- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Identification of Intrinsic RNA Binding Specificity of Purified Proteins by in vitro RNA Immunoprecipitation (vitRIP)
Published: Vol 11, Iss 5, Mar 5, 2021 DOI: 10.21769/BioProtoc.3946 Views: 4675
Reviewed by: Matilda F. ChanLifeng LiuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
RNA-protein interactions are often mediated by dedicated canonical RNA binding domains. However, interactions through non-canonical domains with unknown specificity are increasingly observed, raising the question how RNA targets are recognized. Knowledge of the intrinsic RNA binding specificity contributes to the understanding of target selectivity and function of an individual protein.
The presented in vitro RNA immunoprecipitation assay (vitRIP) uncovers intrinsic RNA binding specificities of isolated proteins using the total cellular RNA pool as a library. Total RNA extracted from cells or tissues is incubated with purified recombinant proteins, RNA-protein complexes are immunoprecipitated and bound transcripts are identified by deep sequencing or quantitative RT-PCR. Enriched RNA classes and the nucleotide frequency in these RNAs inform on the intrinsic specificity of the recombinant protein. The simple and versatile protocol can be adapted to other RNA binding proteins and total RNA libraries from any cell type or tissue.
Graphic abstract: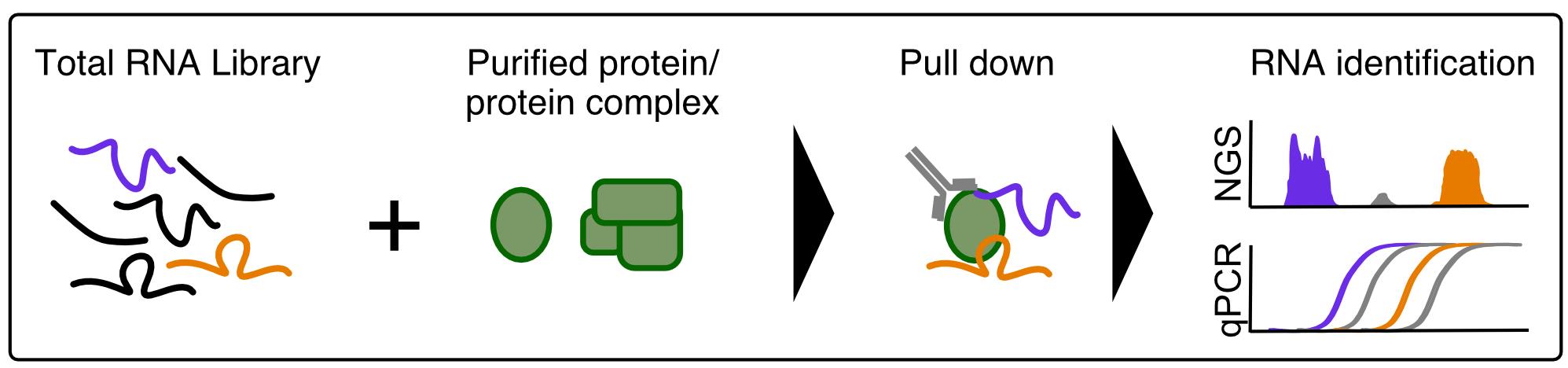
Figure 1. Schematic of the in vitro RNA immunoprecipitation (vitRIP) protocol
Background
Eukaryotic cells contain a number of different RNA classes with thousands of RNA species and a highly diverse set of proteins interacting with those. RNA-protein interactions can be classified as specific and nonspecific, depending on the definition of the bound RNA sequence or structure and on the protein domains involved in the interaction (Jankowsky and Harris, 2015). RNA interactions through non-canonical RNA binding domains with unknown specificity are increasingly observed, which raises the question how dedicated RNA targets are recognized.
A better understanding of the function of a particular RNA binding protein would thus be facilitated by the identification of its intrinsic RNA binding specificity. Since RNA binding in vivo is often modulated by cooperating factors, intrinsic specificity can only be determined in an in vitro approach (Sloan and Bohnsack, 2018). Several high-throughput techniques, such as RNAcompete (Ray et al., 2013), RNA Bind-n-Seq (Lambert et al., 2014), and in vitro iCLIP (Sutandy et al., 2018) are able to determine the RNA binding profiles of individual proteins in vitro. These methodologies use RNA oligonucleotide libraries or pools of in vitro-transcribed RNA as substrates. Such artificial RNA libraries may be highly complex, but do not represent the cellular RNA pool and may not contain secondary structures.
We developed an in vitro RNA immunoprecipitation assay (vitRIP) that uncovers intrinsic RNA-binding specificities of isolated proteins in the context of the total cellular RNA pool (Figure 1) (Müller et al., 2020). Applying vitRIP to the DExH helicase MLE (maleless) and the male-specific lethal dosage compensation complex (MSL-DCC) from Drosophila melanogaster revealed the mechanism of specific incorporation of the long non-coding roX (RNA on the X) RNAs into the MSL-DCC (Müller et al., 2020). The simple vitRIP methodology identifies transcripts bound to a recombinant protein in native conditions (no crosslinking involved) by deep sequencing or quantitative RT-PCR. vitRIP uses the complex cellular transcriptome as substrate and informs on the intrinsic binding specificity of a given protein outside of its physiological context. Furthermore, the experimental setting can be easily controlled and adapted to the research question. The following protocol details the vitRIP procedure using the RNA helicase MLE and total RNA extracted from male Drosophila S2 cells as an example (Müller et al., 2020). However, we propose that vitRIP can be applied to any RNA binding protein, which can be produced recombinantly and purified to near-homogeneity. Moreover, total RNA extracted from any cell type or tissue can serve as an RNA library.
Materials and Reagents
Note: Please ensure that all reagents and materials are RNase-free.
1.5-ml low-binding tubes (Sarstedt, catalog number: 72.706.700 )
Drosophila melanogaster S2 cells (Drosophila Genomics Resource Center, https://dgrc.bio.indiana.edu/Home) or any cell line/tissue
RNeasy Mini Kit (Qiagen, catalog number: 74104 )
Note: We did not test other RNA extraction kits, but would assume the manufacturer is not critical. Nevertheless, the extractability of various RNA species might differ between different kits.
Anti-FLAG M2 Affinity Gel (Sigma, catalog number: A2220 ) or other appropriate affinity resin, depending on the tag of the recombinant protein
Yeast tRNA solution (Sigma, catalog number: R5636 )
BSA fraction V powder (for blocking) (Sigma, catalog number: 0 5482 )
Nuclease-free water (Thermo Fisher Scientific, catalog number: AM9937 )
DNase I recombinant, RNase-free (Sigma, catalog number:0 4716728001 )
BSA 20 mg/ml solution (for vitRIP) (New England Biolabs, catalog number: B9000S )
Recombinant RNasin RNase Inhibitor (40 U/μl) (Promega, catalog number: N2515 )
Adenosine 5’-triphosphate disodium salt (ATP) (Sigma, catalog number: 10127523001 )
Proteinase K (BioCat, catalog number: BIO-37039-BL )
10% SDS solution
Phenol:chloroform:phenylalcohol ROTI Aqua-P/C/I (Carl Roth, catalog number: X985.1 )
3 M sodium acetate pH 5.2
Glycogen (20 mg/ml) (Sigma, catalog number: 10901393001 )
Ethanol absolute for analysis (Sigma, catalog number: 1009832500 )
SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific, catalog number: 18080051 )
Fast SYBR Green Mastermix (Thermo Fisher Scientific, catalog number: 4385610 )
Quant-iT Qubit RNA HS Assay kit (Thermo Fisher Scientific, catalog number: Q32855 )
Agilent RNA 6000 Pico Kit (Agilent, catalog number: 5067-1513 )
Agilent DNA 1000 Kit (Agilent, catalog number: 5067-1504 ) or Agilent High Sensitivity DNA Kit (Agilent, catalog number: 5067-4626 )
NEBNext rRNA Depletion Kit (Human/Mouse/Rat) (New England Biolabs, catalog number: E6310 )
NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs, catalog number: E7760 )
KCl
NaCl
Na2HPO4
KH2PO4
HEPES
MgCl2
NP40 (IGEPAL CA-630, Sigma, catalog number: I3021 )
Anti-FLAG M2 Monoclonal Antibody (Sigma, catalog number: F3165 )
2× SDS PAGE sample buffer (125 mM Tris HCl pH 6.8, 4% SDS, 20% glycerol, 0.01% bromophenol blue, 200 mM DTT)
1× PBS (see Recipes)
vitRIP-100 buffer (see Recipes)
vitRIP-250 buffer (see Recipes)
Equipment
-80 °C freezer
DeNovix DS-11 spectrophotometer (Biozym, catalog number: 31DS-11 ) or other spectrophotometer with 254 nm and 280 nm wavelength
Refrigerated centrifuge (Eppendorf, model: 5424R )
Eppendorf ThermoMixer C with heated lid (Eppendorf, catalog numbers: 5382000015 , 5308000003 )
Rotator (Neolab, catalog number: 2-1175 )
Qubit fluorometer (Invitrogen)
Bioanalyzer 2100 Instrument (Agilent)
Lightcycler 480 Instrument II, 384-well (Roche, catalog number: 0 5015243001 ) or another Real-Time PCR System
(Access to) Illumina HiSeq 1500 Sequencing Platform
Software
STAR – Spliced Transcript Alignment to a Reference (Alexander Dobin, Cold Spring Harbor Laboratory, https://github.com/alexdobin/STAR) (Dobin et al., 2013)
Samtools (Genome Research Limited, Sanger Institute, http://www.htslib.org) (Li et al., 2009)
Bedtools (Quinlan laboratory, University of Utah, https://bedtools.readthedocs.io/en/latest/)
IGV – Integrative Genomics Viewer and igvtools (IGV Team, Broad Institute and UC San Diego, http://software.broadinstitute.org/software/igv/home) (Robinson et al., 2011)
R (R Core Team, https://www.r-project.org)
RStudio (RStudio, https://rstudio.com)
Bioconductor (Bioconductor Core Team, http://bioconductor.org) (Huber et al., 2015)
Procedure
Notes:
It is essential to perform the experiment in ≥3 biological replicates using different batches of purified protein and total RNA.
Conditions need to be optimized for each experimental system. Protein and/or RNA titration might be required to obtain an optimal signal-to-noise ratio.
Include a vitRIP reaction with experimental RNA, but lacking the recombinant protein, to score RNA background on beads.
Additional control reactions (e.g., total RNA extracted from a different species or protein mutated in its RNA binding domain (if known)) might help to further characterize the specificity of identified protein-RNA interactions.
Work in RNase-free conditions.
Preparation of total RNA
Cultivate Drosophila melanogaster S2 cells under standard conditions. For details, see Drosophila Genomics Resource Center (https://dgrc.bio.indiana.edu/Home).
Collect 5 × 106 exponentially growing cells by centrifugation (500 × g, 5 min, 25 °C) and discard the supernatant.
Wash the cell pellet with 500 µl PBS, collect by centrifugation (500 × g, 5 min, 25 °C) and discard the supernatant.
Proceed directly with RNA extraction or store the cell pellet at -80 °C for a maximum duration of 3 months.
Extract total RNA using the Qiagen RNeasy Mini Kit according to the manufacturer’s instructions. Elute the RNA in 50 µl RNase-free water and determine the concentration spectrophotometrically. The typical yield from 5 × 106 S2 cells ranges between 25-50 µg.
Store extracted RNA at -80 °C until further use (for a maximum duration of 1 year). Avoid frequent freezing/thawing of the RNA sample to prevent degradation.
Note: Any other cell line or animal tissue can be used. We successfully tested several male and female Drosophila cell lines as well as head tissue from adult Drosophila. The methodology for RNA extraction from tissues might require adjustment.
Preparation of highly purified protein
Note: The vitRIP protocol presented here uses the well-characterized RNA helicase MLE as an example. However, vitRIP can be applied to any protein, which can be purified to near-homogeneity. Protein purification strategies have to be determined for each protein individually and cannot be generalized.
Express the protein of interest in an appropriate heterologous expression system (e.g., E. coli or SF21 insect cells) and purify to near-homogeneity. Analyze the purity of the protein sample by SDS-PAGE and Coomassie staining.
Determine the protein concentration by an appropriate method (absorption at 280 nm or Bradford protein assay). If necessary, use spin concentrators to increase the protein concentration. Flash freeze small aliquots (10-20 µl) and store at -80 °C. The required protein concentration for vitRIP strongly depends on the affinity to RNA substrates and needs to be determined empirically by applying protein titration series, as described in Müller et al. (2020). See also Step D, Note 3.
Before use, thaw protein prep on ice, spin down to remove potential aggregates (20,000 × g, 10 min, 4 °C) and use the supernatant for vitRIP.
The purification of full-length FLAG-tagged Drosophila melanogaster MLE after baculovirus-driven expression in SF21 cells is described in detail in Izzo et al. (2008) and Maenner et al. (2013), differing only in the elution buffer for MLE (20 mM HEPES-KOH pH 7.6, 200 mM KCl, 1 mM DTT, 2 mM MgCl2, 10% glycerol). Purified MLE has a typical concentration of 0.5-1 µg/µl and can be used directly for vitRIP.
Preparation of Anti-FLAG M2 Affinity Gel
Note: Prepare the resin always fresh. Any other resin matching the affinity tag can be used. Alternatively, protein-specific antibodies coated to Protein A or Protein G Sepharose may be used.
Use 20 µl Anti-FLAG M2 Affinity Gel (“beads”) for each vitRIP reaction.
Wash the beads: Add 1 ml PBS, invert the tube 10 times, centrifuge for 1 min at 500 × g, 4 °C and discard the supernatant. Repeat this step one more time.
Block the beads: Add 2% BSA/PBS/0.1 mg/ml yeast tRNA to the bead and incubate for 1 h at 4 °C with rotation. Centrifuge for 1 min at 500 × g, 4 °C and discard the supernatant. This step intends to prevent non-specific binding of protein and/or total RNA to the FLAG affinity gel.
Wash the beads: Add 1 ml vitRIP-100 buffer, invert the tube 10 times, centrifuge for 1 min at 500 × g, 4 °C and discard the supernatant. Repeat this step one more time.
With the last washing step, discard as much of the supernatant as possible and keep beads on ice.
Pull down of RNA-protein complexes
Notes:
Pipet all steps on ice and centrifuge at 4 °C. Use low-binding tubes.
The RNA amount for vitRIP depends on the abundance of individual transcripts and on the affinity of the studied protein to RNA. The amount might be determined empirically by applying an RNA titration series. In our hands, 2 µg total RNA per vitRIP reaction (100 µl volume) worked well.
The protein concentration for vitRIP depends on the affinity to RNA and needs to be determined empirically by titration. We recommend to start with 10-250 nM of recombinant protein and 2 µg of total RNA. If no binding is observed, protein amount may be increased. MLE vitRIP was performed with 5-50 nM protein.
Per reaction, dilute 2 µg of total RNA with vitRIP-100 buffer to a final volume of 10 µl. 10% of this mix (1 µl) serves as an RNA input sample and is kept on ice.
Mix the remaining 90% of the RNA mix (9 µl) with the purified protein (MLE-FLAG: 5 nM and 50 nM, respectively) in a total volume of 100 µl in vitRIP-100 buffer including 10 µg BSA, 0.1 U/µl RNase-free recombinant DNase I, 0.8 U/µl RNasin RNase inhibitor and 1 mM ATP.
Incubate 30 min at 25 °C (thermoblock with heated lid).
Spin down reaction at 20,000 × g, 1 min, 4 °C and transfer the supernatant to blocked and equilibrated FLAG beads (from Step C5) in a 1.5 ml tube.
Incubate for 30 min at RT (22 °C) with rotation.
Centrifuge at 500 × g, 1 min, 4 °C and discard the supernatant (= unbound fraction).
Wash beads with 1 ml vitRIP-100 buffer: Add 1 ml buffer, rotate 1 min at room temperature, centrifuge for 1 min at 500 × g, 4 °C and discard the supernatant.
Wash beads with 1 ml vitRIP-250 buffer: Add 1 ml buffer, rotate 1 min at room temperature, centrifuge for 1 min at 500 × g, 4 °C and discard the supernatant.
Wash beads with 1 ml vitRIP-100 buffer: Add 1 ml buffer, rotate 1 min at room temperature and distribute the beads in two tubes for protein and RNA extraction: 75% (750 µl) for RNA extraction and 25% (250 µl) for protein extraction. Centrifuge for 1 min at 500 × g, 4 °C and discard the supernatant.
Protein extraction for Western Blot analysis
To each sample (25% bead material, from Step D10), add 20 µl of 2× SDS sample buffer. Incubate at 95 °C, 5 min, with agitation.
Centrifuge for 1 min at 500 × g to pellet beads and analyze the supernatant by SDS-PAGE and Western Blot for successful protein pull down.
Note: vitRIP of FLAG-tagged MLE is analyzed by Western blot analysis using anti-FLAG antibody at 1:5,000 (v/v) dilution. MLE is visible as a single band corresponding to a molecular weight of 145 kDa.
RNA extraction for RNA identification
Input samples: Mix 10% RNA input (1 µl), 5 µl 10% SDS, 10 µl Proteinase K (10 mg/ml) and 84 µl vitRIP-100 buffer. Incubate in a thermoblock (with heated lid) at 55 °C, 45 min with agitation at 1,000 rpm.
IP samples (75% bead material, from step D10): Add 5 µl 10% SDS, 10 µl Proteinase K (10 mg/ml) and 85 µl vitRIP-100 buffer. Incubate in a thermoblock (with heated lid) at 55 °C, 45 min with agitation (1,000 rpm).
Centrifuge tubes for 1 min at 500 × g at room temperature to pellet beads of IP samples.
Transfer the supernatant (contains eluted RNA) and transfer to a fresh 1.5 ml tube.
To each sample (Input and IP), add 1 volume (around 100 µl) of acidic phenol:chloroform:isoamylalcohol. Vortex each tube thoroughly for 10 sec and centrifuge 5 min at 20,000 × g at room temperature.
Safety note: Always use a fume hood when handling phenol:chloroform:isoamylalcohol and appropriately dispose of phenol-containing waste.
Transfer the upper, aqueous phase (contains RNA; around 100 µl) to a fresh 1.5-ml tube and add 1/10 volume 3 M sodium acetate pH 5.2, 20 µg RNase-free glycogen and 2.5 volumes of ice-cold 100% EtOH.
Incubate at -20 °C for > 12 h to precipitate the RNA.
Centrifuge at 20,000 × g (full speed), 4 °C for 30 min. Carefully remove and discard the supernatant with a pipet tip without disturbing the RNA pellet, which is small and might be invisible.
Wash pellet by adding 1 ml of ice-cold 70% EtOH.
Centrifuge at 20,000 × g (full speed), 4 °C for 15 min. Carefully remove and discard the supernatant.
Remove as much liquid as possible with a 20 µl pipet and air-dry the pellet (10-15 min).
Resuspend the RNA pellet in 20 µl RNase-free water. Pipet up and down 10 times, followed by a quick spin. For complete solubilization, incubate the samples for 15 min at 25 °C with gentle agitation (500 rpm).
Quantify 2 µl of the purified RNA using a Qubit fluorometer and the Qubit RNA High Sensitivity (HS) Assay kit.
Note: The RNA concentration of IP samples might very low and not in the range of the Qubit RNA HS Assay kit standard. Our experience showed that RNA sequencing libraries could still be prepared with the NEB Ultra II Directional RNA Library Prep Kit. Other RNA-Seq library prep kits dealing with low-input samples may be employed.
Store RNA at -80 °C until further use.
RT-PCR
Notes:
If one or several RNA targets of the protein of interest are known, vitRIP can be analyzed by quantitative RT-PCR using gene-specific primers. It is important to include a non-bound transcript (e.g., housekeeping genes) as the negative control.
In MLE vitRIP, the enrichment of the known MLE target roX2 RNA is quantified using roX2-specific primers and represented as the bound fraction relative to the input. The abundant 7SK RNA serves as the negative control. For details on primer sequences, see Müller et al. (2020). An example is given in Figure 2A.
DNase I digest
Mix 8 µl RNA, 1 µl 10× DNase I reaction buffer, 0.5 µl DNase I and 0.5 µl RNasin.
Incubate for 1 h at 37 °C. Store on ice.
cDNA synthesis using the SuperScript III First-Strand Synthesis System
Use 10 µl DNase I-treated RNA and follow the manufacturer’s instructions for cDNA synthesis using random hexamer priming.
Dilute the cDNA 1:5 to 1:50 (depending on the transcript abundance) with RNase-free water and analyze by qRT-PCR using transcript-specific primers and FAST SYBR Green Mastermix. Follow the recommendations specific for your Real-Time PCR machine.
RNA-Seq Library preparation
Note: The library preparation protocol depends on the experimental strategy. The high content of rRNA in total RNA will interfere with the analysis and should be removed before the library prep. In MLE vitRIP samples, rRNA is depleted using rRNA probes specific for human/mouse/rat rRNA, which also work reasonably well for Drosophila rRNA. This strategy allows for the identification of various RNA species in vitRIP samples (coding and non-coding). Alternatively, polyadenylated RNAs can be enriched using oligo d(T) beads if the experimental strategy aims at identifying exclusively enriched polyadenylated RNAs.
Starting material for library preparation
Quality control: Analyze 1 µl of purified input and immunoprecipitated RNA on a Bioanalyzer using the RNA 6000 Pico Kit.
Choose an appropriate RNA quantity according to the recommendations of the RNA-Seq library prep kit. For MLE vitRIP library preparation with the NEB Ultra II Directional RNA Library Prep Kit, 30-40 ng RNA served as starting material.
rRNA depletion
Use 30-40 ng RNA as quantified by Qubit fluorometer for rRNA depletion using the NEB rRNA Depletion Kit.
Follow the manufacturer’s protocol for rRNA depletion (NEB # E6310 ).
Analyze 1 µl of rRNA-depleted sample on a Bioanalyzer using the RNA 6000 Pico Kit. The major 18S and 28S rRNA peaks (Drosophila) should be largely eliminated. It is not possible to obtain an RNA Integrity Number (RIN) for Drosophila RNA samples.
Library preparation
Use the rRNA-depleted sample for library preparation following the instructions of the NEB Ultra II Directional RNA Library Prep Kit (NEB # E7760 ).
Assess quality and quantity of 1 µl of each library on a Bioanalyzer using the DNA 1000 or DNA High Sensitivity Kit. A single peak with a maximum of approximately 300 bp should be observed (Figure 2B).
Sequence on an Illumina HiSeq1500 instrument in 50 bp paired-end mode. Sequencing run settings were 50bp-8bp-8bp-50bp (read1-index1-index2-read2), where the second index file was not used.
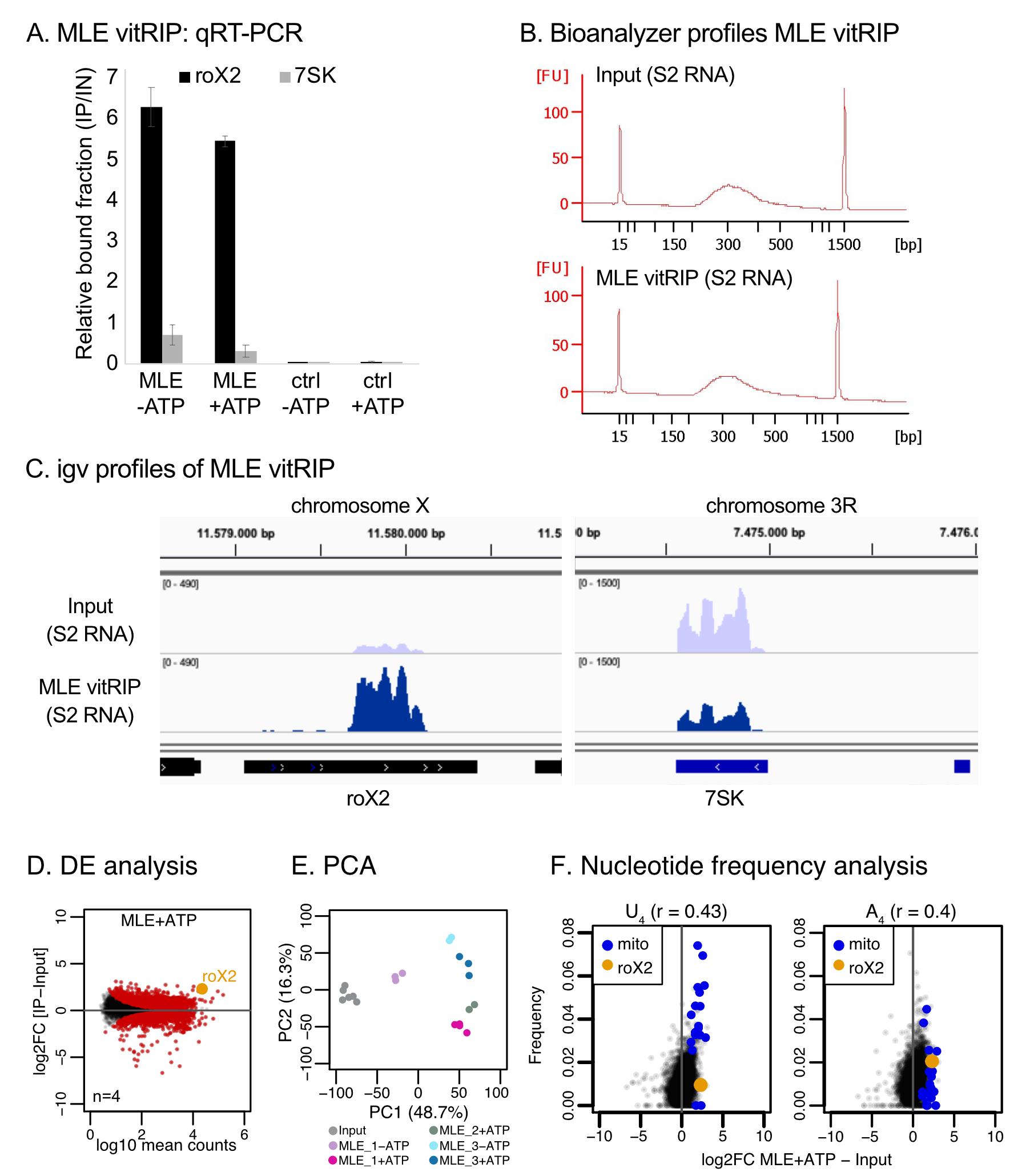
Figure 2. Representative examples of vitRIP analysis. For details, see the original publication (Müller et al., 2020). A. MLE vitRIP with S2 RNA in absence or presence of ATP. Enrichment of roX2 and 7SK was analyzed by qRT-PCR and is displayed relative to the input. Ctrl indicates RNA-only vitRIP samples lacking recombinant MLE. B. Representative Bioanalyzer profiles of RNA sequencing libraries prepared of input and MLE vitRIP samples. Libraries were analyzed on a DNA 1000 chip and show a single peak with an expected maximum at approximately 300 bp. C. Genome browser views of representative vitRIP profiles of MLE on roX2 and 7SK in comparison to S2 input. Genomic coordinates are given above the graph. D. MA plot representing differential expression analysis of MLE vitRIP. E. Principal Component Analysis (PCA) of MLE vitRIP-seq data at different conditions. X-axis shows the first (PC1) and y-axis the second (PC2) principal component. F. Comparison of U-/A-tetranucleotide frequencies of RNA in MLE vitRIP relative to the input. RNA classes of interest (roX2, mitochondrial RNA) are highlighted. Spearman´s correlation (r) is indicated.This section describes the RNA-Seq analysis of MLE vitRIP as published in Müller et al., 2020. Adjustment of the procedure to the chosen model system might be required.
Analyze ≥3 biological replicates to ensure valid interpretation of the data.
Simplified example code chunks are shown, see details:https://github.com/tschauer/vitRIP_2020.
Processing of Illumina sequencing reads
Map paired-end reads (50 bp) to the reference genome (Drosophila melanogaster version dm6) and count reads per gene using the STAR aligner (version 2.5.3a) and the Flybase annotation (ftp://ftp.flybase.net/releases/FB2017_04/dmel_r6.17/gtf/dmel-all-r6.17.gtf.gz) while filtering out reads with multiple alignments.
STAR --runThreadN 8--readFilesCommand gunzip -c
--quantMode GeneCounts
--genomeDir ${path_to_star_index}
--sjdbGTFfile ${gtf_file}
--readFilesIn ${fastq_file}_1.txt.gz ${fastq_file}_2.txt.gz
--outFileNamePrefix ${fastq_file}.
--outSAMtype BAM SortedByCoordinate
--limitBAMsortRAM 5000000000
--outFilterMultimapNmax 1
Count the total number of aligned reads using samtools (version 1.7) and generate normalized (reads per million) bedgraph files using genomeCoverageBed (bedtools version 2.27.1).
total=`samtools view -c ${bam_file}`scaler=`echo "scale=10; 1/(${total}/1000000)" | bc`
genomeCoverageBed -ibam ${bam_file} -bg -split -scale ${scaler} > ${bedgraph_file}
Convert bedgraph to tdf files using igvtools (version 2.3.98).
igvtools toTDF -z 5 ${bedgraph_file} ${tdf_file} ${path_to_fasta}Visualize tdf files in the IGV browser (Figure 2C).
Differential expression analysis
Load and merge count tables (*ReadsPerGene.out.tab files generated by STAR) into R (version 3.6.1) and keep genes with at least 1 read count present in 75% of the samples.
filter <- apply(count_table, 1, function(x) length(x[x>1]) >= ncol(count_table)/1.333)count_table_filtered <- count_table[filter,]
Use the DESeq2 package (version 1.26.0) for differential expression analysis while adding replicate information as batch variable.
dds <- DESeqDataSetFromMatrix(countData = count_table_filtered,colData = column_data,
design = ~batch+sample)
dds <- DESeq(dds)
Note: Samples that should be directly compared to each other have to be fitted in the same DESeq2 model.
Obtain log2FoldChange estimates and adjusted p-values using the results function (DESeq2). Set the adjusted p-value cutoff to 0.01.
res <- results(dds,contrast = c("sample", "IP", "Input"),
independentFiltering = FALSE)
res.sign <- subset(res, padj < 0.01)
Visualize results as MA plot, label significant genes in red and example genes in orange (Figure 2D).
plot(res$baseMean, res$log2FoldChange,log="x", xlim = c(1, 1e6), ylim = c(-10,10),
xlab = "log mean counts", ylab = "log2 fold change",
col = rgb(0,0,0,0.1), pch = 19, cex = 0.25)
points(res.sign$baseMean, res.sign$log2FoldChange,
col = rgb(0.8,0,0,0.5), pch = 19, cex = 0.25)
favorite_gene <- rownames(res) %in% c("FBgn0019660","FBgn0019661")
points(res$baseMean[favorite_gene],res$log2FoldChange[favorite_gene],
col = rgb(0.9,0.6,0,1), pch = 19, cex = 1)
Principal Component Analysis (PCA)
Correct for batch effect using the ComBat function (sva package version 3.32) on the normalized read counts.
batch_var <- colData(dds)$batch
modcombat <- model.matrix(~sample, data = colData(dds))
log2_corrected_counts <- ComBat(dat = log2(counts(dds, normalized = TRUE)+1),
batch = batch_var, mod = modcombat,
par.prior = TRUE, prior.plots = FALSE)
Perform and plot PCA on the batch-corrected counts (Figure 2E).
pca <- prcomp(t(log2_corrected_counts), scale. = TRUE)percentVar <- round(pca$sdev^2/sum(pca$sdev^2)*100,1)[1:10]
plot(pca$x[,1], pca$x[,2],
col = color_palette[colData(dds)$sample], pch = 16, cex = 0.7,
xlab = paste("PC1 (", percentVar[1], "%)", sep=""),
ylab = paste("PC2 (", percentVar[2], "%)", sep=""))
Nucleotide frequency analysis
Set up annotation by converting FlyBase annotation to TxDb object (GenomicFeatures package version 1.36.4) and extract genic sequences using the BSgenome package (version 1.52).
txdb <- makeTxDbFromGFF("dmel-all-r6.17.gtf", format="gtf")
seqlevelsStyle(txdb) <- "UCSC"
seqlevels(txdb) <- gsub("mitochondrion_genome","chrM", seqlevels(txdb))
chromosomes <- c("chrX","chr2L","chr2R","chr3L","chr3R","chr4","chrM","chrY")ranges_genes <- genes(txdb)
ranges_genes <- keepSeqlevels(ranges_genes, chromosomes, pruning.mode = "coarse")
genic_seq <- BSgenome::getSeq(BSgenome.Dmelanogaster.UCSC.dm6, ranges_genes)Calculate genic nucleotide frequencies using the oligonucleotideFrequency function (Biostrings package version 2.52). Set oligonucleotide width to either 4, 5, or 6, sliding window step to 1 and as.prob to TRUE.
genic_4merfreq <- oligonucleotideFrequency(genic_seq, width = 4, step = 1, as.prob = T)
rownames(genic_4merfreq) <- names(genic_seq)
Select genes, which are significantly enriched (adjusted p-value < 0.01 and log2[IP/Input] > 0).
res.sign_enriched <- subset(res, padj < 0.01 & res$log2FoldChange > 0)
select_enriched <- rownames(genic_4merfreq) %in% rownames(res.sign_enriched)
select_notenriched <- !(rownames(genic_4merfreq) %in% rownames(res.sign_enriched))
freqs_enriched <- genic_4merfreq[select_enriched,]
freqs_notenriched <- genic_4merfreq[select_notenriched,]
Visualize nucleotide frequencies for genes enriched or not enriched as boxplots. Sort sequences by their median frequency.
freq_order <- order(apply(freqs_enriched, 2, median), decreasing = T)freqs_enriched <- freqs_enriched[,freq_order]
freqs_notenriched <- freqs_notenriched[,freq_order]
freqs_merged <- c(as.list(data.frame(freqs_enriched)),as.list(data.frame(freqs_notenriched)))
boxplot(freqs_merged[rep(1:25, each=2)+c(0,ncol(freqs_enriched))],col =c("darkred","darkgrey"), las=2, outline=F,
ylab = "Frequency")
Visualize association between vitRIP enrichment (i.e., log2[IP/Input]) and nucleotide frequencies as scatterplot. Calculate Spearman´s correlation to assess the relationship (Figure 2F).
res_freq <- merge(res, genic_4merfreq, by="row.names")
plot(res_freq$log2FoldChange, res_freq[,"TTTT"],main = "", xlab = "log2 Fold Change", ylab = "Frequency UUUU",
xlim = c(-8,8), col = rgb(0,0,0,0.1), pch = 19, cex = 0.25)
abline(v=0, col="grey32")
corr <- cor(res_freq$log2FoldChange, res_freq[,"TTTT"], method = "spearman")title(paste("cor =", round(corr,2)), line = 0.5, cex.main=1)
Evaluation of enriched/not-enriched RNA classes
Extract annotations (e.g., exons, introns, 5’ and 3’ UTRs as well as for snRNAs, snoRNAs, and tRNAs) from the GTF annotation (i.e., dmel-all-r6.17.gtf).
anno_gtf <- import.gff("dmel-all-r6.17.gtf")
genes_3UTR <- anno_gtf[anno_gtf$type == "3UTR",]genes_3UTR_length <- aggregate(width(genes_3UTR),
by = list(genes_3UTR$gene_id), FUN = max)
select_q95 <- genes_3UTR_length[,2] > quantile(genes_3UTR_length[,2], 0.95)
class.long3UTR <- genes_3UTR_length[,1][select_q95]
Note: long 3’ UTRs transcripts were selected as transcripts with the longest 5% of 3’ UTRs. In cases, when a gene was annotated with 3’ UTR isoforms, the isoform with the longest 3’ UTR was taken.
Extract annotation of Drosophila snoRNA classes from the SnOPY database (snoRNA orthological gene database; http://snoopy.med.miyazaki-u.ac.jp) (Yoshihama et al., 2013). Convert gene names to gene ids using org.Dm.eg.db package (version 3.8.2)
snoRNA_snopy <- read.table("snoRNA_snopy.txt", header = T, sep = "\t", stringsAsFactors = F)
snoRNA_HACA <- snoRNA_snopy$snoRNA.name[snoRNA_snopy$Box == "H/ACA"]class.snoRNA_HACA <- mapIds(org.Dm.eg.db, keys = snoRNA_HACA,
keytype = "SYMBOL", column = "FLYBASE")
Extract information on adenosine-to-inosine edited RNAs from published data (St Laurent et al., 2013). Read the excel sheet to R (gdata package version 2.18.0) and select unique gene ids from the GTF annotation.
editedRNAs_symbols <- read.xls("nsmb.2675-S2.xlsx", sheet = 4, stringsAsFactor = FALSE)editedRNAs_symbols <- editedRNAs_symbols[,1]
editedRNAs_symbols <- gsub("-R.*", "", editedRNAs_symbols[-1:-2])
class.editedRNAs <- anno_gtf$gene_id[anno_gtf$gene_symbol %in% editedRNAs_symbols]class.editedRNAs <- unique(class.editedRNAs)
Use Fisher exact test to evaluate the enrichment of selected classes of RNAs.
class.mito <- ranges_genes[seqnames(ranges_genes) == "chrM"]$gene_id
Sign.inClass <- sum((rownames(res) %in% class.mito) &res$padj < 0.01 & res$log2FoldChange > 0)
NS.inClass <- sum((rownames(res) %in% class.mito) &res$padj >= 0.01 & res$log2FoldChange > 0)
Sign.notClass <- sum(!(rownames(res) %in% class.mito) &res$padj < 0.01 & res$log2FoldChange > 0)
NS.notClass <- sum(!(rownames(res) %in% class.mito) &res$padj >= 0.01 & res$log2FoldChange > 0)
fisher.test(matrix(c(Sign.inClass,Sign.notClass,
NS.inClass,
NS.notClass), nrow = 2, byrow = T))
1× PBS
140 mM NaCl
2.7 mM KCl
10 mM Na2HPO4
1.8 mM KH2PO4
vitRIP-100 buffer
25 mM HEPES-NaOH pH 7.6
100 mM NaCl
0.05% (v/v) NP40
3 mM MgCl2
vitRIP-250 buffer
25 mM HEPES-NaOH pH 7.6
250 mM NaCl
0.05% (v/v) NP40
3 mM MgCl2
Data analysis
Notes:
Recipes
Note: Prepare all buffers in RNase-free conditions.
Acknowledgments
This work was supported by the German Research Foundation through grant Be1140/9-1. This protocol was originally published in Müller et al. (2020).
Competing interests
No competing interests to declare.
References
- Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T. R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1): 15-21.
- Huber, W., Carey, V. J., Gentleman, R., Anders, S., Carlson, M., Carvalho, B. S., Bravo, H. C., Davis, S., Gatto, L., Girke, T., Gottardo, R., Hahne, F., Hansen, K. D., Irizarry, R. A., Lawrence, M., Love, M. I., MacDonald, J., Obenchain, V., Oles, A. K., Pages, H., Reyes, A., Shannon, P., Smyth, G. K., Tenenbaum, D., Waldron, L. and Morgan, M. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 12(2): 115-121.
- Izzo, A., Regnard, C., Morales, V., Kremmer, E. and Becker, P. B. (2008). Structure-function analysis of the RNA helicase maleless. Nucleic Acids Res 36(3): 950-962.
- Jankowsky, E. and Harris, M. E. (2015). Specificity and nonspecificity in RNA-protein interactions. Nat Rev Mol Cell Biol 16(9): 533-544.
- Lambert, N., Robertson, A., Jangi, M., McGeary, S., Sharp, P. A. and Burge, C. B. (2014). RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol Cell 54(5): 887-900.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Maenner, S., Muller, M., Frohlich, J., Langer, D. and Becker, P. B. (2013). ATP-dependent roX RNA remodeling by the helicase maleless enables specific association of MSL proteins. Mol Cell 51(2): 174-184.
- Müller, M., Schauer, T., Krause, S., Villa, R., Thomae, A. W. and Becker, P. B. (2020). Two-step mechanism for selective incorporation of lncRNA into a chromatin modifier. Nucleic Acids Res 48(13): 7483-7501.
- Ray, D., Kazan, H., Cook, K. B., Weirauch, M. T., Najafabadi, H. S., Li, X., Gueroussov, S., Albu, M., Zheng, H., Yang, A., Na, H., Irimia, M., Matzat, L. H., Dale, R. K., Smith, S. A., Yarosh, C. A., Kelly, S. M., Nabet, B., Mecenas, D., Li, W., Laishram, R. S., Qiao, M., Lipshitz, H. D., Piano, F., Corbett, A. H., Carstens, R. P., Frey, B. J., Anderson, R. A., Lynch, K. W., Penalva, L. O., Lei, E. P., Fraser, A. G., Blencowe, B. J., Morris, Q. D. and Hughes, T. R. (2013). A compendium of RNA-binding motifs for decoding gene regulation. Nature 499(7457): 172-177.
- Robinson, J. T., Thorvaldsdottir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G. and Mesirov, J. P. (2011). Integrative genomics viewer. Nat Biotechnol 29(1): 24-26.
- Sloan, K. E. and Bohnsack, M. T. (2018). Unravelling the Mechanisms of RNA Helicase Regulation. Trends Biochem Sci 43(4): 237-250.
- St Laurent, G., Tackett, M. R., Nechkin, S., Shtokalo, D., Antonets, D., Savva, Y. A., Maloney, R., Kapranov, P., Lawrence, C. E. and Reenan, R. A. (2013). Genome-wide analysis of A-to-I RNA editing by single-molecule sequencing in Drosophila. Nat Struct Mol Biol 20(11): 1333-1339.
- Sutandy, F. X. R., Ebersberger, S., Huang, L., Busch, A., Bach, M., Kang, H. S., Fallmann, J., Maticzka, D., Backofen, R., Stadler, P. F., Zarnack, K., Sattler, M., Legewie, S. and Konig, J. (2018). In vitro iCLIP-based modeling uncovers how the splicing factor U2AF2 relies on regulation by cofactors. Genome Res 28(5): 699-713.
- Yoshihama, M., Nakao, A. and Kenmochi, N. (2013). snOPY: a small nucleolar RNA orthological gene database. BMC Res Notes 6: 426.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Müller, M., Schauer, T. and Becker, P. B. (2021). Identification of Intrinsic RNA Binding Specificity of Purified Proteins by in vitro RNA Immunoprecipitation (vitRIP). Bio-protocol 11(5): e3946. DOI: 10.21769/BioProtoc.3946.
Category
Biochemistry > Protein > Interaction > Protein-RNA interaction
Biochemistry > RNA > RNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.