusu-Ansah et al., 2008; Kalyanaraman et al., 2012; Wang et al., 2013). An alternative approach, however, is to incorporate these disease-related biochemical elements into pathology-sensitive genetically encoded switches. Such switches enable rapid characterization of critical, early stage events in neurodegeneration using cellular and animal models of disease (Ganesh et al., 2016). Researchers are currently faced with limited options for studying the formation of cofilin-actin rods (a form of cytoskeletal dysregulation that occurs in oxidatively and energetically stressed neurons during the progression of neurodegenerative disease) in living cells. Endogenous populations of cofilin and actin can be coaxed into forming rods through various chemical treatments of cells (azide, glutamate, etc.), but these methods require cell fixation and immunostaining for later characterization (Minamide et al., 2010). Furthermore, isolation and extracellular characterization of cofilin-actin rods is challenging, as they are not stable in the presence of detergents (Bamburg and Bernstein, 2016). Overexpression of cofilin, in particular cofilin-GFP, provides a more reliable means of monitoring rod formation, but is hampered by the tendency of overexpressed cofilin to spontaneously form cofilin-actin rods in the absence of cellular stress (Kim et al., 2009). Most recently, a mutant of cofilin, cofilin R21Q, was combined with red fluorescent protein (CofR21Q-mRFP), and used to monitor the induction of cofilin-actin rods in real time (Mi et al., 2013). This reagent represents an important advance in the study of rod formation and longevity. However, there are drawbacks to its implementation, including irreversibility in the presence of energetic stress and a long half-life (30-60 min) once the cell culture medium is restored to homeostasis. We recently reported the CofActor system, which capitalizes on the formation of cofilin-actin rods, by coupling cofilin and actin to the light-responsive Cry2/Cib protein pair (Salem et al., 2020). The CofActor optogenetic system enables the induction of cofilin-actin rods rapidly and reversibly in response to light and stress. We have also created gain-of-function CofActor mutants (actin S14V) that induce rods in the absence of harsh manipulation of the glycolytic pathway and oxidative environment of the cell. We have previously demonstrated that the CofActor system responds to elevated stress levels in immortalized cell lines and in cultured hippocampal neurons from mice. Rodent neuron cultures originating from different brain areas (cortex, hippocampus, striatum, dorsal root ganglia and retina) have been established and used extensively in studies of protein-protein interactions and localization (Kaech and Banker, 2006). In this protocol, we describe in detail the procedures required to transfect and manipulate an optogenetic tool we recently developed (CofActor system) in dissociated hippocampal neuron cultures, prepared from newborn mice. The procedures described here can also be used for preparation of postnatal rodent neuron cultures from other brain areas, such as the somatosensory cortex and can be applied for expression of other optogenetic tools with low toxicity for neurons. Dissociated hippocampal neuron cultures from C57BL/6 newborn mice were prepared according to a modified standard protocol (Szatmari et al., 2005; Yang et al., 2017). The number of mice from a litter that is used for culture preparation depends on the number of planned experimental conditions and replicates. Using the described method, the CofActor system and other optogenetic tools can be applied to studies of neuronal processes associated with synaptic plasticity and intracellular transport.
Materials and Reagents
Tissue culture dishes-35 mm (VWR, catalog number: 10861-656)
Tissue culture dishes-60 mm (VWR, catalog number: 10861-658)
#1.5 thickness round cover glass (VWR, catalog number: 72230-01)
Serological Pipet, Sterile 2 ml (Fisher Scientific, catalog number: 13-675-17)
Serological Pipet, Sterile 5 ml (Fisher Scientific, catalog number: 13-675-22 )
Serological Pipet, Sterile 10 ml (Fisher Scientific, catalog number: 13-675-17)
Serological Pipet, Sterile 25 ml (Fisher Scientific, catalog number: 13-675-30)
Conical centrifuge tube, 15 ml (Fisher Scientific, catalog number: 12-565-269)
Conical centrifuge tube, 50 ml (Fisher Scientific, catalog number: 12-565-270 )
Sterile syringe filters with 0.45 µm pore size (VWR, catalog number: 28145-505 )
Sterile bottle filters with 0.8 μm pore size (Fisher Scientific, catalog number: 09-740-1C)
10 ml sterile syringes (Fisher Scientific, catalog number: 14-817-54 )
C57BL/6 newborn mice (postnatal day 0-1; P0-P1)
Na2SO4 (Fisher, catalog number: S421), stored at room temperature (RT)
K2SO4 (Avantor, catalog number: 3282), stored at RT
MgCl2 (Fisher, catalog number: AM9530G), stored at RT
HEPES (Gibco, catalog number: 15630080 ), stored at 4 °C
Glucose (Gibco, catalog number: 15023-021 ), stored at RT
0.5% Phenol Red (Sigma, catalog number: P0290-100ml ), stored at RT
NaOH (Fisher, catalog number: SS266-1), stored at RT
Kynurenic acid (Sigma, catalog number: K3375 ), stored at RT
Laminin from mouse (Corning, catalog number: 354232 ), stored at -80 °C
Poly-D-Lysine (Gibco, catalog number: A3890401), stored at 4 °C
Pen/Strep (Thermo Fisher, catalog number: 15140122 ), stored at -20 °C
GlutaMAX (Gibco, catalog number: 35050-061 ), stored at RT
Bovine Calf Serum (BCS), heat inactivated (Gibco, catalog number: 16030074 ), stored at -20 °C
L-(+)-Cysteine Hydrochloride (Thermo Fisher, catalog number: C562-25), stored at RT
Papain latex (Worthington Biochemical Corporation, catalog number: LS003126 ), stored at 4 °C
Trypsin Inhibitor from chicken egg white (Millipore Sigma, catalog number: T2011), stored at 4 °C
Basal Medium Eagle (BME; Gibco, catalog number: 21-010-046), stored at 4 °C
Neurobasal A (Gibco, catalog number: 10888022), stored at 4 °C
B27 plus (Gibco, catalog number: 17504044 ), stored at -20 °C
70% Ethanol (VWR, catalog number: 97064-768 )
ActinS14V.CIB.GFP plasmid construct (Addgene, Plasmid #133620), stored at 4 °C
Cry2PHR-mCh-Cof plasmid construct (Addgene, Plasmid #133619), stored at 4 °C
Lipofectamine 2000 (Thermo Fisher, catalog number: 12566014 ), stored at 4 °C
Sodium Azide (Fisher Scientific, catalog number: 26628-22-8), stored at RT
2-Deoxy-D-glucose (DDG) (Fisher Scientific, catalog number: 154-17-6) stored at RT
Opti-MEM (Thermo Fisher, catalog number: 31985062 ), stored at 4 °C
Tissue Culture water (TC water; autoclaved Milli-Q water), stored at RT
Ice (for keeping working solutions at 4 °C)
Stock Solutions (see Recipes)
Stock Dissociation medium, stored at 4 °C
10× Ky.Mg stock, stored at -20 °C
Stock coating solutions (PDL stock, stored at -20 °C and laminin stock stored at -80 °C)
1½ solution, stored at -20 °C
Cysteine.HCl stock solution, stored at -20 °C
2.5 M Glucose solution, stored at 4 °C
Working Solutions (see Recipes)
Working dissociation medium (DM/Ky.Mg) – prepare 100 ml/culture
Growth medium with serum (sGM) – for washing (prepare 50 ml/culture) and plating (needs 2 ml/35 mm culture dish)
Serum-free Neurobasal A-B27 growth medium (GM) – needs 2 ml/35 mm culture dish
Optimem/Glucose – needs 50 ml/culture
Enzyme (Papain) Solution – needs 10 ml/culture
Trypsin Inhibitor solution – needs 10 ml/culture
Transfection medium (TM) – needs 200 µl/35 mm culture dish
Working PDL solution
Laminin-PDL coating solution
Equipment
Large metal scissors (Fine Science Tools, catalog number: 14001-18 )
Small (Bonn) scissors (Fine Science Tools, catalog number: 14184-09)
Vannas Spring Scissors (Fine Science Tools, catalog number: 15000-00 )
Forceps (Roboz, catalog number: RS-5050 )
Curved Forceps (Fine Scientific, catalog number: 91197-00 )
Spatula (Fine Science Tools, catalog number: 10090-13 )
P20 PIPETMAN (Fisher Scientific, catalog number: F123600G)
P200 PIPETMAN (Fisher Scientific, catalog number: F123601G)
P1000 PIPETMAN (Fisher Scientific, catalog number: F123602G)
Drummond Pipet-Aid (Fisher Scientific, catalog number: 13-681-08)
4 °C refrigerator
Laxco Zoom Stereo Zoom Microscope, 0.67 × 4.5 × zoom (Fisher Scientific, catalog number: Z220PLS200L)
Laxco iLED Series LED Light Source (Fisher Scientific, catalog number: AMPSILED22)
Water bath, 37 °C (VWR, catalog number: WB02)
Metallic beads for water bath (VWR, catalog number: 10158-552)
Biological safety cabinet (Fisher Scientific, catalog number: 13-261-334)
Water jacketed CO2 tissue culture incubator set to 5% CO2 and 37 °C (Fisher Scientific, catalog number: 11-676-600)
Tissue culture microscope, such as EVOS Floid Cell imaging station (Fisher Scientific, catalog number: 4471136)
Swing bucket centrifuge and rotor (Fisher Scientific, catalog number: 75-410-885)
pH meter (Fisher Scientific, catalog number: 13-636-AE153)
Keyence (BZ-X800) digital microscope for live cell imaging
Theraband (red, Superior Medical Equipment, catalog number: H20130)
Sterilization pouches (Santa Cruz Biotechnology, catalog number: SC-363641)
Software
Fiji (Schindelin et al., 2012) equipped with the Bio-Formats package (Linkert et al., 2010)
GraphPad Prism (Version 8.0 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com)
Procedure
Prepare 5-6 days in advance
Cover glass cleaning and sterilizing procedure: wash the cover glass for 2 days in 100% ethanol, then repeat the wash for 2 days in Milli-Q water; dry cover glasses individually on filter paper, then place in a small glass beaker, cover with aluminum foil, then autoclave; store at RT.
Check the sharpness of scissors used for decapitation by making a cut on red Theraband band.
Autoclave dissection tools individually in sterilization pouches.
Prepare 24 h prior culture preparation
Place 4 micro cover glasses in each tissue culture plate and coat plates with laminin-PDL coating solution (use 1 ml/35 mm dish) for at least 24 h (48 h is better) before use and place them in the water jacketed CO2 incubator.
Remove the coating solution and wash the plates on the day of the culture twice with TC water (autoclaved Milli-Q water); then let them dry under the hood (if more plates are made than are needed for the culture; wrap them in parafilm and store in a 4 °C refrigerator for up to one month after coating). If using previously-coated plates stored in the 4 °C refrigerator, bring plates to RT by placing them in the biological safety cabinet; to further minimize the possibility of contamination, UV sterilize opened plates in biological safety cabinet for 15 min.
Prepare on the day of culture
Prepare working Dissociation Medium (DM/Ky.Mg). You will need a minimum of 60 ml per culture (store at 4°C).
Prepare fresh Growth Medium with serum (sGM) before starting the neuron culture procedure.
Prepare fresh Growth Medium (GM) right before starting the neuron culture procedure.
Prepare Enzyme (Papain) solution right before beginning culture procedure.
Prepare Trypsin Inhibitor solution right before beginning culture procedure.
Macro-dissection (Figures 1 and 2)
Run UV in the biological safety cabinet for 15 min.
Wipe the dissection table and dissection microscopes with 70% ethanol, then place a small biohazard bag and tissue paper on the dissection table.
Place autoclaved dissection tools in the biological safety cabinet.
Pipette 5 ml of DM/Ky.Mg in a 60 mm Petri dish and place dish on ice.
Pipette 2 ml of DM/Ky.Mg in a 35 mm Petri dish and keep this dish on ice as well.
Bring newborn mice (postnatal day 0-1; P0-P1) to tissue culture room in approved containers.
Note: The number of mice used for culture depends on the size of litter and the planned number of experimental conditions and the number of repeats.
Spray the mouse with ethanol, then rapidly euthanize by decapitation.
Using a scalpel, expose the skull by making an incision from the nasal bone to the occipital surface.
Once the skull is exposed, use scissors or scalpel to make a T-shaped cut along the nasal bone and midline.
Cut the skull from the starting between the eyes on the nasal bone to the occipital bone along the sagittal suture.
Gently remove the parietal and interparietal bones with forceps from both sides.
The brain should then become visible; use extreme caution to avoid damaging the brain tissue when removing the bone.
Use the curved spatula to isolate the brain and place it immediately into the 60 mm Petri dish with ice cold DM/Ky.Mg.
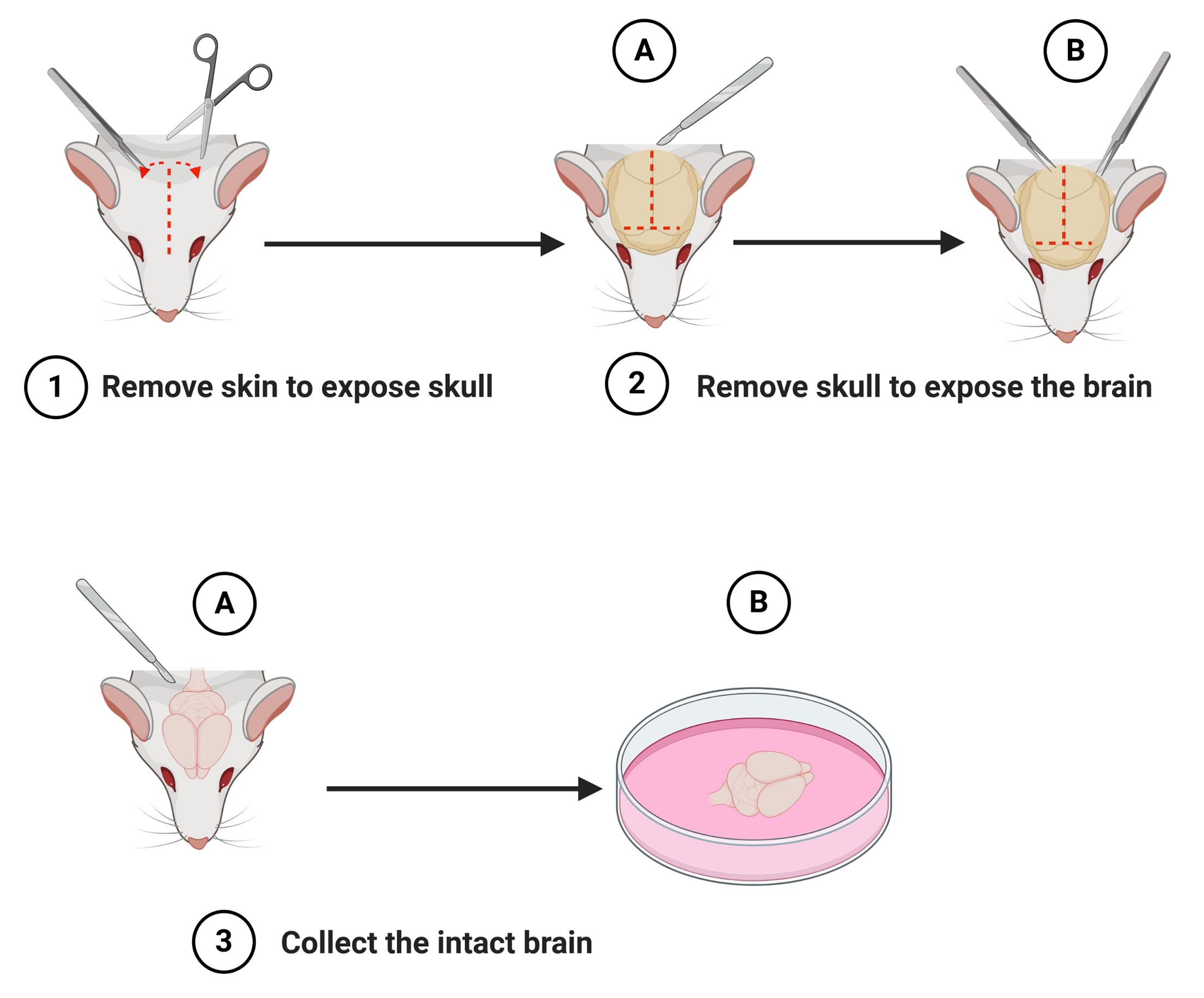
Figure 1. Schematic representation of the steps to isolate the brain (macro-dissection). After decapitation of the newborn mouse, remove the skin to expose the skull (step 1). Cut the skull along the midline using a scalpel (step 2A) and partially remove the skull using a pair of fine tipped tweezers (step 2B). Lift out the intact brain with a scalpel or spatula (step 3A) and place it into a 60 mm Petri dish containing ice-cold dissection medium (DM/Ky.Mg) (step 3B). Schematic representation was created with BioRender.com.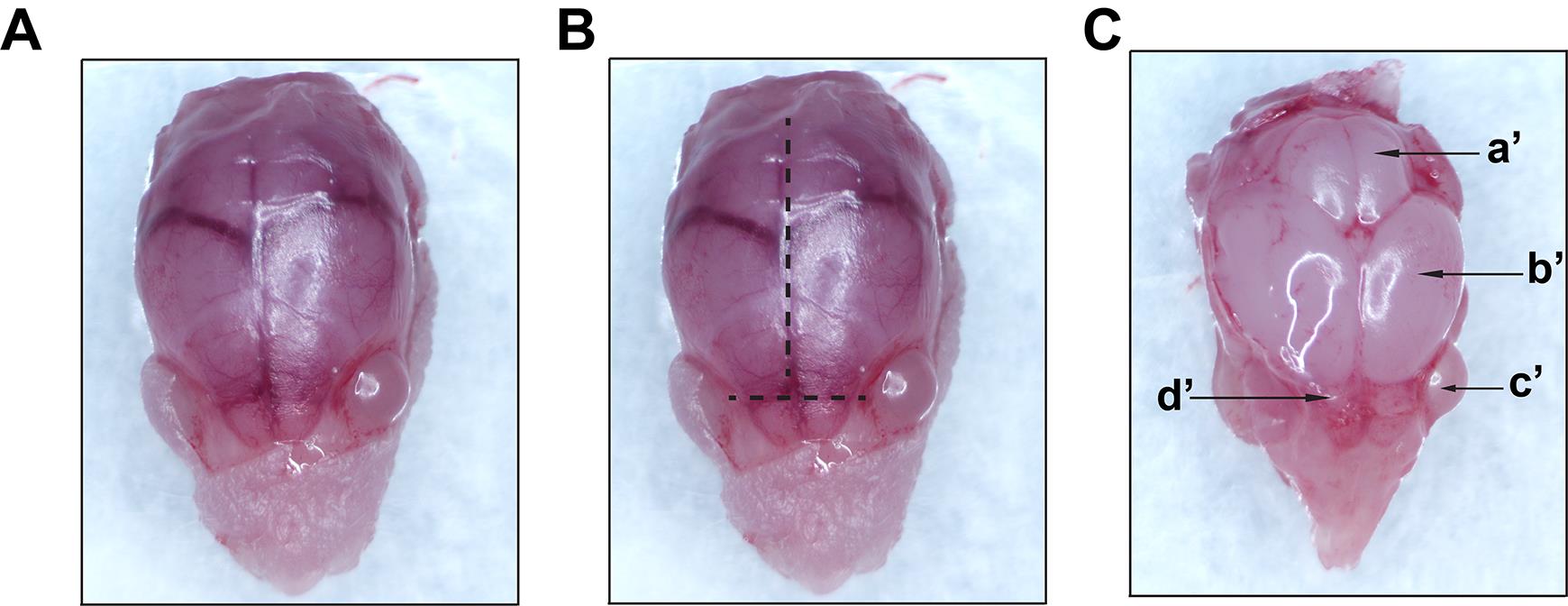
Figure 2. Representative images taken during macro-dissection. A. Exposed skull following skin removal (step 1 from Figure 1). B. Lines showing the direction of cuts along the midline to expose the brain (step 2 from Figure 1). C. Exposed intact brain (step 3A from Figure 1); a’-cerebellum; b’-cerebral hemisphere; c’-eye; d’-olfactory bulb. Images were recorded using an AmScope SM-4TZ-144A Trinocular Stereo Microscope, equipped with an 18MP color CMOS microscope camera.
Micro-dissection (Figures 3 and 4)
Remove the cerebellum and separate the two hemispheres.
Place the 60 mm dish with the brain in ice cold DM/Ky.Mg under the stereo microscope for the following steps.
Carefully expose and remove the hippocampi; then place them into the 35 mm dish containing ice cold DM/Ky.Mg; repeat these steps for every brain used for culture.
If the microscope doesn’t have a cooling function, you can perform the micro-dissection on the top of a clean 10 cm dish with ice; change the ice frequently when performing this procedure on multiple brains.
Begin warming up the Papain (Enzyme) solution on the water bath with beads set to 37 °C.
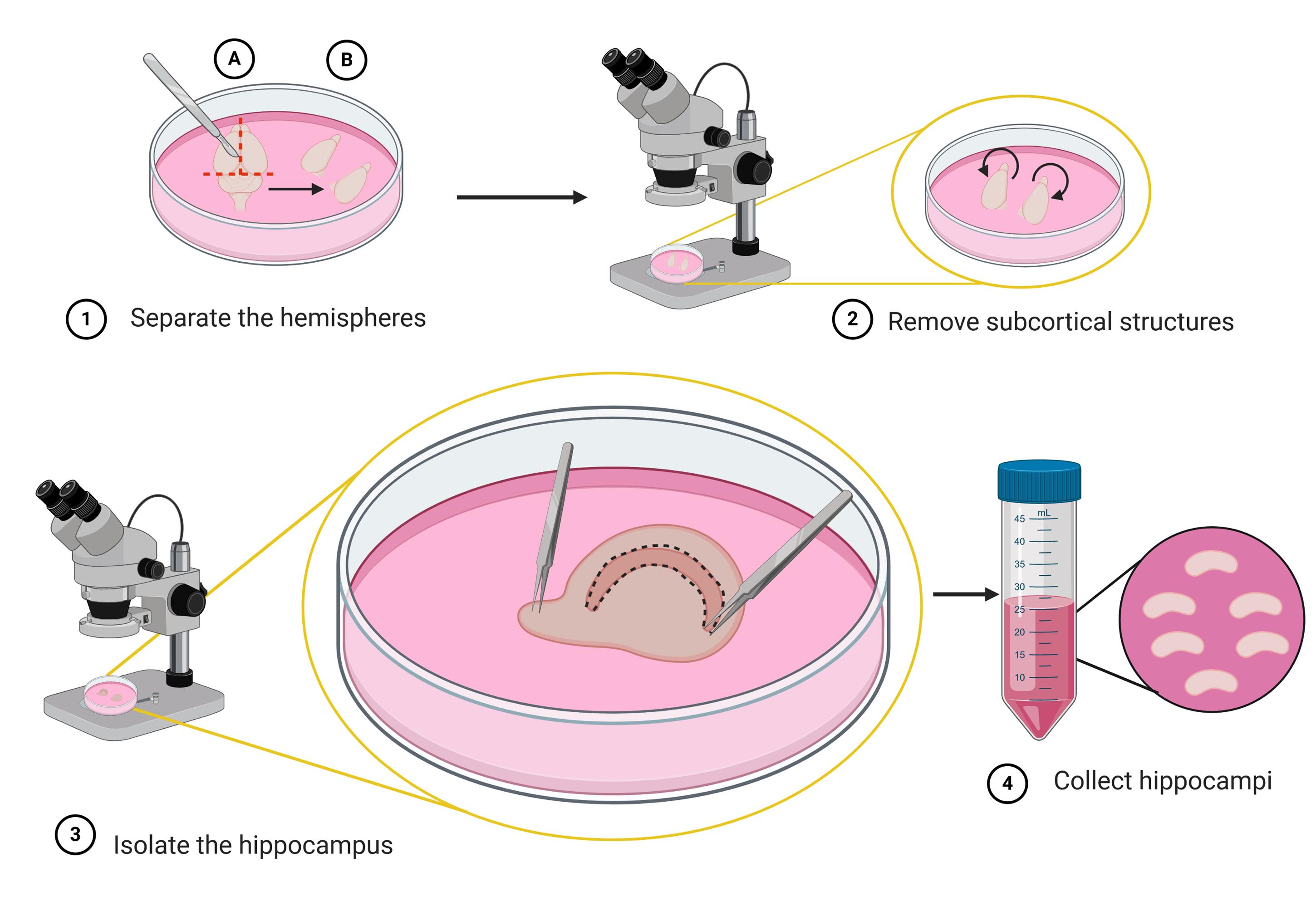
Figure 3. Schematic representation of the steps to isolate the hippocampus (micro-dissection). Use a scalpel to make a coronal incision to remove the cerebellum (step 1A), then separate the two hemispheres by a sagittal cut along the midline (step 1B). Under a stereo microscope turn the hemispheres with the medial surface facing up; then expose the hippocampus by removing subcortical structures (striatum, thalamus, and brainstem) using fine tweezers (step 2). The hippocampus appears as a crescent shaped structure located below the in-folded cortex (step 3). With one tweezer clamped tight, pin down the brain by the olfactory bulb, while with another tweezer gently peel of the meninges above the hippocampus. Use fine tweezer snip along to dissect out the hippocampus (step 3). Collect the isolated hippocampi into a 35 mm Petri dish with ice-cold DM/Ky.Mg or directly into a 50 ml sterile centrifuge tube containing 10 ml of DM/Ky.Mg (step 4). Schematic representation was created with BioRender.com.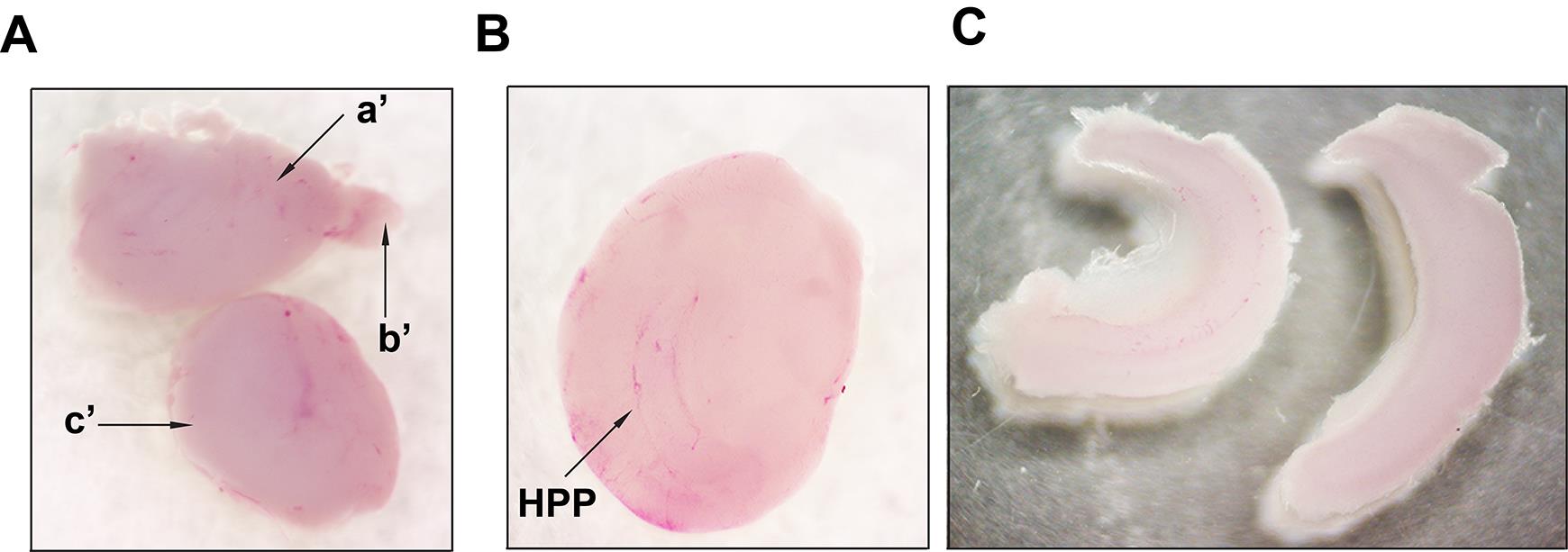
Figure 4. Representative images taken during isolation of the hippocampi. A. Separated cerebral hemispheres (step 1 from Figure 3). Upper image: a'- cerebral hemisphere; b’- olfactory bulb. Lower image shows a hemisphere turned with medial surface facing up (step 2 from Figure 3) and subcortical structures (c’). B. Hemisphere with the medial surface facing up and the exposed hippocampus (HPP) after removal of subcortical structures (striatum, thalamus, and brainstem). C. Isolated hippocampi (step 3 from Figure 3). Images were recorded using an AmScope SM-4TZ-144A Trinocular Stereo Microscope, equipped with an 18MP color CMOS microscope camera.
Digestion with Papain
Transfer the isolated hippocampi into a 50 ml sterile centrifuge tube containing 10 ml of DM/Ky.Mg.
Wash the hippocampi 3 times with 10 ml of ice-cold DM/Ky.Mg using the Pipette-Aid; avoid touching the hippocampi with the pipette.
After the 3rd wash, the DM/Ky.Mg should be kept in the biological safety cabinet at RT.
Add 5 ml of warmed Enzyme solution to the 50 ml sterile tube with hippocampi and incubate for 15 min in the water bath at 37 °C.
Remove the Enzyme solution, avoiding touching the hippocampi and add another 5 ml of Enzyme solution to the tube and perform another 15 min of incubation.
Wash the hippocampi 3 times with 5 ml of DM/Ky.Mg.
Enzyme inhibition
Begin warming up the Trypsin inhibitor solution and Optimem/Glucose solution in a 37 °C water bath.
Add 3 ml of the warm Trypsin inhibitor solution to the hippocampi and incubate for 3 min in the 37 °C water bath.
Repeat this step 2 more times with 3 ml of fresh Trypsin inhibitor solution.
Remove Trypsin inhibitor solution and wash hippocampi 3 times with 5 ml of sGM.
Trituration
Remove the sGM used for washing and add 5 ml of fresh sGM.
With a 5 ml pipette, pipette the hippocampi up and down about 15-30 times or until large chunks disappear and the medium becomes cloudy.
Do not seal the pipette tip opening against the bottom of the tube to avoid bubbling, this leads to changes in pH; let the undissociated tissue settle (about 1 min).
Transfer cell suspension into a 50 ml tube with 25 ml of warm Optimem/Glucose solution.
Add 1 ml of fresh sGM to the pellet and dissociate further by pipetting up and down 10-15 times using a 1 ml pipette.
Let the undissociated tissue settle (for about 1 min), then transfer the cell suspension into the 50 ml tube with 25 ml of Optimem/Glucose solution.
Discard undissociated tissue, as it contains mostly tissue debris.
Centrifuge the cell suspension in Optimem/Glucose for 10 min at 1,000 × g at RT in a swinging-bucket centrifuge.
Carefully remove supernatant and resuspend cells in warmed up sGM.
Plating
Resuspend pelleted cells in warmed up sGM at 1 million cells/ml density.
The yield from one newborn mouse or 2 hippocampi is 5 million cells; therefore use 5 ml of sGM/mouse to achieve a suspension at 1 million cells/ml density.
Plate cultures at 1 million cells/ml density in a 35 mm Petri dish with coverslips: use 2 ml suspension/35 mm Petri dish; higher density leads to higher rate of survival and better transfection efficiency.
On the second day in vitro (DIV2) replace serum containing GM with Neurobasal-B27 plus medium based GM.
Transfection of primary dissociated neuron cultures with the CofActor system on DIV5:
Prepare transfection solutions A and B in sterile conditions, in the biological safety cabinet:
A: 100 µl Optimem + 6 µg plasmid DNA (2 µg Actin-GFP + 4 µg mCherry-Cofilin)
B: 100 µl Optimem + 12 µl Lipofectamine 2000
Mix solution A and B and incubate the resulting transfection mix (AB) for 15 min at RT in the biological safety cabinet.
Replace conditioned medium (CM) on neuron cultures with warm transfection medium (TM); save CM.
Add 200 µl of transfection mix AB/35 mm Petri dish (or 10% of the CM, if using a different size dish) and incubate for 2 h in tissue culture incubator.
Spin CM at 1,000 × g for 10 min and discard pellet; place CM in incubator.
After 2 h, replace TM containing AB with CM.
24-48 h later, check transfection efficiency using a tissue culture microscope; transfected neurons should express both constructs (signal in green and red channels).
Live cell imaging and stimulation of neurons expressing the CofActor system (Figure 5)
Place glass coverslips with transfected neurons facing down into the glass bottom dish with PBS and transfer to Keyence microscope.
Locate transfected neurons, mark their positions (x- and y-axis) and take images before stimulation.
Treat neurons with 10 µl of 10 mM Azide and 6 µl of 6 mM DDG for 10 min without disturbing the position of the culture dish in the Keyence microscope.
Stimulate neurons with blue light for 30 s without disturbing the position of the glass bottom culture dish.
Return to pre-marked positions to image the neurons of interest and record their response at 1-10 min following stimulation.
Note: Length of treatment and imaging is determined by user.
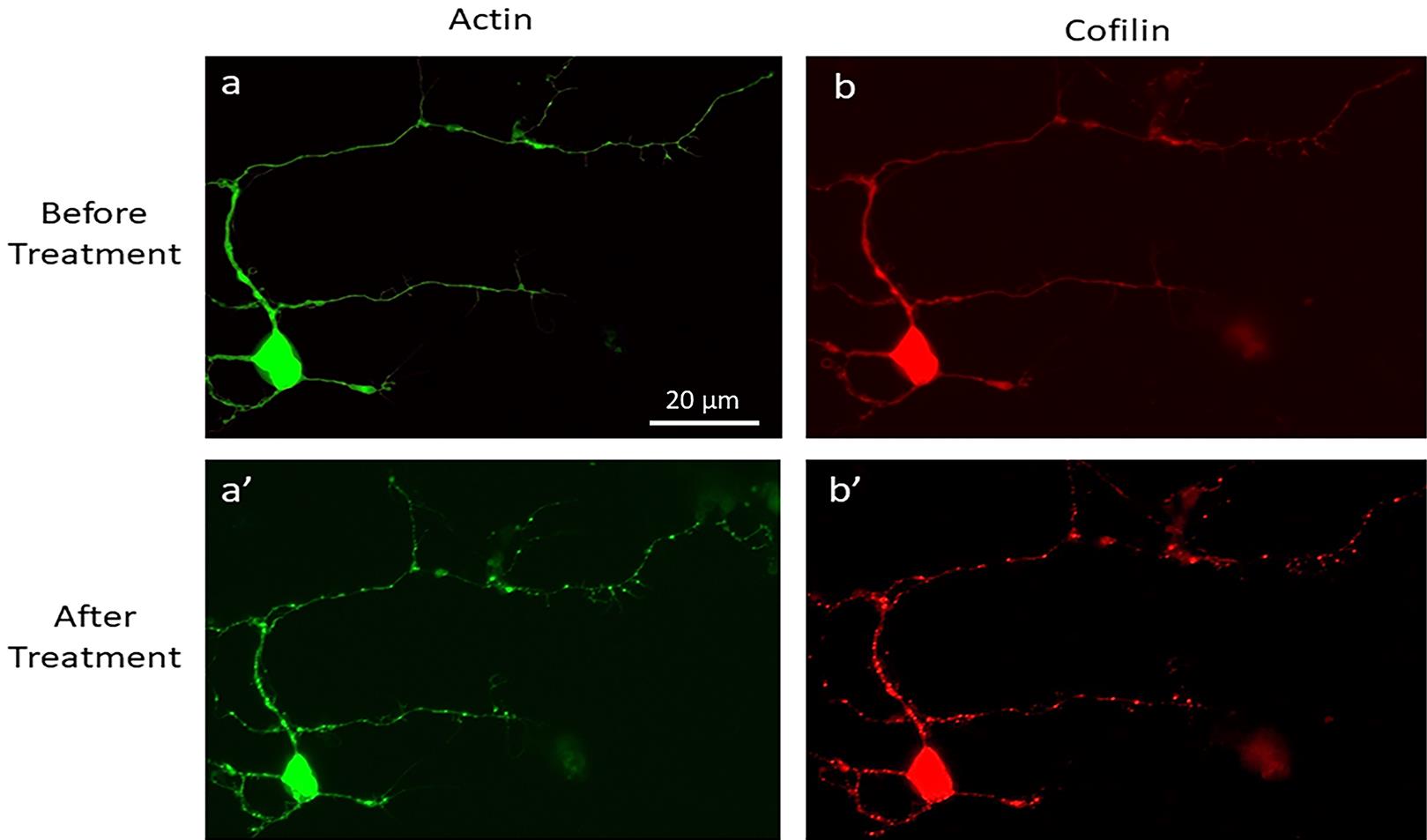
Figure 5. Validation of CofActor in primary hippocampal neuron cultures. Representative images of a primary hippocampal neuron transfected with the CofActor system (40× magnification), before (a, b) and after (a’, b’) exposure to ATP depletion and illumination sequence conditions.
Image analysis using Particle Counting (quantification of actin-cofilin rods formation) (Figure 6)
Use Fiji equipped with the Bio-Formats package.
Count particles using the Analyze Particles feature.
Restrict particle size to 20-200 pixels and restrict circularity to 0.20-1.00.
Draw custom shapes around the neuronal soma and neuronal processes on the images of neurons before treatment and perform particle counting.
Save the shapes and place them on the same area of the soma and neuronal process on the images taken after treatment and stimulation and perform particle counting.
Report particle counts as average number of particles/neuron.
Use GraphPad Prism 8 to perform paired t-test on particle count data.
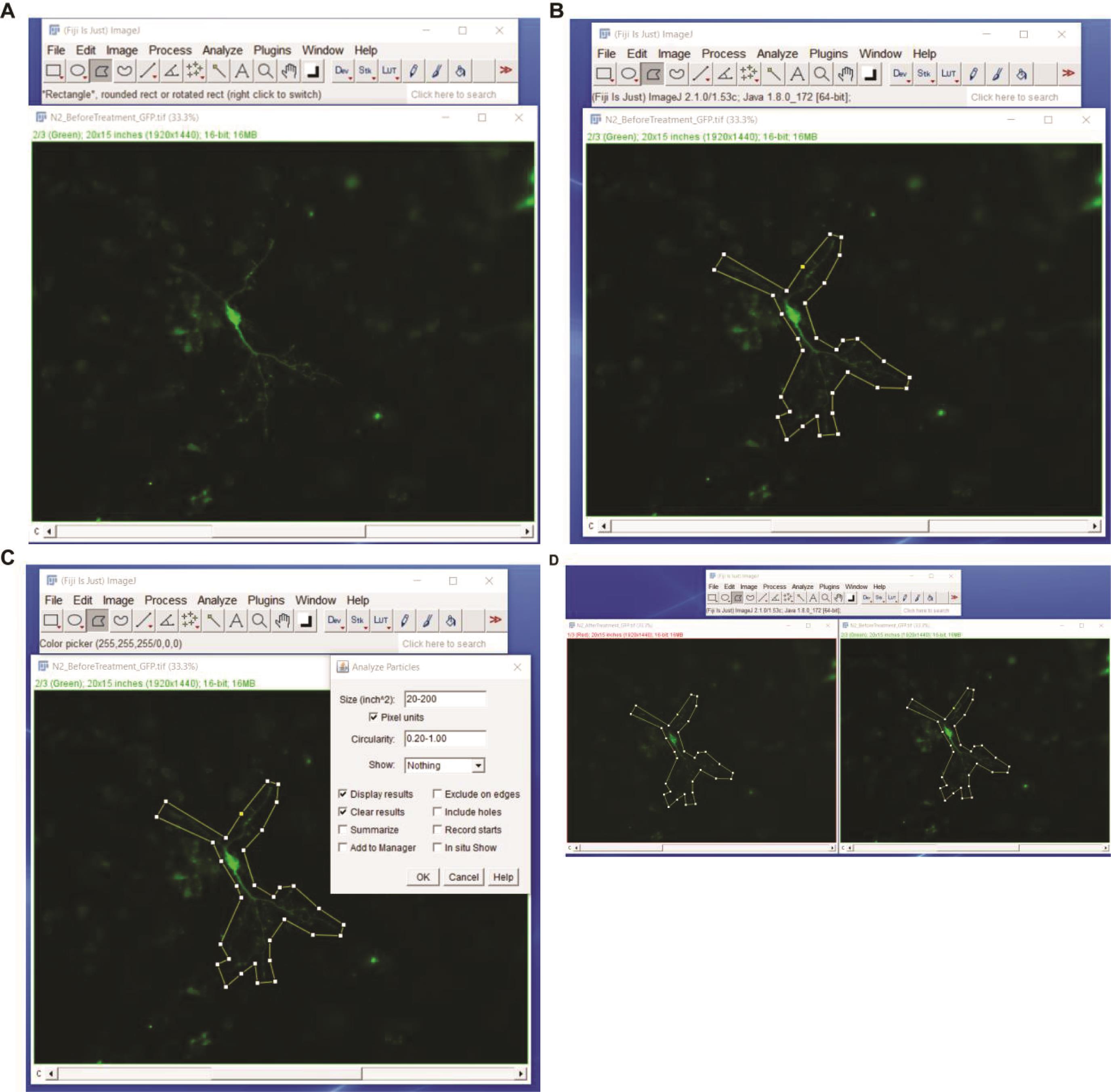
Figure 6. Fiji steps to measure actin/cofilin rod formation. Upload the pre-light treatment image to Fiji (A). Use the polygon out tool to cover the area of the neuron to be measured (B). Select Analyze > Analyze Particles on the Fiji control panel. Select a size of 20-200 pixel units and restrict the circularity to 0.20-1.00 (C) and press OK to analyze. With the original pre-light treatment image with the polygon still outlining it, open the post-light treatment neuron image. With the post-light treatment neuron selected press Shift > E; this will transfer the polygon made in the pre-light treatment step. Align this over the same area in the post-light treatment step (D). Then re-analyze the post-light treatment using the same steps from C.
Recipes
Stock solutions
Stock Glucose solution (2.5 M; for 500 ml)
Glucose 225.2 g
H2O up to 500 ml
Mix until fully dissolved; then filter sterilize and store at 4 °C
Stock dissociation medium (stock DM)
Stock solution For 250 ml
1 M Na2SO4 20.45 ml
0.5 M K2SO4 15 ml
1 M MgCl2 1.45 ml
100 mM CaCl2 0.63 ml
1 M HEPES 0.25 ml
2.5 M Glucose 2 ml
0.5% Phenol Red 0.5 ml
0.1 N NaOH 0.4-0.5 ml
TC H2O 209.72 ml
Filter sterilize and store at 4 °C up to 1 month
Cover with aluminum foil to protect from light
10× Ky.Mg stock (10 mM Kynurenic acid/100 mM MgCl2) (500 ml)
H2O 340 ml
Kynurenic acid 757 mg
0.5% Phenol red 2 ml
1 N NaOH about 3.5 ml drop while stirring
Stir until dissolved (it might take 2-3 h), then add:
1 M HEPES 2 ml
1 M MgCl2 40 ml
H2O about 400 ml
Filter sterilize and store at -20 °C in 15 ml aliquots
Stock coating solutions
Prepare 1 mg/ml Poly-D-Lysine (PDL) stock in TC water
Make 10 ml aliquots of PDL stock solution and store at -20 °C
Prepare 1 mg/ml laminin stock solution in TC water
Aliquot laminin stock solution in 50 or 100 µl aliquots and store at -80 °C
1½ solution (48 ml)
2.5 M Glucose 28 ml
GlutaMAX 10 ml
Pen/Strep 10 ml
Aliquot into 5 ml aliquots in 15 ml sterile centrifuge tubes and store at -20 °C
Cysteine.HCl
Dilute Cysteine.HCl at a concentration of 45 mg/ml in BME (optional: filter sterilize)
Aliquot into 100 µl aliquots and store at -20 °C
Use one aliquot for 10 ml enzyme solution
Working solutions
Working dissociation medium (DM/Ky.Mg) (100 ml)
Stock DM (dissociation medium) 90 ml
10× Ky.Mg 10 ml
Growth medium (GM) with serum (100 ml) for plating
BME 87.6 ml
BCS 10.0 ml
1½ 2.4 ml
Neurobasal A-B27 growth medium for postnatal neurons (20 ml)
Neurobasal A 19.2 ml
GlutaMAX 0.2 ml
Pen/Strep 0.2 ml
B27 plus 0.4 ml
Optimem/Glucose
Optimem 500 ml
2.5 M Glucose 4 ml
Enzyme (Papain) solution
DM/Ky.Mg 9.7 ml
1 N NaOH 22-25 µl
Papain latex 100 units
Cysteine.HCl stock solution 100 µl (1 aliquot)
Add Cysteine.HCl to the ice-cold DM/Ky.Mg; then adjust the pH to 7.4 with NaOH
Add the papain latex and incubate at 37 °C for 5 min
Sterile filter and keep on ice until before needed
Warm up before incubating neurons
Trypsin Inhibitor solution
DM/Ky.Mg 9.7 ml
Trypsin Inhibitor 100 mg
1 N NaOH 22-25 µl
Mix well to dissolve
Adjust the pH to 7.4 with NaOH, then sterile filter
Keep on ice until needed
Warm up before incubating neurons
Transfection medium (TM)
Freshly prepared GM without antibiotics
Working PDL solution
50 µg/ml PDL, prepared with TC water
Laminin-PDL coating solution
2 µg/ml laminin in working PDL solution
Acknowledgments
The authors thank East Carolina University for funding support; to Denys Bashtovyy and members of the Szatmari and Hughes laboratories for reading the manuscript and providing constructive feedback; to Heather Sorenson for laboratory management support; to Jennifer Lynn Johnson and members of ECU Department of Comparative Medicine for support and compassionate animal care and to Azeez Aileru (ECU School of Dental Medicine) for sharing instrumentation used in this work. This protocol was adapted from Szatmari et al. (2005) and Kaech and Banker (2006) and published in Salem et al. (2020).
Competing interests
Authors declare no competing interests.
Ethics
All mice were housed in the Animal Resource Facility of East Carolina University (ECU), compliant with the US National Institutes of Health Guide for Care and Use of Laboratory Animals. All animal experiments were ECU IACUC approved (AUP# P105).
References
- Anoop, A., Singh, P. K., Jacob, R. S. and Maji, S. K. (2010). CSF Biomarkers for Alzheimer's Disease Diagnosis. Int J Alzheimers Dis 2010.
- Bamburg, J. R. and Bernstein, B. W. (2016). Actin dynamics and cofilin-actin rods in alzheimer disease. Cytoskeleton (Hoboken) 73(9): 477-497.
- Blennow, K. and Zetterberg, H. (2009). Cerebrospinal fluid biomarkers for Alzheimer's disease. J Alzheimers Dis 18(2): 413-417.
- Ganesh, H. V. S., Chow, A. M. and Kerman, K. (2016). Recent advances in biosensors for neurodegenerative disease detection. TrAC-Trends Anal Chem 79: 363-370.
- Halliwell, B. and Whiteman, M. (2004). Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br J Pharmacol 142(2): 231-255.
- Johansson, P., Mattsson, N., Hansson, O., Wallin, A., Johansson, J. O., Andreasson, U., Zetterberg, H., Blennow, K. and Svensson, J. (2011). Cerebrospinal fluid biomarkers for Alzheimer’s disease: diagnostic performance in a homogeneous mono-center population. J Alzheimers Dis 24(3): 537-546.
- Kaech, S. and Banker, G. (2006). Culturing hippocampal neurons. Nat Protoc 1(5): 2406-2415.
- Kalyanaraman, B., Darley-Usmar, V., Davies, K. J., Dennery, P. A., Forman, H. J., Grisham, M. B., Mann, G. E., Moore, K., Roberts, L. J., 2nd and Ischiropoulos, H. (2012). Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med 52(1): 1-6.
- Kanazawa, I. (2001). How do neurons die in neurodegenerative diseases? Trends Mol Med 7(8): 339-344.
- Kim, J. S., Huang, T. Y. and Bokoch, G. M. (2009). Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol Biol Cell 20(11): 2650-2660.
- Kitamura, Y., Usami, R., Ichihara, S., Kida, H., Satoh, M., Tomimoto, H., Murata, M. and Oikawa, S. (2017). Plasma protein profiling for potential biomarkers in the early diagnosis of Alzheimer's disease. Neurol Res 39(3): 231-238.
- Laske, C., Sohrabi, H. R., Frost, S. M., Lopez-de-Ipina, K., Garrard, P., Buscema, M., Dauwels, J., Soekadar, S. R., Mueller, S., Linnemann, C., Bridenbaugh, S. A., Kanagasingam, Y., Martins, R. N. and O'Bryant, S. E. (2015). Innovative diagnostic tools for early detection of Alzheimer's disease. Alzheimers Dement 11(5): 561-578.
- Lee, V. M., Goedert, M. and Trojanowski, J. Q. (2001). Neurodegenerative auopathies. Annu Rev Neurosci 24: 1121-159.
- Linkert, M., Rueden, C., Allan, C., Burel, J., Moore, W., Patterson, A., Loranger, B., Moore, J., Neves, C., MacDonald, D., Tarkowska, A., Sticco, C., Hill, E., Rossner, M., Eliceiri, K. and Swedlow, J., (2010). Metadata matters: access to image data in the real world. J Cell Biol 189(5): 777-782.
- López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. and Kroemer, G. (2013). The hallmarks of aging.Cell 153(6): 1194-1217.
- McKhann, G. M., Knopman, D. S., Chertkow, H., Hyman, B. T., Jack, C. R., Jr., Kawas, C. H., Klunk, W. E., Koroshetz, W. J., Manly, J. J., Mayeux, R., Mohs, R. C., Morris, J. C., Rossor, M. N., Scheltens, P., Carrillo, M. C., Thies, B., Weintraub, S. and Phelps, C. H. (2011). The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 7(3): 263-269.
- Mi, J., Shaw, A. E., Pak, C. W., Walsh, K. P., Minamide, L. S., Bernstein, B. W., Kuhn, T. B. and Bamburg, J. R. (2013). A genetically encoded reporter for real-time imaging of cofilin-actin rods in living neurons. PLoS One 8(12): e83609.
- Minamide, L. S., Maiti, S., Boyle, J. A., Davis, R. C., Coppinger, J. A., Bao, Y., Huang, T. Y., Yates, J., Bokoch, G. M. and Bamburg, J. R. (2010). Isolation and characterization of cytoplasmic cofilin-actin rods. J Biol Chem 285(8): 5450-5460.
- NIA, NIH, U.S. Department of health and human services. (2015). 2014-2015 Alzheimer’s Disease Progress Report: Advancing Research Toward a Cure.
- Owusu-Ansah, E., Yavari, A. and Banerjee, U. (2008). A protocol for in vivo detection of reactive oxygen species. Protoc Exch doi:10.1038/nprot.2008.23.
- Polidori, M. C., Griffiths, H. R., Mariani, E. and Mecocci, P. (2007). Hallmarks of protein oxidative damage in neurodegenerative diseases: focus on Alzheimer's disease. Amino Acids 32(4): 553-559.
- Salem, F. B., Bunner, W. P., Prabhu, V. V., Kuyateh, A. B., O'Bryant, C. T., Murashov, A. K., Szatmari, E. M. and Hughes, R. M. (2020). CofActor: A light- and stress-gated optogenetic clustering tool to study disease-associated cytoskeletal dynamics in living cells. J Biol Chem 295(32): 11231-11245.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J.-Y., White, D.J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nature Methods 9(7): 676-82.
- Sweeney, P., Park, H., Baumann, M., Dunlop, J., Frydman, J., Kopito, R., McCampbell, A., Leblanc, G., Venkateswaran, A., Nurmi, A. and Hodgson, R. (2017). Protein misfolding in neurodegenerative diseases: implications and strategies. Transl Neurodegener 6: 6.
- Szatmari, E., Habas, A., Yang, P., Zheng, J. J., Hagg, T. and Hetman, M. (2005). A positive feedback loop between glycogen synthase kinase 3β and protein phosphatase 1 after stimulation of NR2B NMDA receptors in forebrain neurons. J Biol Chem 280(45): 37526-37535.
- Wang, X., Fang, H., Huang, Z., Shang, W., Hou, T., Cheng, A. and Cheng, H. (2013). Imaging ROS signaling in cells and animals. J Mol Med (Berl) 91(8): 917-927.
- Yang, Q., Ke, Y., Luo, J. and Tang, Y. (2017). Protocol for culturing low density pure rat hippocampal neurons supported by mature mixed neuron cultures. J Neurosci Methods 277: 38-45.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Bunner, W. P., Dodson, R., Hughes, R. M. and Szatmari, E. M. (2021). Transfection and Activation of CofActor, a Light and Stress Gated Optogenetic Tool, in Primary Hippocampal Neuron Cultures . Bio-protocol 11(8): e3990. DOI: 10.21769/BioProtoc.3990.
- Salem, F. B., Bunner, W. P., Prabhu, V. V., Kuyateh, A. B., O'Bryant, C. T., Murashov, A. K., Szatmari, E. M. and Hughes, R. M. (2020). CofActor: A light- and stress-gated optogenetic clustering tool to study disease-associated cytoskeletal dynamics in living cells. J Biol Chem 295(32): 11231-11245.
Category
Neuroscience > Basic technology
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.