- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Defined Mutant Library Sequencing (DML-Seq) for Identification of Conditional Essential Genes
(*contributed equally to this work) Published: Vol 11, Iss 5, Mar 5, 2021 DOI: 10.21769/BioProtoc.3943 Views: 4670
Reviewed by: Srujana Samhita YadavalliThirugnanasambandam RajendranAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Transposon insertion sequencing (TIS) is an emerging technique which utilizes a massive transposon mutant library to screen specific phenotype and determine the conditional essential genetic requirements for bacterial fitness under distinct conditions combined with high-throughput parallel sequencing technology. Compared with a massive mutant library in traditional TIS, the defined mutant library sequencing (DML-Seq) has advantages as: 1) efficient mutagenesis; 2) low bottleneck effects; 3) avoid hotpots caused by screening; 4) can be directly used in the following experiments. Here, we described an optimized procedure of DML-Seq for fitness screen to supply classical TIS using the marine pathogenic bacterium Edwardsiella piscicida as an example.
Keywords: TISBackground
Transposon insertion mutagenesis coupled with next-generation sequencing (NGS) has been proven to be a powerful method to investigate gene function under a multitude of conditions (Chao et al., 2016; Price et al., 2018). In general, TIS analysis consists of the massive parallel sequencing of transposon insertion sites and statistical analysis of the abundance of the insertion events.
TIS-based screens can provide high-resolution maps of the fitness contributions of individual loci and domains based on the bacterial fitness of highly saturated transposon mutant libraries under a multitude of conditions (Chao et al., 2016). The insertion frequency at each locus or the relative abundance of corresponding mutants is generally inversely correlated with the locus’s contribution to fitness following the imposition of a selective pressure, such as that imposed by hosts and antibiotics (Chao et al., 2016). The principles of this methodology underlie a variety of related approaches, including TIS, transposon sequencing (TnSeq), insertion sequencing (INSeq), transposon-directed insertion site sequencing (TraDIS), high-throughput insertion tracking by deep sequencing (HITS), and transposon insertion site sequencing (TIS-Seq) (Gawronski et al., 2009; Langridge et al., 2009; Chao et al., 2016; Veeranagouda et al., 2017; Price et al., 2018). These approaches are revolutionizing microbiological studies by facilitating unprecedented and deep genome-wide explorations of a wide range of bacterial species.
With the aim to generate highly saturated mutant libraries for further analysis, the insertion events caused by transposon should be unbiased and the transposon will be delivered into recipient cells several times. However, this will lead to a large amount of nonsense mutation and the complexity of mutant libraries may cause bottleneck effect during the following screens. Compared with this, the defined mutant libraries containing mutants with a single insertion at a known genomic location will be more useful and facilitated (Fu et al., 2013; Abel et al., 2015). The first human pathogenic bacterium defined mutant library for Pseudomonas aeruginosa was constructed in 2003. Subsequently, many defined mutant libraries of human pathogens were also generated, such as a library of Francisella tularensis in 2007, Vibrio cholera in 2008, Burkholderia thailandensis in 2013, Chlamydia and Mycobacterium in 2015 (Jacobs et al., 2003; Gallagher et al., 2007 and 2013; Cameron et al., 2008; Kokes et al., 2015). With these defined mutant libraries, a myriad of important researches were completed, such as the identification of ciprofloxacin resistome from P. aeruginosa library, a novel T6SS regulator TsrA from V. cholera library, and the exploration of functional genomics with Desulfovibrio alaskensis G20 library (Breidenstein et al., 2008; Zheng et al., 2008 and 2010; Kuehl et al., 2014). Hence, such defined libraries may serve as valuable resources because they often consist of collections of insertion mutants in almost all non-essential loci for an organism of interest and have drawn more attention to the exploration of functional genomics of microbe.
In order to optimize TIS approach, we developed the DML-seq method which adopts the mixed defined mutant libraries for conditional essential fitness screenings (Figure 1) (Wei et al., 2019a and 2019b). Compared with massive mutant library in traditional TIS, the DML library has advantages as: 1) insertions within the 80% of the ORF to ensure the effective mutagenesis; 2) low complexity and few isogenes to ensure low bottleneck effects; 3) the mixture of an equal amount of individual mutants to avoid hotpots caused by screening; 4) can be directly used in the following experiments.
Compared to human pathogenic bacterium, the pathogenesis of marine pathogenic bacterium is poorly understood. In our model organism Edwardsiella piscicida, which causes Edwardsiellosis in farmed fish and leads to severe economic losses in aquaculture worldwide (Leung et al., 2019), DML-seq has been employed to identify conditional essential genes as well as those crucial for survival in different environments (Wei et al., 2019b), colonization and infection in host cells (Wei et al., 2019a). Briefly, we constructed MKGR, a derivative of Himar1 mariner transposon based on MAR2xT7 (Liberati et al., 2006), to generate a library of random transposon insertion mutants of E. piscicida EIB202 (Table 1). In total, 2,806 genes were disrupted out of the 3,599 predicted genes with an average of 5.04-fold coverage on E. piscicida chromosome. After that, 5 non-redundant subset libraries were created manually. The 1st and 2nd subset libraries contained a single mutant for each disrupted ORF in the parental library and they were paralleled for each other. The 3rd subset library contains transcriptional fusion mutants to eliminate the location effect on transcription. The 4th subset library is an intergenic library and the 5th library was a composited library, which was generated by mixing the 1st, 2nd, 4th subset libraries into a pool.
Here, we will focus on the specific steps for DML-seq because the excellent protocols deTAILing TIS analyses are reviewed and available elsewhere (Chao et al., 2016; Yamaichi et al., 2017). In principle, the procedure described here can be applied to conditional essential screens and facilitate the exploration of functional genomics of microbe.
Table 1. The statistic values of defined mutant library of E. piscicida EIB202

Figure 1. A defined transposon mutant library construction. A. pMKGR, a derivative of Himar1 mariner transposon, was constructed to build the defined transposon mutant library of E. piscicida EIB202. B. Work flow of a defined mutant library construction. E. coli SM10 is used as a donor for conjugation with recipient strain E. piscicida EIB202. Conjugation is processed between EIB202 and SM10 on LB plates for 3 h at 30 °C. Later, conjugators are separated on LB plates containing 20 µg/ml polymyxin and 15 µg/ml gentamicin. After 24 h of incubation at 30 °C, monoclonal will be picked into 96-well plates and incubated at 30 °C for about 16 h. Before storing at -80 °C, glycerol was added into every well and mix to make the final 20% glycerol concentration to preserve strains. C. Schematic of TAIL-PCR amplification used for determination of the Tn insertion loci on the genome. The pair of primer Sp1/AB2 is used for the first round of PCR and another pair of primer Sp2/ABS is used for the second round of PCR. Finally, primer Seq2 is used for sequencing of the amplified PCR products. D. The number of new disrupted genes increased linearly with new mutants and the number of genes hit approached a plateau.
Materials and Reagents
Handling Bacteria
Large square Petri dish (500 cm2) (Thermo Fisher Scientific, catalog number: R80115TS )
Cellulose filter membrane (0.45 μm pore size, Merck Millipore)
Eppendorf tube (Axygen, catalog number: MCT-150-C )
PCR tube (Axygen, catalog number: PCR-02-A )
Corning 50 ml centrifuge tube (Merck, catalog number: CLS430828-500EA )
96-Well Deep Well Plates (Thermo Fisher Scientific, catalog number: A43075 )
Cryotube (Thermo Fisher Scientific, catalog number: 5000-1020 )
Wild type E. piscicida EIB202 (ColR); “recipient” cells
E. coli SM10 λpir harboring pMKGR for delivering Tn via conjugation (AmpR, KmR); “donor” cells (see Note 1)
Luria-Bertani (Miller's LB Broth Base, Thermo Fisher Scientific, catalog number: 12795027 ) and agar media (Merck, catalog number: 05040 )
Colistin (Col, 20 mg/ml, BBI)
Ampicillin (Amp, 100 mg/ml, BBI)
Gentamicin (Gm, 15 mg/ml, BBI)
Glycerol (Titan, catalog number: 56-81-5 )
Thermal Asymmetric Interlaced PCR (TAIL-PCR)
Genomic DNA (gDNA) Purification
Library Construction
Qiagen Gel purification kit (QIAGEN, catalog number: 28104 )
VAHTS HiFi Amplification Mix (Vazyme Biotech, catalog number: 616-01/02 )
Agarose (Thermo Fisher Scientific, catalog number: 16500500 ) and Electrophoresis apparatus (BIO-RAD, catalog number: 1704489EDU )
QubitTM dsDNA HS Assay kit (Invitrogen, catalog number: Q32851 )
Oligo DNAs (See Note 2) (HPLC purified, BGI)
“EIB202-F”: 5’-TCATCGCACATACAGAATAAACGCC-3’
“EIB202-R”: 5’-CCGTAACATTTCTTACAACACTGCG-3’
“pMKGR-F”: 5’-AGGTGATGCTACATACGGAAAG-3’
“pMKGR-R”: 5’-AGCGCATGAACTCCTTGATG-3’
“Tn-F”: 5’-AAAAGTCCGCTGGCAAAG-3’
“Tn-R”: 5’-CCCTTCAAGAGCGATACAAC-3’
“Sp1-F”: 5’-GCTCCGTAGTAAGACATTCATCGCG-3’
“Sp2-F”: 5’-GCTTACGTTCTGCCCAAGTTTGAG-3’
“ABS-R”: 5’-GGCCACGCGTCGACTAGTAC-3’
“AB2-R”: 5’-GGCCACGCGTCGACTAGTACNNNNNNNNNNCCTGG-3’
“Seq2-F”: 5’-CAATTCGTTCAAGCCGAGATCG-3’
“AD_fork truncated NH2”: 5’- TACCACGACCA-3’
“AD_Index Fork R”: 5’- GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTATGGTCGTGGT-3’
“1st_seq-out-psc189”: 5’- ACTCTGGGGTACGCGTCTAG-3’
“1st_PCR_Index ‘R’ primer”: 5’-GTGACTGGAGTTCAGACGTGTG-3’
“P5 spacer primers”: equimolar mixture of the following 6 oligos:
5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGACTTATCAGCCAACCTGT-3’
5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTCGACTTATCAGCCAACCTGT-3’
5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTATGACTTATCAGCCAACCTGT-3’
5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTTGTCGACTTATCAGCCAACCTGT-3’
5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTTCGACGACTTATCAGCCAACCTGT-3’
5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGCAGCGACGACTTATCAGCCAACCTGT-3’
“P7 barcode primers”
CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC (vary from sample to sample)
Equipment
Multichannel pipette (Thermo Fisher Scientific)
High speed centrifuge (Eppendorf, model: 5415R )
Thermal cycler (EASTWIN, model: ETC811 )
Water bath (Thermo Fisher Scientific, catalog number: TSGP02 )
High speed centrifuge (Eppendorf, model: 5415R )
Bath sonicator (Diagenode, catalog number: B01020001 )
Thermal cycler (EASTWIN, catalog number: ETC811 )
VAHTS Universal End preparation Module for Illumina (Vazyme Biotech, catalog number: N203-01 )
VAHTS Universal Adapter Ligation Module for Illumina (Vazyme Biotech, catalog number: N204-01 )
QubitTM 4 Fluorometer (Invitrogen, catalog number: Q33238 ), or equivalent fluorescent-based DNA quantification apparatus
Procedure
Parental Library Construction
Inoculate “donor” (in LB broth with Amp, 100 μg/ml) and “recipient” cells (in LB broth with Col, 20 μg/ml) at 37 °C with shaking and culture overnight.
Prepare LB agar plate(s) without antibiotics. Let them dry well and place 0.45 μm membrane filters on top of the plates (four membranes can be applied to each plate) (see Note 3).
Passage E. piscicida, 2 ml in 50 ml (4%), add CaCl2 to 50 mM (500 μl, 10 M), Rotate 200 rpm at 30 °C, the OD600 will reach ~0.6-0.8 after 1 h 15 min culture (see Note 4).
Measure the OD600 of all bacteria, which are expected to be 0.8, for SM10 and E. piscicida, respectively.
Move the culture into 50 ml corning tubes, spin 5,000 × g at 10 °C for 5 min, then wash with 10 ml LB and spin 5,000 × g for 10 min.
Resuspend in LB with the following volume to reach final OD600 = 4.
Mix equal volume of SM10 and E. piscicida in an Eppendorf tube, and spot 4*80 μl on the filter prepared in Step A2. Let drop adsorb into the agar and dry.
Place these plates into a 30 °C incubator with care not to let the cell suspension spill outside the membrane (do not flip the plate).
After 3 h of incubation, remove the filter into a 50-ml centrifuge tube by forceps. Resuspend cells from filters in 1 ml of LB broth by pipetting up/down and vortexing.
Make serial dilutions of resuspended cells and plate 100 µl of 1:1, 1:10, and 1:100 dilutions onto LB agar plates containing Col and Gm (to provide an estimate of transposon insertion efficiency, as the actual library plates can become too crowded to pick colonies). Incubate overnight at 30 °C.
Examine ~100 ColR GmR colonies by colony PCR with 3 pairs of universal primers EIB202-F/R, pMKGR-F/R, and Tn-F/R (see Note 5).
Pick monoclonal into 96 deep well plates, rotate 200 rpm at 30 °C overnight.
Mix 900 µl of cells with 300 µl of 80% glycerol into cryotube to store a recoverable library at -80 °C.
Thermal Asymmetric Interlaced PCR (TAIL-PCR)
For each monoclonal, prepare the first reaction system in a PCR tube:
0.5 µl bacterial culture (from Subheading 3.1).
1 µl 10 µM “Sp1-F”.
1 µl 10 µM “AB2-R”.
10 µl Tiangen 2× Taq PCR MasterMix.
Add H2O to 20 µl.
Run on the PCR Thermo cycler under the program.
1 = 94 °C for 4 min.
2 = 94 °C for 30 s.
3 = 42 °C for 30 s.
[option] -1 °C per cycle.
4 = 72 °C for 3 min.
5 = go to 2, 6 times.
6 = 94 °C for 30 s.
7 = 58 °C for 30 s.
8 = 72 °C for 1 min.
9 = go to 6, 25 times.
10 = 72 °C for 3 min.
Dilute the PCR product ten-fold, prepare the second reaction system in a PCR tube:
1 µl Dilute PCR products.
2 µl 10 µM “Sp2-F”.
2 µl 10 µM “ABS-R”.
25 µl Tiangen 2× Taq PCR MasterMix.
Add H2O to 50 µl.
Run on the PCR Thermo cycler under the program.
1 = 94 °C for 3 min.
2 = 94 °C for 30 s.
3 = 64 °C for 30 s.
4 = 72 °C for 1 min.
5 = go to 2, 30 times.
6 = 72 °C for 3 min.
Collect the PCR product and sequence with primer “Seq2”.
Non-redundant Subset Library Construction (see Note 6, E. piscicida EIB202 used as an example)
Sequence results are mapped to recipient EIB202 genome.
Saturation curve is drawn to ensure the saturation of the library.
According to identified insertion sites, non-redundant subset libraries were generated manually to facilitate a genome-wide screen.
According to different experimental purposes, non-redundant subset libraries would be used for the screen.
gDNA Extraction
Use commercial gDNA extraction kit, refer to relevant protocol.
gDNA Shearing by Sonication
Dilute gDNA in fresh 0.5 ml individual tubes to a final concentration of 50 ng/µl in 100 µl H2O.
Sonicate DNA to yield fragments: 30s ON/ 90s OFF, for 12 cycles.
Run a sample (5 µl) of the digest on an agarose gel (2%). Most of the DNA should fall in the range of 200-500 bp, which will be fine for downstream ligations and PCR.
End Repair and A-TAILing
Prepare the reaction in a PCR tube:
VAHTS Turbo End Prep Enzyme Mix: 3.0 µl.
VAHTS Turbo End Prep Reaction Buffer (10×): 6.5 µl.
Fragmented DNA (50 ng/µl, add 1 µg): 20 µl.
Add H2O to 65.0 µl.
Carry out PCR using the following cycling conditions.
1 = 20 °C for 30 min.
2 = 65 °C for 30 min.
Adapter Ligation
Prepare the following adapter mixture in a PCR tube.
5 µl 100 µM “AD_fork truncated NH2”.
5 µl 100 µM “AD_Index Fork R”.
0.4 µl 2 mM MgCl2.
Run on the PCR Thermo cycler under the program.
1 = 95 °C for 4 min.
2 = 95 °C for 1 min.
[option] -1 °C per cycle.
3 = go to 2, 75 times.
4 = End.
Dilute to half concentration: Add the same volume of H2O.
Prepare the following ligation reaction in a PCR tube.
A-TAILed DNA (from above Subheading 3.5): 65 µl.
VAHTS Turbo T4 DNA Ligase: 2.0 µl.
VAHTS Turbo Ligation Enhancer: 30.5 µl.
Adapter for Tn-seq (from above Subheading 3.6, step 2): 2.5 µl.
Add H2O to 100 µl.
Incubate the reaction at 20 °C for 30 min.
Column-purify DNA, elute with 35 µl pre-warmed H2O.
Amplification of Tn-associated gDNA
Prepare the following PCR reaction in an Eppendorf tube.
50 µl VAHTS HiFi Amplification Mix.
200 ng Ligated DNA (from Subheading 3.6).
5 µl 10 µM “1st_seq-out-psc189 primer”.
5 µl 10 µM “1st_PCR_Index ‘R’ primer”.
Add H2O to 100 µl.
Aliquot into 2 PCR tubes (50 µl each) and carry out PCR using the following cycling conditions.
1 = 98 °C for 30 s.
2 = 98 °C for 10 s.
3 = 53 °C for 30 s.
4 = 72 °C for 30 s.
5 = go to 2, 29 times in all.
6 = 72 °C for 5 min.
Pool the 2 PCR reactions and then column-purify the PCR products, elute with 35 µl pre-warmed H2O.
Second PCR to Add barcodes, Illumina Attachment (P5, P7) and Variability Sequences
Prepare the following PCR reaction in an Eppendorf tube.
300 ng purified PCR product (from Subheading 3.7).
75 µl VAHTS HiFi Amplification Mix.
7.5 µl 10 µM “P5 spacer primers”.
7.5 µl 10 µM P7 Index barcode (should vary sample by sample).
Add H2O to 150 µl.
Aliquot into 3 PCR Tubes (50 µl each) and carry out PCR using the following cycling conditions.
1 = 98 °C for 30 s.
2 = 98 °C for 10 s.
3 = 55 °C for 30 s.
4 = 72 °C for 30 s.
5 = go to 2, 17 times total.
6 = 72 °C for 7 min.
Pool the 3 PCR reactions and then column-purify the PCR products, elute with 35 µl pre-warmed H2O.
Size Selection
Run all the purified PCR products (from Subheading 3.8) on a 2% agarose gel in 0.5× TAE buffer.
Size select by cutting out the smear between 200 and 500 bp. Make sure not to include the visible head-to-head primer dimers around 200 bp.
Column-purify DNA using a standard gel extraction procedure, elute with 50 µl pre-warmed H2O.
Quantify the DNA concentration by Qubit. Now the DNA is ready to be run on an Illumina sequencer.
Conditional Essential Genes analysis
The transposition loci and the abundance of the insertions are determined using the EL-ARTIST pipeline (Chao et al., 2013). Based on the neutral base pair model, a random distribution of transposon insertions, a library containing n mutants, and the total genes number on the genome is k, the number of genes uncovered by transposon is N. The probability of gene j getting insertion pj, the uncovered genes number by transposon is
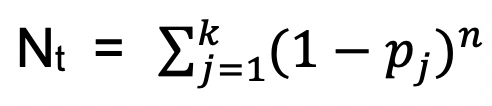 The essential genes number is set as k1. As neutral base pair model, the probability of essential genes uncovered
The essential genes number is set as k1. As neutral base pair model, the probability of essential genes uncovered 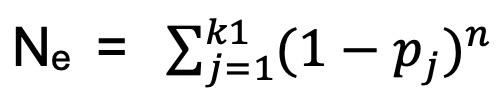 and the predicted essential genes number k1 = N - Nt + Ne.
and the predicted essential genes number k1 = N - Nt + Ne.
Notes
pMKGR was constructed based on Himar1 mariner transposon. The promoterless kanamycin resistance (Kanr) cassette and gene egfp with ribosomal binding site (RBS) were amplified. The RBS sequence used in this study was “AAGGAGG”, which originated from the E. piscicida 16S rRNA 3’ terminal conserved sequence “CCUCCUU”. A triple terminal sites “TGACTAGCTAA” and 48-bp T7 terminator sequence were introduced into pMKGR. The constructed pMKGR was transformed into E. coli SM10 and validated by PCR and sequencing.
The pMKGR, a derivative of Himar1 mariner transposon and strain E. piscicida EIB202 are used as examples in this protocol; for different transposons and strains, all primer sequences need to be modified accordingly.
The prepared membranes should be as many as conjugation reactions. With more and more picked colonies for sequencing, the number of disrupted genes is increasing and the increasing rate becomes horizontal gradually and reaches a plateau in Step C2. It means that plenty of mutagenesis has already been generated and collected mutants are able to assemble a comprehensive and saturated library.
The optimal growth temperature of E.piscicida EIB202 is 30 °C. The following incubation temperature needs to be modified according to different recipient strains.
When antibiotics are used as selective markers, there is a small chance to observe colonies that are not truly resistant and can grow in the presence of antibiotics even without containing a transposon insertion (phenotypic resistance). To ensure successful transposition, it is suggested to randomly restreak ~100 colonies on fresh plates and perform colony PCR with 3 pairs of universal primers which are designed for transposon, its vector plasmid and “recipient” cells. For colony PCR, 1) Pick a single colony with a sterile flat toothpick or pipette tip and swirl in sterile water. 2) Lyse the bacteria to release DNA by either simply boiling the sample before use or directly adding a small volume of the sample to the PCR reaction. The bacteria will be lysed at the initial heating step of the conventional PCR procedure. 3) The cycling conditions are: 98 °C, 1 min; followed by 30 cycles of 98 °C, 5 s; 55 °C, 5 s; and 72 °C, 40 s. 4) The size of PCR products is determined by electrophoresis and then PCR products are verified with Sanger Sequencing.
Five subset libraries were separated from the parental library. The 1st and parallel 2nd subset libraries include a single mutant for each ORF, respectively and insertion sites at 0.4-0.6 percentage within each ORF 5’-3’ end were given priority. Once the ORF contains only one insertion site, these mutants were selected prior into 1st subset library. The 3rd subset library is a transcriptional fusion library in which each mutant carries the same orientation of MKGR with local ORF and the priority was given to insertion sites near the 5’ end of each ORF. Similar to the 1st subset library criteria, the 4th library was generated and includes no more than two mutants for each intergenic region. The 5th compound library is the mixture of all the mutants from the 1st, 2nd and 4th subset libraries and takes 2 x 107 CFU for each corresponding mutant.
Acknowledgments
The described protocol is adapted from Wei et al., 2019. This work was supported by grants from the National Natural Science Foundation of China (32002436 to S.S., 82001033 to L.W.), National Key Research and Development Program of China (2018YFD0900504 to Q.W.), Shanghai Sailing Program (20YF1411100), China Postdoctoral Science Foundation (2020M671034), Ministry of Agriculture of China (CARS-47), and the Science and Technology Commission of Shanghai Municipality (17391902000).
Competing interests
There are no conflicts of interest or competing interest.
References
- Abel, S., Abel zur Wiesch, P., Chang, H. H., Davis, B. M., Lipsitch, M. and Waldor, M. K. (2015). Sequence tag-based analysis of microbial population dynamics. Nat Methods 12(3): 223-226.
- Breidenstein, E. B., Khaira, B. K., Wiegand, I., Overhage, J. and Hancock, R. E. (2008). Complex ciprofloxacin resistome revealed by screening a Pseudomonas aeruginosa mutant library for altered susceptibility. Antimicrob Agents Chemother 52(12): 4486-4491.
- Cameron, D. E., Urbach, J. M. and Mekalanos, J. J. (2008). A defined transposon mutant library and its use in identifying motility genes in Vibrio cholerae. Proc Natl Acad Sci U S A 105: 8736-8741.
- Chao, M. C., Abel, S., Davis, B. M. and Waldor, M. K. (2016). The design and analysis of transposon insertion sequencing experiments. Nat Rev Microbiol 14: 119-128.
- Chao, M. C., Pritchard, J. R., Zhang, Y. J., Rubin, E. J., Livny, J., Davis, B. M. and Waldor, M. K. (2013). High-resolution definition of the Vibrio cholerae essential gene set with hidden Markov model-based analyses of transposon-insertion sequencing data. Nucleic Acids Res 41(19): 9033-9048.
- Fu, Y., Waldor, M. K. and Mekalanos, J. J. (2013). Tn-Seq analysis of Vibrio cholerae intestinal colonization reveals a role for T6SS-mediated antibacterial activity in the host. Cell Host Microbe 14(6): 652-663.
- Gallagher, L. A., Ramage, E., Jacobs, M. A., Kaul, R., Brittnacher, M. and Manoil, C. (2007). A comprehensive transposon mutant library of Francisella novicida, a bioweapon surrogate. Proc Natl Acad Sci U S A 104(3): 1009-1014.
- Gallagher, L. A., Ramage, E., Patrapuvich, R., Weiss, E., Brittnacher, M. and Manoil, C. (2013). Sequence-defined transposon mutant library of Burkholderia thailandensis. mBio 4(6): e00604-00613
- Gawronski, J. D., Wong, S. M., Giannoukos, G., Ward, D. V. and Akerley, B. J. (2009). Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc Natl Acad Sci U S A 106:16422-16427.
- Jacobs, M. A., Alwood, A., Thaipisuttikul, I., Spencer, D., Haugen, E., Ernst, S., Will, O., Kaul, R., Raymond, C., Levy, R., Chun-Rong, L., Guenthner, D., Bovee, D., Olson, M. V. and Manoil, C. (2003). Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 100(24): 14339-14344.
- Kokes, M., Dunn, J. D., Granek, J. A., Nguyen, B. D., Barker, J. R., Valdivia, R. H. and Bastidas, R. J. (2015). Integrating chemical mutagenesis and whole-genome sequencing as a platform for forward and reverse genetic analysis of Chlamydia. Cell Host Microbe 17: 716-725.
- Kuehl, J. V., Price, M. N., Ray, J., Wetmore, K. M., Esquivel, Z., Kazakov, A. E., Nguyen, M., Kuehn, R., Davis, R. W., Hazen, T. C., Arkin, A. P. and Deutschbauer, A. (2014). Functional genomics with a comprehensive library of transposon mutants for the sulfate-reducing bacterium Desulfovibrio alaskensis G20. mBio 5(3): e01041-01014.
- Langridge, G. C., Phan, M. D., Turner, D. J., Perkins, T. T., Parts, L., Haase, J., Charles, I., Maskell, D. J., Peters, S. E., Dougan, G., Wain, J., Parkhill, J. and Turner, A. K. (2009). Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res 19(12): 2308-2316.
- Leung, K. Y., Wang, Q., Yang, Z. and Siame, B. A. (2019). Edwardsiella piscicida: A versatile emerging pathogen of fish. Virulence 10(1): 555-567.
- Liberati, N. T., Urbach, J. M., Miyata, S., Lee, D. G., Drenkard, E., Wu, G., Villanueva, J., Wei, T. and Ausubel, F. M. (2006). An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A 103(8): 2833-2838.
- Price, M. N., Wetmore, K. M., Waters, R. J., Callaghan, M., Ray, J., Liu, H., Kuehl, J. V., Melnyk, R. A., Lamson, J. S., Suh, Y., Carlson, H. K., Esquivel, Z., Sadeeshkumar, H., Chakraborty, R., Zane, G. M., Rubin, B. E., Wall, J. D., Visel, A., Bristow, J., Blow, M. J., Arkin, A. P. and Deutschbauer, A. M. (2018). Mutant phenotypes for thousands of bacterial genes of unknown function. Nature 557(7706): 503-509.
- Veeranagouda, Y. and Didier, M. (2017). Transposon Insertion Site Sequencing (TIS-Seq): An Efficient and High-Throughput Method for Determining Transposon Insertion Site(s) and Their Relative Abundances in a PiggyBac Transposon Mutant Pool by Next-Generation Sequencing. Curr Protoc Mol Biol 120: 21 35 21-21 35 11.
- Wei, L., Qiao, H., Sit, B., Yin, K., Yang, G., Ma, R., Ma, J., Yang, C., Yao, J., Ma, Y., Xiao, J., Liu, X., Zhang, Y., Waldor, M. K. and Wang, Q. (2019). A Bacterial Pathogen Senses Host Mannose to Coordinate Virulence. iScience 20: 310-323.
- Wei, L., Wu, Y., Yang, G., Xu, R., Liu, X., Liu, Q., Zhang, Y., Ma, Y. and Wang, Q. (2019). Genome-Wide Identification of Fitness Factors in Seawater for Edwardsiella piscicida. Appl Environ Microbiol 85(10): e00233-19.
- Yamaichi, Y. and Dorr, T. (2017). Transposon Insertion Site Sequencing for Synthetic Lethal Screening. Methods Mol Biol 1624: 39-49.
- Zheng, J., Shin, O. S., Cameron, D. E. and Mekalanos, J. J. (2010). Quorum sensing and a global regulator TsrA control expression of type VI secretion and virulence in Vibrio cholerae. Proc Natl Acad Sci U S A 107(49): 21128-21133.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Shao, S., Wei, L., Xia, F., Zhang, Y. and Wang, Q. (2021). Defined Mutant Library Sequencing (DML-Seq) for Identification of Conditional Essential Genes. Bio-protocol 11(5): e3943. DOI: 10.21769/BioProtoc.3943.
Category
Molecular Biology > DNA > Mutagenesis
Microbiology > Microbe-host interactions > Bacterium
Microbiology > Microbial genetics > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.