- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Investigate Synaptic Vesicles Mobility in Neuronal Culture Axons by FRAP Imaging
Published: Vol 11, Iss 6, Mar 20, 2021 DOI: 10.21769/BioProtoc.3962 Views: 6282
Reviewed by: Chiara AmbrogioAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2019
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Synaptic vesicles (SVs) are clustered in the presynaptic terminals and consistently trafficking along axons. Based on their release features, SVs are classified into different “pools”. Imaging of SVs that are traveling among multiple presynaptic terminals has helped define a new pool named “SV super-pool”. Here we describe a Fluorescent Recovery After Photobleaching (FRAP) approach to elucidate the relationship between SVs from the super-pool with SV clusters at presynaptic terminals. This method is powerful to investigate SV mobility regulation mechanisms.
Keywords: Synaptic vesicleBackground
Synaptic vesicles (SVs) are key organelles involved in neurotransmission through the storage and release of neurotransmitters. SVs are mostly identified in a cluster adjacent to the active zone of the presynaptic terminals. SVs have a homogeneous appearance with a diameter of 40-50 nm under electron microscopy (EM) (Landis et al., 1988; Korogod et al., 2015). While, to our knowledge, there is no significant biochemical distinction between SVs. Under different stimulation paradigms, they show different release properties. Thus, SVs were classified into different functional pools: the reserve pool, the recycling pool and the readily releasable pool (Figure 1) (Denker and Rozzoli, 2010). The detailed synapse structure, SV localization, SV release mechanisms were intensively studied with EM. SVs were found linked to one or two neighboring vesicles by filaments, and synapsins were thought to be part of the connector and maintain the SVs in the reserve pool (Siksou et al., 2007). The dissection of molecular steps for SVs docking and fusion was also revealed by ultrastructure study (Imig et al., 2014). The fusion of SV with the plasma membrane exposes the acidic lumen (pH around 5.0) to the neutral extracellular medium (pH of 7.4) (Südhof, 1995). Therefore, SVs recycling and related molecular mechanisms were also intensively studied by labeling SVs with pH sensors (Sankaranarayanan et al., 2000; Soykan et al., 2017).
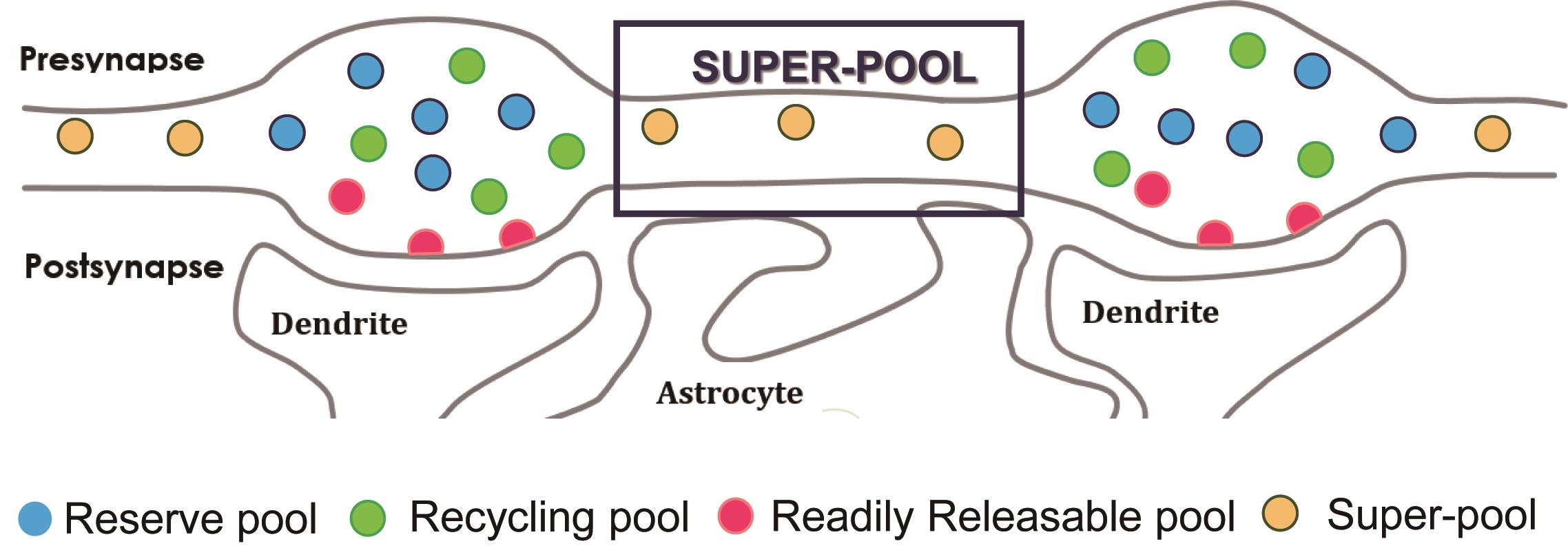
Figure 1. Synaptic vesicle pools. The classical model of synaptic vesicle (SV) pools are the reserve pool, the recycling pool, and the readily releasable pool. A newly defined “super-pool” comprises SVs that share among en-passant presynaptic boutons along the axon.
For many years, it was thought that SVs recycle inside a single presynaptic terminal without major exchange with neighboring en-passant boutons of the same axon, while terminals may function with significant autonomy from the distant cell soma. In the past decade, SVs were observed trafficking and sharing between multiple en-passant presynaptic boutons along the axon. This axonal SV population was designated as “super-pool” (Figure 1) (Denker and Rozzoli, 2010). This super-pool was observed through both in vitro and in vivo paradigms (Staras et al., 2010; Herzog et al., 2011; Zhang et al., 2019). Yet, how SVs in super-pool contribute to the neurotransmission remains largely unknown. We could recently show that, changes in the super-pool size impact spontaneous release frequency (Zhang et al., 2019).
FRAP is normally used for determining the kinetics of cell membrane diffusion or protein binding (Axelrod et al., 1976; Qin et al., 2008), while the kinetics of SV mobility is unlike these cases. However, as described above, a certain number of SVs are clustered in each presynaptic bouton and consistently refresh with SVs in super-pool. FRAP allows to measure diffusion of SVs between the presynaptic bouton and the axon. Here, we detail a protocol for FRAP imaging that is able to elucidate the exchange of SVs between the super-pool and presynaptic SV clusters. SVs are labeled with a synaptic vesicle protein tagged with a fluorescent protein, such as VGLUT1venus or Synaptobrevin 2EGFP (Figures 2A-2D). This FRAP method can be applied to both in vitro and in vivo systems. In this paper, we will describe the detailed protocol applied to dissociated hippocampal neuronal culture. This FRAP imaging method will be useful for further explorations of SV mobility related mechanisms.

Figure 2. FRAP sequences acquisition and analysis. In neurons, SVs are labeled by fluorescent protein Venus/EGFP. A presynaptic bouton contains a cluster of SVs, this is shown as a bright individual bouton. A. The experiment work flow. The virus transduction is performed on DIV 1/2 of the culture, and the FRAP imaging is acquired from DIV 17 to 21. Detailed FRAP imaging procedure is showed in the yellow box. B. Randomly select a field of culture for imaging. In the field, select 5 boutons (labeled as red circles) that are distant from each other as ROIs for bleaching. C. The frame acquired just after photobleaching. The fluorescent intensity of 5 selected boutons is significantly decreased due to the photobleaching. D. A frame acquired 1 h after photobleaching, the ROIs fluorescent intensity is gradually recovered. E. Analyze the FRAP sequence with FRAP Analysis Macro. Following menus on the pop-up windows, select the regions of bleached boutons (red circles), the cells in the field for photobleaching correction (yellow region), and the background area for background subtraction (blue region).
Materials and Reagents
Round cover glass, #1 thickness, 18 mm (Warner Instruments, catalog number: 64-0384 )
35 mm Glass bottom dish with 10 mm micro-well (Cellvis, catalog number: D35-10-1-N )
15 ml Conical centrifuge tubes (FalconTM, catalog number: 14-959-53A )
200 µl pipette tips (QuickRack, catalog number: NC9640144 )
C57/BL6J mice (The Jackson Laboratory, catalog number: 000664 )
Leibovitz’s L-15 medium (Gibco, catalog number: 11415064 ), store at 4 °C
0.05% trypsin-EDTA (Gibco, catalog number: 25300054 ), store at -20 °C
Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, catalog number: 61965026 ), store at 4 °C
Fetal bovine serum (Eurobio, catalog number: CVFSVF001 ), store at -20 °C
Penicillin-streptomycin (Gibco, catalog number: 15140122 ), store at -20 °C
Poly-L-lysine (Sigma, catalog number: P2636 ), store at -20 °C
Neurobasal A medium (Gibco, catalog number: 12349105 ), store at 4 °C
B27 supplement (Gibco, catalog number: 17504044 ), store at -20 °C
Glutamax (Gibco, catalog number: 35050038 ), store at -20 °C
MycoZap plus-PR (Lonza, catalog number: VZA2021 ), store at -20 °C
HEPES buffer (Stemcell, catalog number: 0 7200 ), store at 4 °C
BrainPhysTM without phenol red medium (Stemcell, catalog number: 05791), store at 4 °C
F(syn)W-RBN::VGLUT1-venus (PMID: 23581566, available upon request to Dr. Etienne Herzog), store at -80 °C
F(syn)W-RBN::Synaptobervin2-EGFP (PMID: 23581566, available upon request to Dr. Etienne Herzog), store at -80 °C
Complete DMEM medium (see Recipes)
Complete Neurobasal A medium (see Recipes)
Complete BrainPhys medium (see Recipes)
Equipment
Leica DMI 6000 microscope (Leica Microsystems, Wetzlar, Germany)
Spinning disk confocal head Yokogawa CSU-X1(Yokogawa Electric Corporation, Tokyo, Japan)
EM-CCD QuantEM camera (Photmetrics, Tucson, USA)
iLAS FRAP scanner system (Roper Scientific, Evry, France)
Piezo nanofocusing scanner P721.LLQ (Physik Instrumente, Karlsruhe, Germany)
Thermal incubator (Life Imaging Services, Switzerland)
Software
MetaMorph microscopy automation and image analysis software (Molecular Devices, Sunnyvale, USA, https://www.moleculardevices.com/products/cellular-imaging-systems/acquisition-and-analysis-software/metamorph-microscopy)
ImageJ (NIH, USA, https://imagej.nih.gov/ij)
FRAP Analysis Macro (Fabrice P. Cordelieres, https://github.com/fabricecordelieres/IJ-Macro_FRAP-MM)
Procedure
Prepare dissociated hippocampal neuronal culture for live imaging
Coat the glass coverslips or glass bottom dishes with 0.1 mg/ml poly-L-lysine overnight, and rinse with ddH2O 3 times before using.
Dissect hippocampi from P0 (Post-natal day 0) C57/BL6 mice in ice-cold Leibovitz’s L-15 medium.
Collected all hippocampi in a 15 ml Falcon tube filled with ice-cold Leibovitz’s L-15 medium.
Remove Leibovitz’s L-15 medium. Incubate tissues in 5 ml of 0.05% trypsin-EDTA solution at 37 °C for 15 min. Trypsin will digest extracellular proteins to facilitate cell dissociation.
Remove 0.05% trypsin-EDTA solution, and replace with complete DMEM medium to stop the reaction.
Then remove the DMEM medium, and wash the tissue with 5 ml complete Neurobasal A medium.
Add 1 ml complete Neurobasal A medium in the tube. Dissociate cells mechanically by pipetting up and down with a 200 µl tip for 10-15 strokes.
Incubate the cell suspension for 3 min to allow the sedimentation of large tissue debris. Take the 800 µl upper suspension and avoid sampling of tissue clusters.
Measure cell density and plate cells to pre-coated poly-L-lysine glass coverslips/glass bottom dishes at a density of 20,000 cells/cm2.
Note: Cells are grown in 2 ml complete Neurobasal A medium in the well of 12-well plate, or 3 ml medium in a 35 mm glass bottom dish.
Cells are grown in complete Neurobasal A medium for 5 days in-vitro (DIV).
From DIV 5-6, replace half conditioned medium (1 ml for each well of 12-well plate, and 1.5 ml for 35 mm glass bottom dishes) with complete BrainPhys medium every 2-3 days.
Viral transduction of reporter gene
On DIV 1 or 2, transduce cells with the lentivirus vector allowing for the expression of a fluorescent reporter of synaptic vesicles. We use F(syn)W-RBN::Synaptobrevin2-EGFP or F(syn)W-RBN::VGLUT1-venus. For each vector batch, Western-blot and fluorescent imaging are performed to adjust the dilution to limit protein overexpression to 2-fold of wild-type levels.
The lenti vector dilution and protein expression are measured by Western-blot and fluorescent imaging:
Cells are grown in 6 cm dishes that are pre-coated with poly-L-lysine with the same cell density as described above.
On DIV 1 or 2, lentivirus (titer range in ~×108 TU/ml) is diluted to 1/100 with Neurobasal A medium. Add 20/40/80 µl diluted virus to each dish of the culture for transduction. Return the culture to the incubator.
On DIV17, the cells are scraped and collected from the dishes. The targeted protein expression level in each sample is measured by Western-blot. VGLUT1-venus or Synaptobrevin2-EGFP has ~27 kDa higher molecular weight, thus can be easily distinguished from the corresponding endogenous wildtype protein. The increased virus transduction normally will result a linearized increase of protein expression.
Compare the expression of VGLUT1-venus/Synaptobervin2-EGFP with the wildtype VGLUT1/Synaptovervin2. Calculate the right dilution of virus which results less than 2-fold of protein overexpression. Normally, the fluorescent intensity of the presynaptic boutons is bright enough and is suitable for FRAP imaging with this protein expression level.
Based on the cell amount, proportionally add the right dilution of virus to the 12-well plates or 35 mm glass bottom dishes culture on DIV 1 or 2 that are prepared for FRAP imaging (Figure 2A).
FRAP Imaging
Note: FRAP imaging is performed with a spinning disk confocal microscope that is controlled by MetaMorph microscopy automation software. Take DIV 17-21 cell culture for imaging, normally neurons are mature and synaptic networks are well established at this time point. Incubate the cultures in conditioned culture medium (cells are grown in) containing HEPES (40 mM) and at physiological temperature (37 °C) during the imaging procedure.
Use MetaMorph to control the entire FRAP procedure.
Randomly select one field of the cell culture for imaging with a 63×/1.4 numerical aperture oil-immersion objective (Figure 2B).
Select up to 5 fluorescent boutons that are distant from each other as Regions of Interest (ROIs) for bleaching (Figure 2B). Selecting too many boutons that are closely related on the same axon would affect the measurement. Before bleaching, monitor the boutons fluorescence every 30 s for 3 min (Figure 2A). Image as a Z-stack of 4.8 µm thickness with 0.8 µm step interval. ROIs should be at the midplane of the stack. Stack imaging allows for a more robust measurement of bouton intensity and to buffer fluctuations due to small Z-directed movements.
Apply three passes of 491 nm laser (40 mW) to bleach the Syb2EGFP labeling boutons; two laser passes of 491 nm laser (30 mW) and the 405 nm laser (10 mW) to bleach the VGLUT1venus labeling boutons (Figure 2B). After bleaching, the fluorescence of ROIs should remain around 40%-60% of the initial fluorescence intensity (Figure 2C). VENUS and EYFP related dyes require a stimulation at 405 nm to prevent fluorescence recovery from a reversible dark state of the fluorescent proteins (McAnaney et al., 2005; Herzog et al., 2011).
Monitor the fluorescence recovery after bleaching every 30 s during the first 3 min and then every 5 min during the next 70 min (Figure 2A).
Data analysis
Open the stack sequence with ImageJ and perform a sum Z projection that will generate a 32 bits/pixel sequences (Video 1).
 Video 1. An example of image processing with ImageJ FRAP analysis Macro
Video 1. An example of image processing with ImageJ FRAP analysis MacroUse Image J FRAP Analysis Macro under “plugin” (https://github.com/fabricecordelieres/IJ-Macro_FRAP-MM) to automate image analysis (Video 1).
The macro commands apply x-y realignment to each individual stack.
Then extract the integrated fluorescence intensity of bleached boutons, the cells in the field for photobleaching correction, and the background area for background subtraction (Figure 2E).
The Macro will output all the raw data and processed data in a table named “Results” (Figure 3A) and a plot named “Values” that shows all ROIs’ intensity value
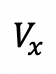 (x stands for time point) as a function of time (Figure 3B).
(x stands for time point) as a function of time (Figure 3B).
The boutons intensity slightly varies in each frame before bleaching. Take the average value before bleaching (
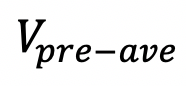 )as prebleaching reference. Normalize boutons intensity at different time point
)as prebleaching reference. Normalize boutons intensity at different time point 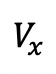 in the sequence as:
in the sequence as: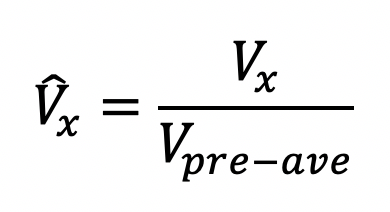
Filter out rejected ROIs from the final quantification which may correspond to the following cases:
The bouton (ROI) fluorescence
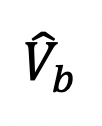 (b stands for the frame that right after bleaching) are over-bleached or less-bleached, which means
(b stands for the frame that right after bleaching) are over-bleached or less-bleached, which means 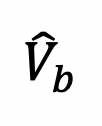 < 20% or > 70% (Figure 3B, the bouton with black curve). Indeed,
< 20% or > 70% (Figure 3B, the bouton with black curve). Indeed, 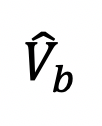 below 20% the risk of photodamage to the sample is high, and above 70% the recovery will not be measured accurately.
below 20% the risk of photodamage to the sample is high, and above 70% the recovery will not be measured accurately.Because axonal exchange is not entirely linear and continuous, some boutons may receive or loose massive fluorescent clusters during the fluorescence recovery time (Figure 3B, the bouton with green curve). This was extensively described in our former paper (Herzog et al., 2011).
Calculate the percentage of recovered fluorescence of each bouton. Normalize the boutons average intensity before bleaching as 1, right after bleaching as 0. The total bleached fluorescence is 1 -
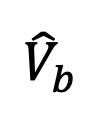 , the recovered fluorescence at different time point is
, the recovered fluorescence at different time point is 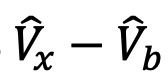 .
.
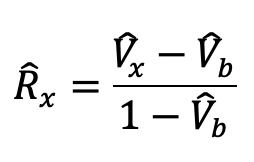
The recovered fluorescent value
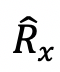
Summarize the normalized bouton fluorescence data in your favorite software for statistics and plotting (Figure 3C). Fit the average of all normalized FRAP traces with a double exponential function (Figure 3D).
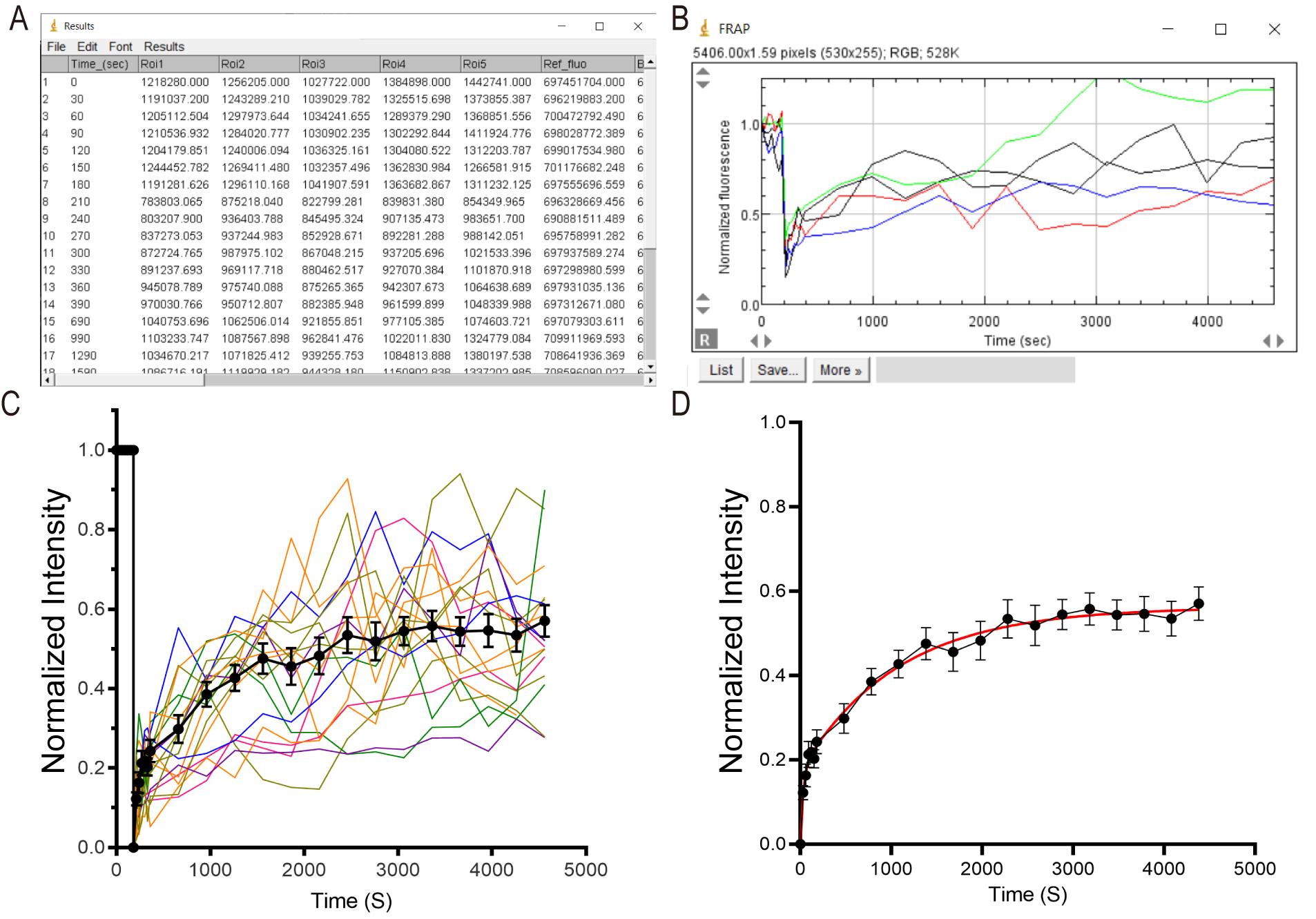
Figure 3. FRAP Data analysis. A. When FRAP Analysis Macro analysis is done, a results file with the ROIs’ raw data and normalized data is generated. B. And a plot presenting the corrected normalized values as a function of time for all ROIs is generated. C. An example figure showed the summary of normalized FRAP traces. One colorful trace stands for an individual bouton fluorescence variation in the FRAP experiment. The black dots stand for the mean value of all the ROIs at different time points. D. A double exponential trace is fitted to the average FRAP curve.
Notes
Use serum-free medium for mixed glial-neuronal culture. Because it is important to keep the cell morphology stable during the whole acquisition. Neurons grown on a layer of astrocytes will not be suitable for imaging, because of movements during the imaging procedure. Mixed culture containing few glia cells or banker culture are suitable for this long-time FRAP imaging.
Select a field with nice neuronal network but with at least one empty region (no fluorescent material) for imaging. The dark region is essential to be taken as a background reference for later imaging analysis.
Apply certain amount of 405 nm laser for Venus/EYFP fluorescence bleaching. As previously reported, YFP has a photochemical reversible dark state representing roughly 20% of YFP/VENUS molecules. This dark pool of fluorescent proteins is emptied by photoactivation at 405 nm (McAnaney et al., 2005; Herzog et al., 2011). Thus, accompanying bleaching with UV light significantly reduces the unwanted recovery.
Set the right laser power for the bouton fluorescence bleaching. The bleach will result a ~50% fluorescence reduction. To avoid the bleaching variance caused by a single laser pass, repeat a mild laser pass for 2-3 times to the selected boutons is recommended. The appropriate laser power needs to be tested based on your own culture before experiments.
Recipes
Complete DMEM medium
450 ml DMEM medium
50 ml FBS
and 5 ml Penicillin-streptomycin
store at 4 °C
Complete Neurobasal A medium
50 ml Neurobasal A medium
1 ml B27 supplement
125 µl Glutamax
And 100 µl Mycozap plus-PR
Store at 4 °C
Complete BrainPhys medium
50 ml BrainPhys medium
1 ml B27 supplement
125 µl Glutamax
And 100 µl Mycozap plus-PR
Store at 4 °C
Acknowledgments
We thank Bordeaux Imaging Center for their excellent technical support and equipment for the experiments. This work is supported by the Erasmus Mundus ENC program and the LabEx BRAIN extension grant (ANR-10-LABX-43 BRAIN), Agence Nationale de la Recherche (ANR-12-JSV4-0005-01 VGLUT-IQ ; ANR-10-LABX-43 BRAIN; ANR-10-IDEX-03-02 PEPS SV-PIT) and Fondation pour la Recherche Médicale (ING20150532192). This protocol introduced here was detailed from past studies (Herzog et al., 2011; Zhang et al., 2019).
Competing interests
The authors declare no financial or non-financial competing interests related to this work.
Ethics
The experimental design and all procedures were performed in accordance with the European guide for the care and use of laboratory animals and approved by the ethics committee of Bordeaux University (CE50) under the APAFIS n°1692.
References
- Axelrod, D., Koppel, D. E., Schlessinger, J., Elson, E. and Webb, W. W. (1976). Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J 16(9): 1055-1069.
- Denker, A. and Rizzoli, S. O. (2010). Synaptic vesicle pools: an update. Front Synaptic Neurosci 2: 135.
- Herzog, E., Nadrigny, F., Silm, K., Biesemann, C., Helling, I., Bersot, T., Steffens, H., Schwartzmann, R., Nagerl, U. V., El Mestikawy, S., Rhee, J., Kirchhoff, F. and Brose, N. (2011). In vivo imaging of intersynaptic vesicle exchange using VGLUT1 Venus knock-in mice. J Neurosci 31(43): 15544-15559.
- Imig, C., Min, S. W., Krinner, S., Arancillo, M., Rosenmund, C., Sudhof, T. C., Rhee, J., Brose, N. and Cooper, B. H. (2014). The morphological and molecular nature of synaptic vesicle priming at presynaptic active zones. Neuron 84(2): 416-431.
- Korogod, N., Petersen, C. C. and Knott, G. W. (2015). Ultrastructural analysis of adult mouse neocortex comparing aldehyde perfusion with cryo fixation. Elife 4: e05793.
- Landis, D. M., Hall, A. K., Weinstein, L. A. and Reese, T. S. (1988). The organization of cytoplasm at the presynaptic active zone of a central nervous system synapse. Neuron 1(3): 201-209.
- McAnaney, T. B., Zeng, W., Doe, C. F., Bhanji, N., Wakelin, S., Pearson, D. S., Abbyad, P., Shi, X., Boxer, S. G. and Bagshaw, C. R. (2005). Protonation, photobleaching, and photoactivation of yellow fluorescent protein (YFP 10C): a unifying mechanism. Biochemistry 44(14): 5510-5524.
- Qin, K., Sethi, P. R. and Lambert, N. A. (2008). Abundance and stability of complexes containing inactive G protein-coupled receptors and G proteins. FASEB J 22(8): 2920-2927.
- Sankaranarayanan, S., De Angelis, D., Rothman, J. E. and Ryan, T. A. (2000). The use of pHluorins for optical measurements of presynaptic activity. Biophys J 79(4): 2199-2208.
- Siksou, L., Rostaing, P., Lechaire, J. P., Boudier, T., Ohtsuka, T., Fejtova, A., Kao, H. T., Greengard, P., Gundelfinger, E. D., Triller, A. and Marty, S. (2007). Three-dimensional architecture of presynaptic terminal cytomatrix. J Neurosci 27(26): 6868-6877.
- Soykan, T., Kaempf, N., Sakaba, T., Vollweiter, D., Goerdeler, F., Puchkov, D., Kononenko, N. L. and Haucke, V. (2017). Synaptic Vesicle Endocytosis Occurs on Multiple Timescales and Is Mediated by Formin-Dependent Actin Assembly. Neuron 93(4): 854-866 e854.
- Staras, K., Branco, T., Burden, J. J., Pozo, K., Darcy, K., Marra, V., Ratnayaka, A. and Goda, Y. (2010). A vesicle superpool spans multiple presynaptic terminals in hippocampal neurons. Neuron 66(1): 37-44.
- Südhof, T. C. (1995). The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature 375(6533): 645-653.
- Zhang, X. M., Francois, U., Silm, K., Angelo, M. F., Fernandez-Busch, M. V., Maged, M., Martin, C., Bernard, V., Cordelieres, F. P., Deshors, M., Pons, S., Maskos, U., Bemelmans, A. P., Wojcik, S. M., El Mestikawy, S., Humeau, Y. and Herzog, E. (2019). A proline-rich motif on VGLUT1 reduces synaptic vesicle super-pool and spontaneous release frequency. Elife 8: e50401.
Article Information
Copyright
![]() Zhang et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Zhang et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Zhang, X. M., Cordelières, F. and Herzog, E. (2021). Investigate Synaptic Vesicles Mobility in Neuronal Culture Axons by FRAP Imaging. Bio-protocol 11(6): e3962. DOI: 10.21769/BioProtoc.3962.
- Zhang, X. M., Francois, U., Silm, K., Angelo, M. F., Fernandez-Busch, M. V., Maged, M., Martin, C., Bernard, V., Cordelieres, F. P., Deshors, M., Pons, S., Maskos, U., Bemelmans, A. P., Wojcik, S. M., El Mestikawy, S., Humeau, Y. and Herzog, E. (2019). A proline-rich motif on VGLUT1 reduces synaptic vesicle super-pool and spontaneous release frequency. Elife 8: e50401.
Category
Neuroscience > Cellular mechanisms > Synaptic physiology
Biophysics > Biophotonics
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.