- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Monitoring Real-time Temperature Dynamics of a Short RNA Hairpin Using Förster Resonance Energy Transfer and Circular Dichroism
Published: Vol 11, Iss 6, Mar 20, 2021 DOI: 10.21769/BioProtoc.3950 Views: 5326
Reviewed by: Wenrong HeYingnan HouLusheng Fan
Original research article
The authors used this protocol in:

May 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
RNA secondary structures are highly dynamic and subject to prompt changes in response to the environment. Temperature in particular has a strong impact on RNA structural conformation, and temperature-sensitive RNA hairpin structures have been exploited by multiple organisms to modify the rate of translation in response to temperature changes. Observing RNA structural changes in real-time over a range of temperatures is therefore highly desirable. A variety of approaches exists that probe RNA secondary structures, but many of these either require large amount and/or extensive processing of the RNA or cannot be applied under physiological conditions, rendering the observation of structural dynamics over a range of temperatures difficult. Here, we describe the use of a dually fluorescently labelled RNA oligonucleotide (containing the predicted hairpin structure) that can be used to monitor subtle RNA-structural dynamics by Förster Resonance Energy Transfer (FRET) at different temperatures with RNA concentration as low as 200 nM. FRET efficiency varies as a function of the fluorophores’ distance; high efficiency can thus be correlated to a stable hairpin structure, whilst a reduction in FRET efficiency reflects a partial opening of the hairpin or a destabilisation of this structure. The same RNA sequence can also be used for Circular Dichroism spectroscopy to observe global changes of RNA secondary structure at a given temperature. The combination of these approaches allowed us to monitor RNA structural dynamics over a range of temperatures in real-time and correlate structural changes to plant biology phenotypes.
Graphic abstract:
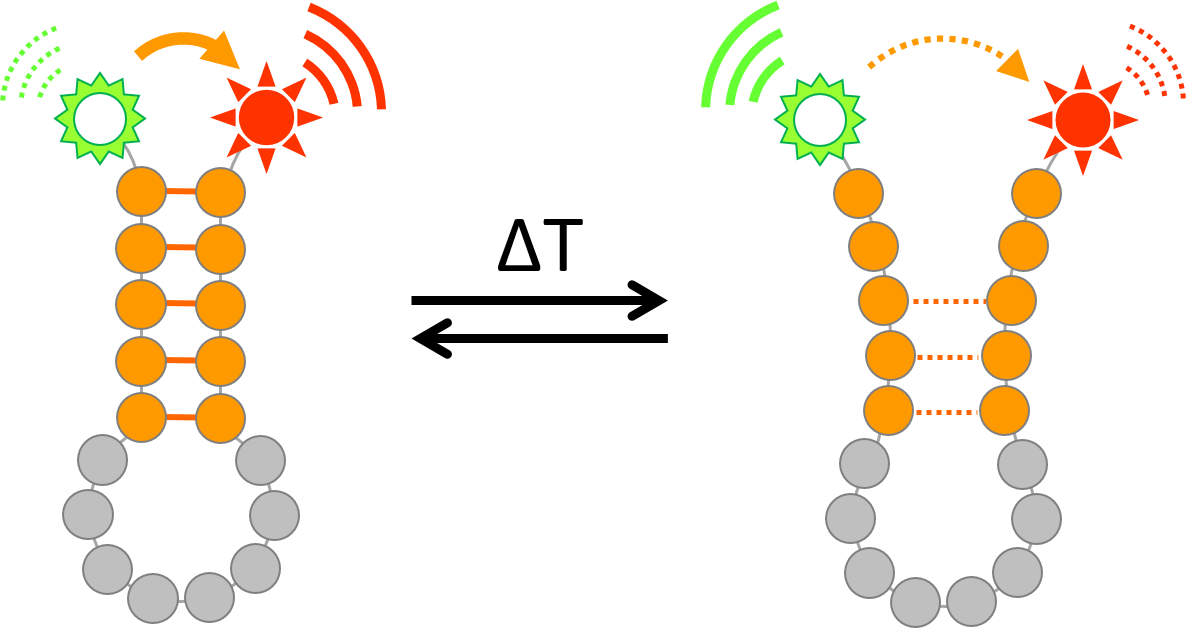
Monitoring temperature-dependent RNA structural dynamics using Förster Resonance Energy Transfer (FRET)
Background
RNA is the central molecule in gene expression in all extant life, serving as the messenger. It is also central to biocatalysis, playing a key role in ribosome assembly and function. Ultimately, RNA function is regulated by its structure and there is a seemingly limitless variety of structures that allows for diverse functions. RNA molecules can form both inter- and intramolecular Watson-Crick and Hoogsteen base pairs, which lead to the formation of secondary structures such as hairpins, apical and internal loops, 3-way-junctions or pseudoknots (Cruz and Westhof, 2009). Such secondary structures can impart catalytic activity, serve as molecular scaffolds as well as provide or occlude binding sites for RNA-binding proteins (RBPs) and other RNAs. Secondary structures on messenger RNA are known to affect a plethora of biological processes such as RNA stability and processing (Soemedi et al., 2017), alternative splicing (Bartys et al., 2019), subcellular localisation (Lazzaretti and Bono, 2017) and translation regulation (Jacobs et al., 2012).
Whilst RNA molecules have the capacity to adopt a multitude of distinct conformations, the most thermodynamically and kinetically favourable structures will be populated under certain conditions. As a consequence, RNA secondary structures can be highly dynamic and interconvert between different structural arrangements as the environment changes (Ganser et al., 2019; Mustoe et al., 2014). For example, binding of RBPs can induce or facilitate structural rearrangement, while riboswitches change their conformation and exert regulatory function upon binding of small ligands. Finally, physical parameters such as temperature and osmolarity strongly affect the distribution and the stability of RNA secondary structures and thereby promote structural transitions. Given the biological relevance of RNA secondary structure formation, several biochemical and biophysical strategies have been developed to monitor RNA structural changes in real-time, including Circular Dichroism, NMR, DMS-footprinting, and Förster Resonance Energy Transfer (FRET).
As RNA secondary structures are dependent on their thermodynamic free energies, temperature is one of the most direct parameters to control such structures for translation regulation. Thermosensitive RNA secondary structures – referred to as RNA thermometers – have been harnessed by multiple organisms to fine-tune translation in a temperature-dependent manner (Kortmann and Narberhaus, 2012; Krajewski and Narberhaus, 2014). A classic example of an RNA thermometer is found in Listeria, where a secondary structure in the prfA transcript controls virulence in response to temperature: At temperatures up to 30 °C, a hairpin structure forms and prevents access to the ribosome binding site; this structure partially melts when temperature rises to 37 °C (the body temperature of the human host), exposing the ribosome binding site and allowing translation to occur (Johansson et al., 2002). Many prokaryotes employ such zipper-like RNA thermometers as a switch to control translation. However, increased temperature may not necessarily lead to a complete loss of secondary structure, but favour the adoption of alternative conformations instead. We recently identified several eukaryotic RNA thermoswitches in the model flowering plant Arabidopsis thaliana, which are activated at warm temperatures (27-32 °C) by adopting a more relaxed hairpin conformation to enhance translation (Chung et al., 2020).
These examples emphasise the importance of investigating RNA structural dynamics in order to rationalize their biological functions. Chemicals or enzymes that react selectively with single or double stranded RNA can be employed to probe secondary structure both in vitro and in vivo (Mailler et al., 2019). However, these approaches are irreversible, leaving the RNA molecules permanently modified and preventing real-time observation of the same molecules’ dynamics over a range of conditions. Several biophysical methods can be used to observe RNA secondary structure as well, but come with their own set of limitations: besides significant requirement of RNA input, nuclear magnetic resonance (NMR) spectroscopy and X-ray crystallography preclude observation under physiological conditions, while cryo electron microscopy still suffers from limited resolution and is thus currently restricted to larger structures (Mailler et al., 2019). In our characterisation of plant RNA thermoswitches, we employed a combination of Förster Resonance Energy Transfer (FRET) measurements and circular dichroism (CD) spectroscopy to observe RNA hairpin dynamics over a range of temperatures in real-time. A detailed protocol of these methods is outlined below.
Materials and Reagents
1.5 ml Eppendorf tube
Semi-micro quartz cell 108F-QS (Sigma-Aldrich, Hellma Analytics, catalog number: HL108-F-10-40 )
1 mm quartz cell 350 ml (Sigma-Aldrich, Hellma Analytics, catalog number: HL110-F-1-40 )
Cacodylic Acid (Sigma-Aldrich, catalog number: C0125-5G ). Store at 4 °C
KOH in pellets, BioXtra (Sigma-Aldrich, catalog number: P5958 )
RNase-free water (Sigma-Aldrich, catalog number: W4502 )
RNA oligonucleotide with 5’-FAM and 3’-TAMRA fluorescent probes (5’-FAM-AAG AGA GCU UAA UUG UCA GUU UAU UCU CUG-3’-TAMRA in case of the PIF7 hairpin); the RNA sequence should cover the entire predicted hairpin-forming sequence, but should not exceed more than 3 nucleotides beyond this sequence on either side to ensure efficient FRET (IBA Gmbh, HPLC Purified). Store at 4 °C
Cacodylate buffer (see Recipes)
1 M KOH (see Recipes)
Equipment
2 L glass beaker
Stirring bar
SevenCompact pH meter S220 (Mettler Toledo, model: 30019029 )
-80 °C freezer
Varian Cary Eclipse Fluorescence Spectrometer fitted with a Varian temperature control unit (Agilent, Varian, Cary Eclipse Fluorimiter)
ThermoMixer C (Eppendorf, model: EP5382000015 ) fitted with SmartBlock 1.5 ml (Eppendorf, model: EP5361000031 )
Applied Photophysics Chirascan (CD) fitted with temperature control unit (Applied Photophysics, ChirascanTM)
Software
Cary Eclipse Kinetics software (Agilent, https://www.agilent.com/en/product/molecular-spectroscopy/fluorescence-spectroscopy/fluorescence-software/cary-eclipse-software)
GraphPad Prism 8 (GraphPad Software, https://www.graphpad.com/scientific-software/prism/)
Procedure
60 mM Cacodylate Buffer Preparation
Notes:
It is important to wear appropriate PPE whilst performing buffer preparation, including nitrile gloves, corrosive resistant lab coat and safety goggles.
Cacodylic Acids contains Arsenic and therefore is toxic and should be disposed in dedicated heavy metal waste collection.
Weight 8.280 g of Cacodylic Acid in a 2 L glass beaker and dissolve it in 500 ml of RNase free water, ensuring all white powder is fully solubilized. If needed, use additional water up to a total volume of 700 ml to ensure full solubilization of the Cacodylic Acid.
Measure the pH of the Cacodylic Acid solution generated in Step A1 with any standard laboratory pH-meter, ensuring that calibration of the pH-meter has been performed on the same day. The measured pH of the starting solution is expected to be ~4.0.
Prepare a 1 M solution of potassium hydroxide (KOH) by dissolving 5.61 g of KOH in 100 ml of RNase free water. Given the highly exothermic reaction caused by KOH, dissolving in water is recommended to perform in an ice bath.
With a glass pipette add 1 M KOH solution dropwise to the Cacodylic Acid solution while stirring the solution with a stirring bar over a magnetic stirring plate. Measure the pH after each drop addition until it stabilizes at 7.4, which is the final pH required for both CD and FRET experiments.
Add RNase-free water to the Cacodylate buffer (pH 7.4) to a total volume of 1 L.
Place the solution at 4 °C for long term storage. Buffer storage at 4 °C should not exceed 3 months. After this time, preparation of fresh buffer is strongly recommended.
RNA Annealling
Note: It is important to anneal the RNA sequence the day before the experiment is performed to allow sufficient time for the correct RNA structure to be properly folded.
Resuspend the dually labelled RNA oligonucleotide in the appropriate volume of RNase-free water to obtain a stock solution with the final concentration of 100 μM. To achieve this, add a volume of water that is 10× with respect to the total numbers of RNA nanomoles. For example, for 5 nmol of RNA add 50 μl of RNase free water. Store this solution at -80 °C for long term storage.
Dilute RNA as follows:
For FRET measurements, dilute 1 μl of the 100 μM RNA stock solution in 499 μl (200 nM final concentration) of 60 mM Cacodylate Buffer (pH 7.4) in a 1.5 ml Eppendorf tube.
For CD measurements, dilute 10 μl of the 100 μM RNA stock solution in 190 μl (5 μM final concentration) of 60 mM Cacodylate Buffer (pH 7.4) in a 1.5 ml Eppendorf tube.
Anneal the RNA solution prepared in Step B2 by heating the Eppendorf tube containing the solution at 90 °C for 10 min using the Eppendorf Thermomixer equipped with the 1.5 ml SmartBlock. Prepare an ice box while the RNA solution is heating and immediately place the RNA-solution in ice after the 10 min heating cycle is completed.
Store the RNA-solution at 4 °C overnight before performing any FRET or CD measurements.
FRET protocol
In this experiment it is possible to gain insight into the distance between the 5’ and the 3’ ends of the RNA sequence using FRET. To achieve this, excite the FAM fluorophore (placed at the 5’- end of the sequence) at 488 nm and record the emission between 500 and 700 nm, covering the emission peaks of both the FAM and the TAMRA (placed at the 3’ end of the sequence) fluorophore.
Transfer 500 μl of the RNA solution prepared in Step B2a in the Semi-micro quartz cell 108F-QS cuvette and insert it in the Cary Eclipse Fluorimiter pre-equilibrated at the temperature of 17 °C.
After equilibrating the cuvette in the fluorimeter for 10 min record the fluorescence emission using the following parameters:
Excitation wavelength 488 nm;
Emission range 500-700 nm;
Excitation slit 5 nm;
Emission slit 5 nm;
Detector Voltage High.
Increase the temperature of the Cary Variant heating block at 27 °C and once the temperature has been reached equilibrate the sample for at least 10 min.
Record the fluorescence emission using the following parameters:
Excitation wavelength 488 nm;
Emission range 500-700 nm;
Excitation slit 5 nm;
Emission slit 5 nm;
Detector Voltage High.
Repeat Steps C2 to C4 as appropriate. Due to the RNA’s relatively low stability this cycle can be repeated for a maximum of 12-14 times. Representative spectra are displayed in Figure 1A.
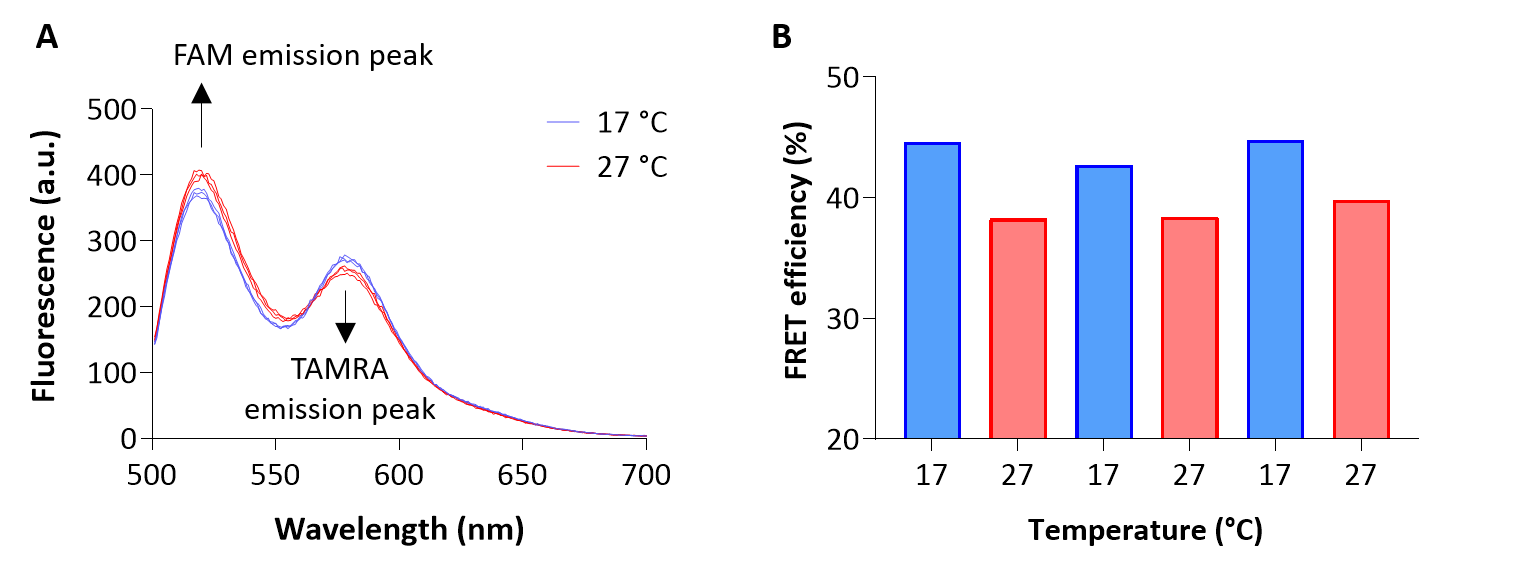
Figure 1. Temperature-dependent hairpin dynamics measured as changes in FRET efficiency. A 5’-FAM and 3’-TAMRA fluorescently labelled 32 nt RNA oligonucleotide harbouring the hairpin sequence was used for the experiment. Emission spectra between 500 and 700 nm (A) were recorded upon excitation at 488 nm over multiple shifts between 17 and 27 °C; peak emissions at 520 and 578 nm were used to calculate FRET efficiencies (B). FRET efficiency is calculated using the following equation: [Fa/(Fa + Fd)] × 100, where Fa is the emission of the acceptor (FAM) and Fd is the emission intensity of the donor (TAMRA).
CD measurement
In this experiment the overall secondary structure adopted by the RNA under different conditions will provide a characteristic CD signature that can be monitored in real time. Spectra with a maximum at ~270 nm and a minimum at ~210 nm are indicative of classical RNA hairpin structures.
Transfer 200 μl of the RNA prepared in Step B2b into a 1 mm quartz cell and place it in the ChirascanTM instrument pre-equilibrated at the temperature of 17 °C.
After equilibrating the cuvette in the Circular Dichroism machine for at least 10 min record the CD intensity (reported as molar ellipticity θ) between 300 and 180 nm using the following parameters:
Record 1 point per nm;
Bandwidth 0.5 nm;
Time per point 1 s.
Acquire the CD spectra at 17 °C for 3 times consecutively.
Raise the ChirascanTM to 27 °C and equilibrate the sample at this temperature for at least 10 min.
Record the CD intensity between 300 and 180 nm using the following parameters:
Record 1 point per nm;
Bandwidth 0.5 nm;
Time per point 1 s.
Acquire the CD spectra at 27 °C for 3 times consecutively.
Repeat Steps D1 to D6 with 200 μl of 60 nM Cacodylate Buffer to record a baseline.
Data analysis
For FRET measurements, divide the recorded emission at 578 nm (TAMRA emission peak) by the sum of the recorded emission at 578 nm and 520 nm (FAM emission peak) for each temperature (Figure 1B); this ratio is inversely correlated with the distance between the two fluorophores (i.e., the ends of the RNA oligonucleotide). A reduction in FRET efficiency thus reflects a (partial) opening or destabilisation of the hairpin structure.
For CD spectroscopy, average the three replicate measurements for each temperature to reduce background noise and subtract the baseline recorded at the respective temperature. Plot the baseline-corrected CD intensity versus wavelength and overlay the two spectra obtained in Steps D3 and D6 to visualize structural differences between the RNA hairpin structures at 17 and 27 °C (Figure 2). Hairpin-type characteristic signals display a detectable maximum at ~270 nm and a minimum ~210 nm, with a more pronounced peak ~210 nm suggesting an increased stability of the hairpin structure.
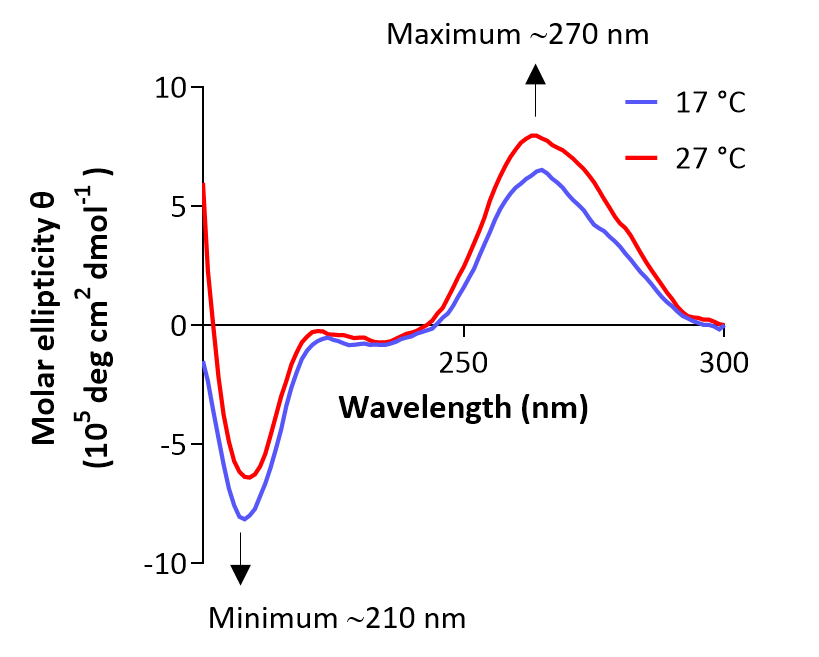
Figure 2. Temperature-dependent hairpin dynamics observed as changes in CD spectra. CD spectra of the hairpin-containing RNA oligonucleotide were recorded at 17 and 27 °C.
Notes
We recommend that RNase-free labware be used throughout the protocol.
The assay temperatures of 17 and 27 °C used in this protocol may be adjusted to the experimenter’s temperature of interest.
Recipes
Cacodylate buffer
60 nM Cacodylic Acid in RNase-free water, pH 7.4 adjusted with 1 M KOH; store at 4 °C (see Section A for details on buffer preparation)
1 M KOH
In RNase-free water
Acknowledgments
M.B. was supported by an EMBO Long-term Fellowship (ALTF 4815), M.D.A and his group are supported by a BBSRC David Phillips Fellowship (BB/R011605/1). B.Y.W.C. and her group are supported by grants from the Wellcome Trust (096082), MRC (MR/R021821/1) and the Royal Society (RGS\R2\192222).
This work is based on the original research paper “An RNA thermoswitch regulates daytime growth in Arabidopsis” by Chung et al. (2020).
Competing interests
We declare that there is no conflict of interest.
References
- Bartys, N., Kierzek, R. and Lisowiec-Wachnicka, J. (2019). The regulation properties of RNA secondary structure in alternative splicing. Biochim Biophys Acta Gene Regul Mech 1862(11-12): 194401.
- Chung, B. Y. W., Balcerowicz, M., Di Antonio, M., Jaeger, K. E., Geng, F., Franaszek, K., Marriott, P., Brierley, I., Firth, A. E. and Wigge, P. A. (2020). An RNA thermoswitch regulates daytime growth in Arabidopsis. Nat Plants 6(5): 522-532.
- Cruz, J. A. and Westhof, E. (2009). The dynamic landscapes of RNA architecture. Cell 136(4): 604-609.
- Ganser, L. R., Kelly, M. L., Herschlag, D. and Al-Hashimi, H. M. (2019). The roles of structural dynamics in the cellular functions of RNAs. Nat Rev Mol Cell Biol 20(8): 474-489.
- Jacobs, E., Mills, J. D. and Janitz, M. (2012). The role of RNA structure in posttranscriptional regulation of gene expression. J Genet Genomics 39(10): 535-543.
- Johansson, J., Mandin, P., Renzoni, A., Chiaruttini, C., Springer, M. and Cossart, P. (2002). An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell 110(5): 551-561.
- Kortmann, J. and Narberhaus, F. (2012). Bacterial RNA thermometers: molecular zippers and switches. Nat Rev Microbiol 10(4): 255-265.
- Krajewski, S. S. and Narberhaus, F. (2014). Temperature-driven differential gene expression by RNA thermosensors. Biochim Biophys Acta 1839(10): 978-988.
- Lazzaretti, D. and Bono, F. (2017). mRNA localization in metazoans: A structural perspective. RNA Biol 14(11): 1473-1484.
- Mailler, E., Paillart, J. C., Marquet, R., Smyth, R. P. and Vivet-Boudou, V. (2019). The evolution of RNA structural probing methods: From gels to next-generation sequencing. Wiley Interdiscip Rev RNA 10(2): e1518.
- Mustoe, A. M., Brooks, C. L. and Al-Hashimi, H. M. (2014). Hierarchy of RNA functional dynamics. Annu Rev Biochem 83: 441-466.
- Soemedi, R., Cygan, K. J., Rhine, C. L., Glidden, D. T., Taggart, A. J., Lin, C. L., Fredericks, A. M. and Fairbrother, W. G. (2017). The effects of structure on pre-mRNA processing and stability. Methods 125: 36-44.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Balcerowicz, M., Di Antonio, M. and Chung, B. Y. W. (2021). Monitoring Real-time Temperature Dynamics of a Short RNA Hairpin Using Förster Resonance Energy Transfer and Circular Dichroism. Bio-protocol 11(6): e3950. DOI: 10.21769/BioProtoc.3950.
Category
Biochemistry > RNA > RNA structure
Plant Science > Plant molecular biology > RNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.