- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Generation and Implementation of Reporter BHK-21 Cells for Live Imaging of Flavivirus Infection
Published: Vol 11, Iss 5, Mar 5, 2021 DOI: 10.21769/BioProtoc.3942 Views: 4391
Reviewed by: Yelena BernadskayaSandeep Kumar UpadhyayAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2020
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The genus Flavivirus within the family Flaviviridae includes many viral species of medical importance, such as yellow fever virus (YFV), Zika virus (ZIKV), and dengue virus (DENV), among others. Presently, the identification of flavivirus-infected cells is based on either the immunolabeling of viral proteins, the application of recombinant reporter replicons and viral genomes, or the use of cell-based molecular reporters of the flaviviral protease NS2B-NS3 activity. Among the latter, our flavivirus-activatable GFP and mNeptune reporters contain a quenching peptide (QP) joined to the fluorescent protein by a linker consisting of a cleavage site for the flavivirus NS2B-NS3 proteases (AAQRRGRIG). When the viral protease cleaves the linker, the quenching peptide is removed, and the fluorescent protein adopts a conformation promoting fluorescence. Here we provide a detailed protocol for the generation, selection and implementation of stable BHK-21 cells expressing our flavivirus genetically-encoded molecular reporters, suitable to monitor the viral infection by live-cell imaging. We also describe the image analysis procedures and provide the required software pipelines. Our reporter cells allow the implementation of single-cell infection kinetics as well as plaque assays for both reference and native strains of flaviviruses by live-cell imaging.
Graphic abstract:
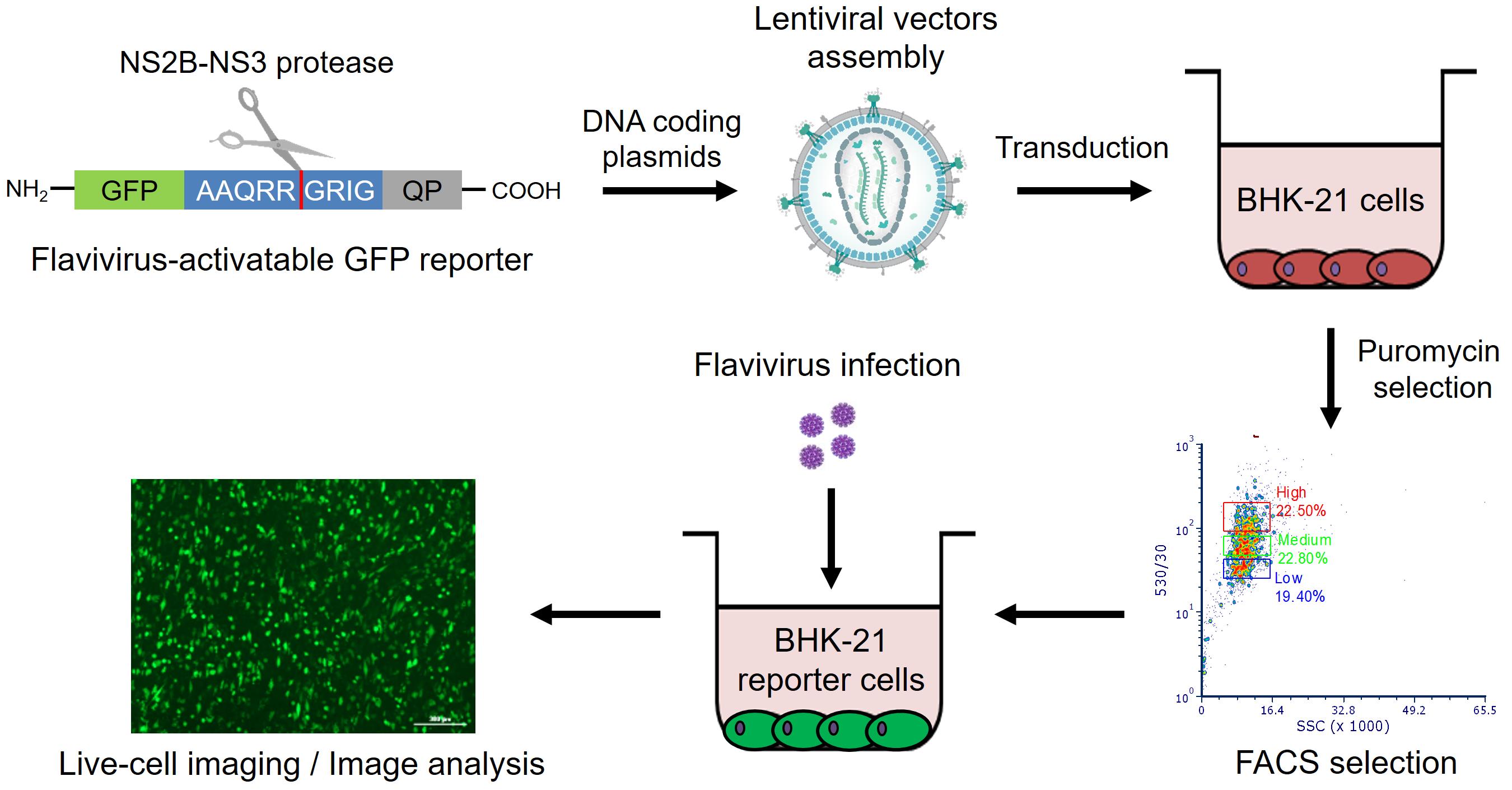
Workflow for the generation and implementation of reporter BHK-21 cells for live imaging of flavivirus infection.
Background
Flaviviruses represent an emerging and re-emerging global threat that cause diseases both in animals and humans, including many medically relevant viruses like yellow fever virus (YFV), West Nile virus (WNV), Japanese encephalitis virus (JEV), dengue virus (DENV), and Zika virus (ZIKV), among others (Gould and Solomon, 2008). At present, the detection of flavivirus-infected cells is based on either the antibody labeling of viral proteins (Balsitis et al., 2008), the use of recombinant reporter replicons and viral genomes (Li et al., 2013; Schmid et al., 2015; Xie et al., 2016; Tamura et al., 2017; Kümmerer, 2018), or the application of genetically-encoded molecular reporters of the flavivirus NS2B-NS3 proteolytic activity (Medin et al., 2015; Hsieh et al., 2017; McFadden et al., 2018). Immunolabeling implies both fixation and permeabilization which hamper their implementation for studies in living cells. Reporter replicons and viral genomes are suitable for live-cell imaging assays, but they are restricted to particular molecular clones mainly derived from reference strains and thus, not applicable when working with clinical isolates or native viral strains. In this context, cell-based molecular reporters of the flaviviral proteases constitute a favorable alternative for the study of native flavivirus strains by live-cell imaging. Based on our recently published flavivirus-activatable GFP (FlaviA-GFP) and flavivirus-activatable mNeptune (FlaviA-mNeptune) reporters (Arias-Arias et al., 2020), here we describe in detail a protocol for the generation, selection, and implementation of stably-transduced reporter BHK-21 cells for live imaging of flavivirus infection in single cells and viral plaques. Furthermore, we provide a rationale for a software-based image analysis approach to demonstrate the capabilities of this reporter cell line for single-cell and viral-plaque tracking. In addition, we include the optimized CellProfiler analysis pipelines for studies employing this or other cell-based reporters. Our approach represents the first fluorescence activatable cell-based reporter system for monitoring the kinetics of infection by both reference and native strains of flaviviruses like DENV, ZIKV, and YFV using live-cell imaging.
Materials and Reagents
Cell culture flasks, 75 cm2 (Greiner Bio-One, CELLSTAR®, catalog number: 658175 )
Cell culture dishes, 100/20 mm (Greiner Bio-One, CELLSTAR®, catalog number: 664160 )
2 ml reaction tubes (Greiner Bio-One, catalog number: 623201 )
3 ml sterile syringes (Ultident Scientific, catalog number: BD-309657 )
Syringe filters 0.2 μm hydrophilic polyethersulfone, 32 mm diameter (Pall, Acrodisc®, catalog number: 4652 )
10 ml sterile syringes (Ultident Scientific, catalog number: BD-302995 )
Syringe filters 0.45 μm hydrophilic cellulose acetate, 28 mm diameter (Sartorius, Minisart®, catalog number: 16555 )
15 ml conical tubes (Greiner Bio-One, CELLSTAR®, catalog number: 188271 )
0.5 ml reaction tubes (Greiner Bio-One, catalog number: 667201 )
Hexadimethrine bromide (Merck, Sigma-Aldrich, catalog number: H9268 )
48-well cell culture plates (Greiner Bio-One, CELLSTAR®, catalog number: 677180 )
1.5 ml reaction tubes (Greiner Bio-One, catalog number: 616201 )
12-well cell culture plates (Greiner Bio-One, CELLSTAR®, catalog number: 665180 )
1.5 ml light protection reaction tubes (Greiner Bio-One, catalog number: 616283 )
Cell culture flasks, 25 cm2 (Greiner Bio-One, CELLSTAR®, catalog number: 690175 )
6-well cell culture plates (Greiner Bio-One, CELLSTAR®, catalog number: 657160 )
96-well black cell culture plates (Greiner Bio-One, µCLEAR®, catalog number: 655096 )
24-well cell culture plates (Greiner Bio-One, CELLSTAR®, catalog number: 662160 )
HEK 293T cells (ATCC, catalog number: CRL-3216 )
Plasmids:
pLenti-FlaviA-GFP-puro (a gift from Jorge L. Arias-Arias, Addgene plasmid #140088)
pLenti-CMV-FlaviA-mNeptune-puro (a gift from Jorge L. Arias-Arias, Addgene plasmid #140091)
pMD2.G (a gift from Didier Trono, Addgene plasmid #12259)
psPAX2 (a gift from Didier Trono, Addgene plasmid # 12260 )
LB agar plates with 100 µg/ml ampicillin (Merck, Sigma-Aldrich, catalog number: L5667 )
LB broth (Miller) (Merck, Sigma-Aldrich, catalog number: L2542 )
100 mg/ml ampicillin solution (Merck, Sigma-Aldrich, catalog number: A5354 )
NucleoSpin plasmid mini kit (Macherey-Nagel, catalog number: 740588.5 0)
DMEM, high glucose, GlutaMAXTM, pyruvate (Thermo Fisher Scientific, Gibco, catalog number: 10569044 )
Antibiotic-antimycotic 100× (Thermo Fisher Scientific, Gibco, catalog number: 15240062 )
Fetal bovine serum (FBS) qualified, heat inactivated (Thermo Fisher Scientific, Gibco, catalog number: 10438-026 )
Polyethylenimine (PEI), linear, MW 25000, transfection grade (Polysciences, PEI 25KTM, catalog number: 23966-1 )
UltraPureTM DNase/RNase-free distilled water (Thermo Fisher Scientific, Invitrogen, catalog number: 10977015 )
Hydrochloric acid, 36.5-38.0%, BioReagent (Merck, Sigma-Aldrich, catalog number: H1758 )
BHK-21 [C-13] (ATCC, catalog number: CCL-10 )
MEM, GlutaMAXTM supplement (Thermo Fisher Scientific, Gibco, catalog number: 41090101 ).
Sodium pyruvate, 100 mM (Thermo Fisher Scientific, Gibco, catalog number: 11360070 ).
PBS, pH 7.4 (Thermo Fisher Scientific, Gibco, catalog number: 10010023 ).
TrypLETM express enzyme (1×), no phenol red (Thermo Fisher Scientific, Gibco, catalog number: 12604013 )
Puromycin dihydrochloride from Streptomyces alboniger (Merck, Sigma-Aldrich, catalog number: P8833 )
Clinical isolate DENV-2/CR/13538/2007 (Instituto Costarricense de Investigación y Enseñanza en Nutrición y Salud, Cartago, Costa Rica) (Soto-Garita et al., 2016)
Vaccine strain YFV/US/17D/1937 (Sanofi Pasteur, YF-VAX®)
FluoroBriteTM DMEM (Thermo Fisher Scientific, Gibco, catalog number: A1896701 )
GlutaMAXTM supplement (Thermo Fisher Scientific, Gibco, catalog number: 35050061 )
Minimum essential medium eagle AutoModTM (Merck, Sigma-Aldrich, catalog number: M0769 )
Carboxymethylcellulose sodium salt (Merck, Sigma-Aldrich, catalog number: C4888 )
Sodium bicarbonate (Merck, Sigma-Aldrich, catalog number: S5761 )
Complete DMEM (see Recipes)
PEI solution (1 mg/ml) (see Recipes)
Polybrene solution (10 mg/ml) (see Recipes)
Complete MEM (see Recipes)
PBS 1% FBS (see Recipes)
Puromycin solution (10 mg/ml) (see Recipes)
FluoroBriteTM DMEM 2% FBS (see Recipes)
Plaque media 2% FBS (see Recipes)
Equipment
Biological safety cabinet (ESCO, Labculture® Class II, Type A2, catalog number: LA2-3A2-E )
CO2 incubator (Thermo Scientific, model: Forma Series II, catalog number: 3110 )
4 °C refrigerator (Thermo Scientific, Value Lab, catalog number: 20LREETSA )
-20 °C freezer (Thermo Scientific, Value Lab, catalog number: 20LFEETSA )
-80 °C freezer (Sanyo, VIP series, catalog number: MDF-U32V )
Water bath (PolyScience, catalog number: WBE20A12E )
Centrifuge (Eppendorf, model: 5810 )
Microcentrifuge (Eppendorf, model: 5418 )
Pipettes (Thermo Scientific, FinnpipetteTM F2 GLP Kit, catalog number: 4700880 )
Pipet filler (Thermo Scientific, S1, catalog number: 9511 )
Hemocytometer (Boeco, Neubauer improved, catalog number: BOE 13 )
Shaking incubator (Shel Lab, catalog number: SSI3 )
ThermoMixer® C block (Eppendorf, catalog number: 5382000015 )
Vortex mixer (Thermo Scientific, MaxiMixTM, catalog number: M16715Q )
Ultraviolet crosslinker (UVP, CL-100, catalog number: UVP95017401 )
NanoDropTM 2000 spectrophotometer (Thermo Scientific, catalog number: ND-2000 )
Steam sterilizer (Yamato, catalog number: SQ510 )
Water purification system (Merck, Milli-Q advantage A10)
Inverted microscope (Nikon, Eclipse, catalog number: TS100 )
Flow cytometer (BD Biosciences, BD AccuriTM C6)
Cell sorter (BD Biosciences, BD FACSJazzTM)
Automated fluorescence microscope (Biotek, Lionheart FX ) with:
CO2 gas controller (Biotek, catalog number: 1210012 )
Humidity chamber (Biotek, catalog number: 1450006 )
4× objective (Biotek, catalog number: 1220519 )
20× objective (Biotek, catalog number: 1220517 )
GFP filter cube (Biotek, catalog number: 1225101 )
465 nm LED cube (Biotek, catalog number: 1225001 )
Cy5 filter cube (Biotek, catalog number: 1225105 )
623 nm LED cube (Biotek, catalog number: 1225005 )
Software
Gen5 Image+ (Biotek, https://www.biotek.com)
CellProfiler 4.0 (Broad Institute, https://cellprofiler.org/releases)
Procedure
Lentiviral vectors assembly and titration
AssemblyPrepare plasmid stocks following standard molecular biology procedures (https://www.jove.com/v/5062/plasmid-purification) and the protocol provided by the manufacturer of the NucleoSpin plasmid mini kit (Macherey-Nagel).
Manually seed 6,000,000 HEK 293T cells in 100/20 mm cell culture dishes with 8 ml of DMEM 10% FBS (Recipe 1) and incubate overnight at 37 °C and 5% CO2 to reach 70-80% confluency. For a detailed procedure of seeding cells please refer to the bio-protocol paper by Freppel et al., 2018 (reference 3).
Using a pipette, replace the medium by removing and discarding all the DMEM 10% FBS in the dishes (~8 ml) and adding 4 ml of fresh DMEM 2% FBS (Recipe 1).
Prepare the transfection mix as follows:
Suspension A: 45 µl PEI 1 mg/ml (Recipe 2) + 955 µl unsupplemented DMEM (without FBS and antibiotic-antimycotic).
Suspension B: 6 µg pLenti-FlaviA-GFP-puro or pLenti-CMV-FlaviA-mNeptune-puro + 6 µg pMD2.G + 6 µg psPAX2 and bring to 1,000 µl with unsupplemented DMEM.
Mix suspensions A with B and incubate for 15 min at room temperature.
Add the 2 ml transfection mix to the cells (dropwise) and incubate overnight at 37 °C and 5% CO2.
Replace the medium with 6 ml of fresh DMEM 2% FBS and incubate for 48 h at 37 °C and 5% CO2.
Harvest culture supernatants and filter them (0.45-μm pore size) to eliminate cells and debris. Use filters with low protein adherence, like cellulose acetate or polyethersulfone (PES). Do not use nitrocellulose filters as they could bind the lentiviral particles.
Prepare 500 µl aliquots and store at -80 °C.
As described above, seed 50,000 BHK-21 cells per well in a 48-well cell culture plate with 500 µl/well of MEM 2% FBS (Recipe 4) and incubate overnight at 37 °C and 5% CO2. Prepare extra wells to be used as control cells.
Thaw in a water bath (37 °C) one of the lentiviral 500 µl stock aliquots and add polybrene (Recipe 3) at a final concentration of 5 µg/ml.
Prepare 10-fold serial dilutions (10-1-10-6) of the lentiviral stock in 1.5 ml reaction tubes. Start mixing 50 µl of the lentiviral particles in 450 µl of DMEM 2% FBS + 5 µg/ml polybrene (10-1). Vortex the tube for 5 s and mix 50 µl of the 10-1 lentiviral dilution in 450 µl of DMEM 2% FBS + 5 µg/ml polybrene (10-2). Repeat this procedure for the other dilutions (10-3-10-6) and incubate for 15 min at room temperature. Change the tip between dilutions to avoid cross-contamination. For a detailed and graphical explanation of 10-fold serial dilutions preparation please refer to the bio-protocol paper by Freppel et al., 2018 (reference 3).
Replace the medium of the cells with 150 µl/well of undiluted lentiviral stock and each serial dilution thereof. Add 150 µl/well of DMEM 2% FBS + 5 µg/ml polybrene to the control cells. To avoid cross contamination, make the inoculation of the samples in order, starting with the control, following with the serial dilutions (from higher to lower dilution), and ending with the undiluted lentiviral stock.
Centrifuge at 300 × g for 2 h at 25 °C for viral adsorption.
Replace the inoculum with 500 µl/well of DMEM 2% FBS and incubate for 48 h at 37 °C and 5% CO2.
Discard the medium, wash the cells once with 100 µl/well of PBS, add 100 µl/well of TrypLETM express (no phenol red), incubate for 5 min at 37 °C, and resuspend the cells with 400 µl/well of PBS 1% FBS (Recipe 5).
Based on the basal background of the reporter proteins and by comparison with the non-transduced control cells (Figure 1), determine by flow cytometry the percentage of GFP+ (488 nm laser - 530/30 nm filter) or mNeptune+ (640 nm laser -675/25 nm filter) cells present in the samples infected with different dilutions of the lentiviral seed. For a detailed procedure about flow cytometry of fluorescent proteins please refer to the protocol by Hawley et al., 2004 (reference 5).
Using the data of the higher dilution with detectable transduced cells, calculate the biological titer in transducing units per milliliter (TU/ml), applying the following formula:
TU/ml = (P × N / 100 × V) × 1/DF
where P = % GFP+ or mNeptune+ cells, N = number of cells at transduction = 50,000, V = volume of inoculum per well = 0.15 ml, and DF = dilution factor = 1 (undiluted), 10–1 (diluted 1/10), 10–2 (diluted 1/100), and so on (Tiscornia et al., 2006).
Reporter cell lines production and selection
Production
As described above, seed 100,000 BHK-21 cells per well in a 12-well cell culture plate with 1 ml/well of MEM 2% FBS and incubate overnight at 37 °C and 5% CO2. Prepare an extra well for the control cells.
Thaw in a water bath (37 °C) one of the 500 µl stock aliquots of lentiviral particles carrying genetic constructs codifying for either the FlaviA-GFP or the FlaviA-mNeptune reporters and add polybrene at a final concentration of 5 µg/ml.
Based on the previously calculated biological titer in TU/ml, prepare 500 µl/well of lentiviral inoculum at a multiplicity of infection (MOI) of 1 (1 TU per cell). As 100,000 cells per well were plated in manteinance medium (MEM 2% FBS), 100,000 TU must be diluted in MEM 2% FBS + 5 µg/ml polybrene to a final volume of 500 μl. Incubate for 15 min at room temperature.
Replace the medium of the cells with the 500 µl/well of lentiviral inoculum. Add 500 µl/well of MEM 2% FBS + 5 µg/ml polybrene to the control cells.
Centrifuge at 300 × g for 2 h at 25 °C for viral adsorption.
Replace the inoculum with 1 ml/well of MEM 2% FBS and incubate for 48 h at 37 °C and 5% CO2.
Based on the basal background of the reporter proteins, monitor the effectiveness of the transduction by fluorescence microscopy in the green/GFP (FlaviA-GFP) and far-red/Cy5 (FlaviA-mNeptune) channels.
Selection
For the antibiotic selection of transduced cells, replace the medium with 750 µl/well of MEM 10% FBS containing 8 µg/ml of puromycin (Recipe 6) and incubate overnight at 37 °C and 5% CO2. Apply the same treatment to the control cells.
Replace the medium of both transduced and control cells with 1 ml/well of MEM 10% FBS containing 4 µg/ml of puromycin. Incubate at 37 °C and 5% CO2 until 100% mortality of the control cells is evidenced by light microscopy (commonly 24-48 h).
Replace the medium of the selected cells with 1 ml/well of MEM 10% FBS + 0.5 µg/ml puromycin and incubate at 37 °C and 5% CO2 until reaching confluency of 80-90%.
Passage the selected cells to 25 cm culture flasks with 5 ml of MEM 10% FBS + 0.5 µg/ml puromycin and incubate at 37 °C and 5% CO2 until reaching a confluency of 80-90% (https://www.jove.com/v/5052/passaging-cells).
For the FACS selection, discard the medium, wash the cells once with 1 ml of PBS, add 500 µl of TrypLETM express (no phenol red), incubate for 5 min at 37 °C, and resuspend the cells with 1 ml of PBS 1% FBS.
Count the cells with a hemocytometer (https://www.jove.com/v/5048/using-a-hemacytometer-to-count-cells) and prepare 2 ml of cell suspensions at 1,000,000 cells/ml in PBS 1% FBS.
Aspirate the cell suspensions into the cell sorter. For the FlaviA-GFP reporter isolate at least two cell subpopulations with different but homogeneous levels of the reporter's basal background using the FL1 detector (488 nm laser - 530/30 nm filter) (Figure 1). Apply the same procedure to the FlaviA-mNeptune reporter but using the FL4 detector (640 nm laser - 675/25 nm filter).
Note: The basal background of the FlaviA-GFP and FlaviA-mNeptune reporters make them sensitive to the levels of cellular expression: If the expression is too high the background will mask the signal produced upon activation of the reporters. If the expression is too low there will not be enough reporter’s signal over the background.
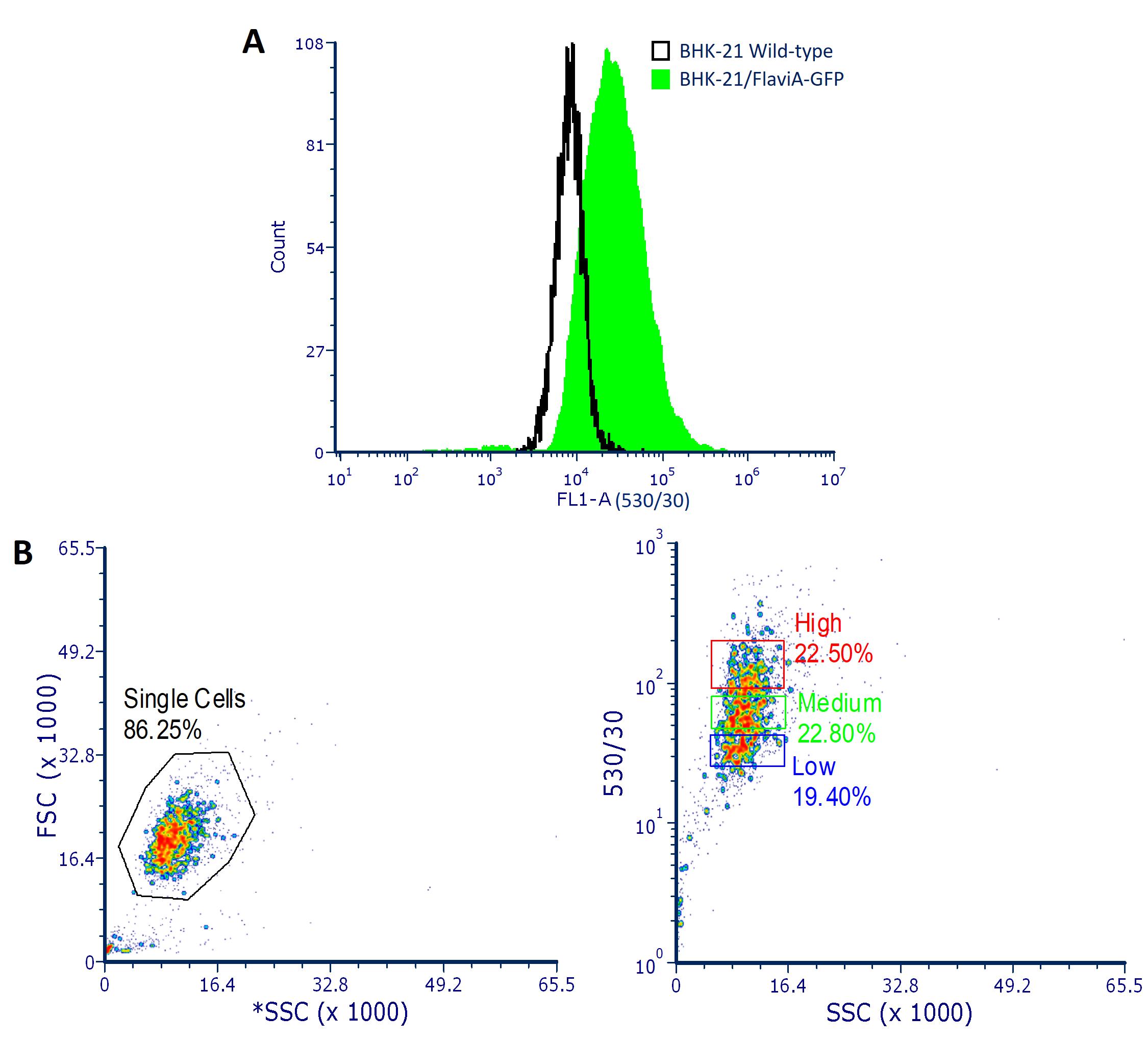
Figure 1. FACS analysis of BHK-21/FlaviA-GFP stable cells. A. Histogram showing the difference in the green fluorescence (FL1 detector- 530/30 nm filter) between wild-type and reporter BHK-21 cells due to the basal background of the FlaviA-GFP reporter proteins. B. Scatter plots showing the heterogeneity in the population of stable BHK-21 cells with different levels of expression of the FlaviA-GFP reporter. For the selection of the best reporter cells (highest signal-to-noise ratio), at least two cell subpopulations with different (low, medium or high) but homogeneous levels of the reporter's basal background must be isolated and tested by live-cell imaging upon flavivirus infection.Seed the isolated cell suppopulations in different wells of a 48-well cell culture plate with 1 ml/well of MEM 10% FBS + 0.5 µg/ml puromycin and incubate at 37 °C and 5% CO2 until reaching a confluency of 80-90%.
Passage the selected cells to a 6-well cell culture plate with 3 ml of MEM 10% FBS + 0.5 µg/ml puromycin and incubate at 37 °C and 5% CO2 until reaching a confluency of 80-90%.
Test the different isolated cell suppopulations by a live-cell imaging flavivirus infection kinetics according to the protocol described in Procedure C.
Note: The best reporter cells will be those with the highest signal-to-noise ratio upon flavivirus infection. The fluorescence signal-to-noise ratio is calculated by dividing the signal of the reporter cells treated with infectious virus by the noise given by the reporter cells treated with UV-inactivated virus at the same post-inoculation time.
Infection kinetics in reporter cells by live-cell imaging
Prepare and titer the flaviviral seed of your choice (e.g., DENV, ZIKV, or YFV) according tostandard virological methodologies (Medina et al., 2012; Freppel et al., 2018). For viral inactivation, place 200 µl/well of the flaviviral seed in a 24-well cell culture plate, remove the lid and apply 5 cycles of UV light (254 nm) exposure at an energy of 400,000 µJ/cm2 into an ultraviolet crosslinker. Between cycles, shake the plate for 5 s using your hands.
As described above, seed 15,000 BHK-21/FlaviA-GFP stable cells per well in a µClear black 96-well plate with 100 µl/well of MEM 2% FBS and incubate overnight at 37 °C and 5% CO2.
Based on the calculated titer of the flaviviral seed in plaque forming units (PFU)/ml, prepare 50 µl/well of inoculum at a low MOI (between 0.1 and 0.25). As 15,000 cells per well were plated in maintenance medium, between 1,500 and 3,750 PFUs must be diluted in MEM 2% FBS to a final volume of 50 μl/well. Multiply for the total number of wells to be inoculated and prepare a single inoculum suspension. Likewise, prepare the UV-inactivated inoculum suspension.
Replace the medium of the cells with 50 µl/well of either the infectious or the UV-inactivated flaviviral inoculum and incubate for 2 h at 37 °C and 5% CO2 for viral adsorption. Using your hands, shake the plate for 5 s every 15 min.
Replace the inoculum with 150 µl/well of FluoroBriteTM DMEM 2% FBS (Recipe 7) and incubate for the desired time of your kinetics (e.g., 120 h) at 37 °C and 5% CO2 into the automated fluorescence microscope. Add 200 µl/well of PBS to the surrounding wells to avoid desiccation.
Using your microscope’s software (e.g., Gen5 Image+), program the image acquisition with the 4× or the 20× objective in the green/GFP channel at the desired post-infection times (Figure 2A).
Note: The acquisition parameters (excitation intensity, exposure time, and camera gain) of the images are variable according to the particular reporter cell subpopulation isolated by FACS. Always include an extra well of control cells for the adjustment of the acquisition parameters as the overexposure to the excitation light may harm the cells of the experimental conditions. Our recommendation is to set those parameters to a level where the background signal of the reporter proteins is just perceptible in the first images of the kinetics, in order to increase the dynamic range of the reporter’s fluorescence upon activation by flaviviral proteases.
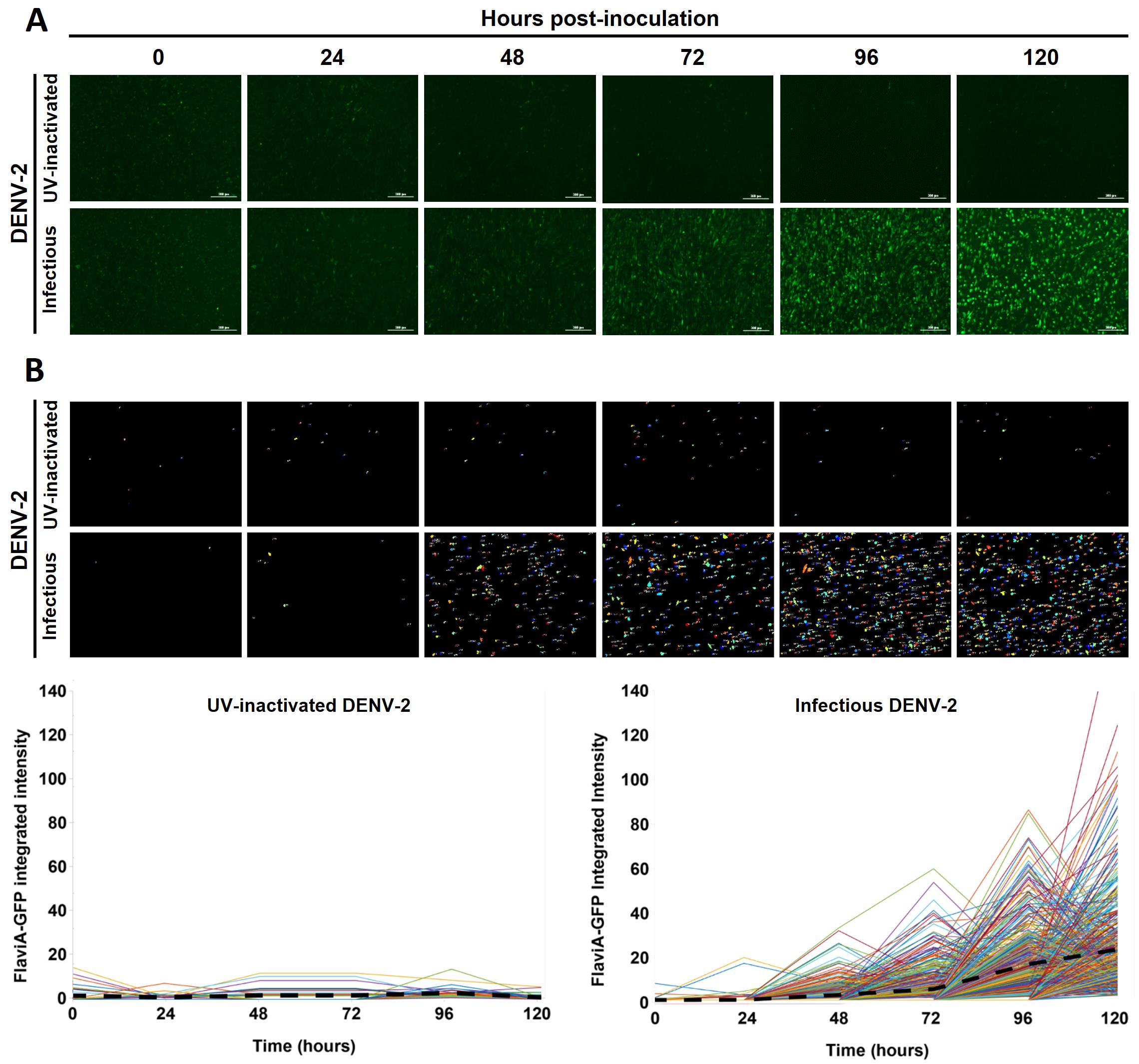
Figure 2. DENV-2 infection kinetics in reporter BHK-21 cells by live-cell imaging. A. Stable BHK-21 cells expressing the FlaviA-GFP reporter were inoculated with either infectious or UV-inactivated DENV-2 13538 at a low MOI of 0.1 and captured by live-cell imaging at the specified time periods. Magnification of 40×, scale bar = 100 µm. B. The image analysis of the infection kinetics with the software CellProfiler 4.0 allowed the tracking of single cells over time based on the reported fluorescence (each colored line in the graphs corresponds to an individual cell). The black dashed lines represent the mean values of cell fluorescence.
Kinetic plaque assay in reporter cells by live-cell imaging
As described above, seed 25,000 BHK-21/FlaviA-mNeptune stable cells per well in a µClear black 96-well plate with 100 µl/well of MEM 10% FBS and incubate overnight at 37 °C and 5% CO2.
Prepare 10-fold serial dilutions (10-1-10-6) of the flaviviral seed in 1.5 ml reaction tubes. Start mixing 50 µl of the flaviviral seed in 450 µl of MEM 2% FBS (10-1). Vortex the tube for 5 s and mix 50 µl of the 10-1 flaviviral dilution in 450 µl of MEM 2% FBS (10-2). Repeat this procedure for the other dilutions (10-3-10-6). Change the tip between dilutions to avoid cross-contamination. For a detailed and graphical explanation of 10-fold serial dilutions preparation please refer to the bio-protocol paper by Freppel et al., 2018 (reference 3).
Replace the medium of the cells with 50 µl/well of each serial dilution of the flaviviral seed. Inoculate a control well with 50 µl of UV-inactivated flaviviral seed. Incubate for 2 h at 37 °C and 5% CO2 for viral adsorption. Using your hands, shake the plate for 5 s every 15 min.
Replace the inoculum with 150 µl/well of plaque media 2% FBS (Recipe 8) and incubate for 120 h at 37 °C and 5% CO2 into the automated fluorescence microscope. Add 200 µl/well of PBS to the surrounding wells to avoid desiccation.
Using your microscope’s software (e.g., Gen5 Image+), program the image acquisition (a montage of the whole well) with the 4× objective in the far-red/Cy5 channel at the desired post-infection times (Figure 3A).
Note: The acquisition parameters (excitation intensity, exposure time, and camera gain) of the images are variable according to the particular reporter cell suppopulation isolated by FACS. Always include an extra well of control cells for the adjustment of the acquisition parameters, as the overexposure to the excitation light may harm the cells of the experimental conditions. Our recommendation is to set those parameters to a level where the background signal of the reporter proteins is just perceptible in the first images of the kinetics, in order to increase the dynamic range of the reporter’s fluorescence upon activation by flaviviral proteases.
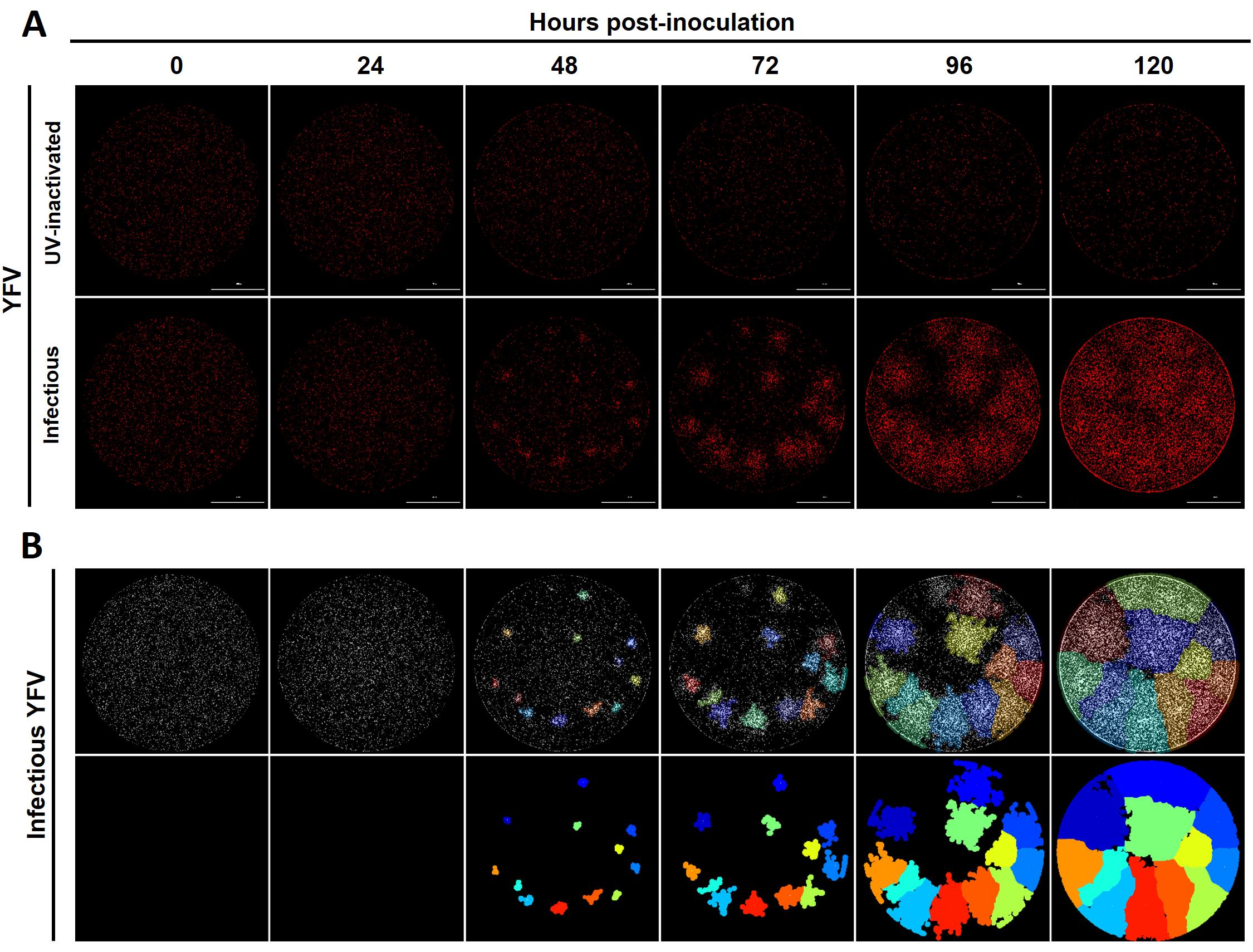
Figure 3. YFV kinetic plaque assay in reporter BHK-21 cells by live-cell imaging. A. Stable BHK-21 cells expressing the FlaviA-mNeptune reporter were inoculated with decimal dilutions (10-1-10-6) of either infectious or UV-inactivated YFV 17D. After addition of plaque media 2% FBS, entire wells of the plate were captured by live-cell imaging at the specified time periods. Magnification of 40×, scale bar = 1,000 µm. B. The image analysis of the kinetic plaque assay with the software CellProfiler 4.0 allowed the identification of single viral plaques (upper panel) and the tracking of those plaques over time (lower panel). For a deeper exemplification of the results obtained with our kinetic plaque assay please watch the live-cell imaging video (Video 1). Video 1. ZIKV plaque assay kinetics on reporter BHK-21 cells
Video 1. ZIKV plaque assay kinetics on reporter BHK-21 cells
Data analysis
Infection kinetics in reporter cells
Single cell tracking of infected cells based on its fluorescence (Figure 2B) was performed using our CellProfiler pipeline for single cell analysis (“SingleCellsTracking.cpproj”, Figure 4). To perform this analysis just drag and drop the images in the Images module and press Analyze Images. In order to modify the pipeline for your cells and conditions, go to Start Test Mode, adjust the parameters described in the modules (one by one) and press Step to see the output of the modifications applied to a particular module. Once all the modifications are ready, press Exit Test Mode and go to Analyze Images. The modules contained in the single cell analysis pipeline are the following:
Images: Simply drag and drop the image files of a time series in the Images module. Make sure that the name of all the files in the dataset do include the name of the channel where the fluorescence was measured (e.g., GFP) and the increasing consecutive numbers for time-lapse microscopy images (e.g., GFP_72h).
NamesAndTypes: Select the rule criteria with an expression contained in the name of all the files in the dataset, in this case, indicating the channel of the fluorescence (e.g., GFP). For each file of the dataset it creates an image called “Sensor”.
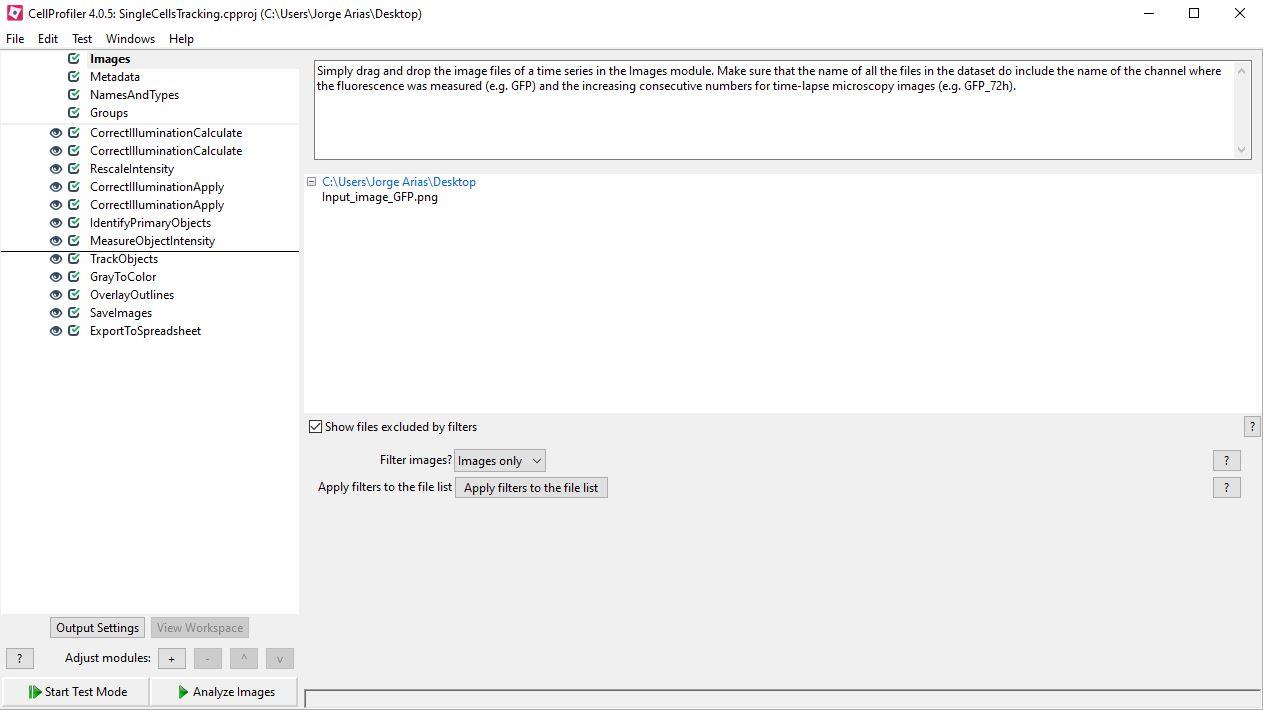
Figure 4. CellProfiler image analysis pipeline for single cell tracking of flavivirus infected BHK-21/FlaviA-GFP reporter cellsCorrectIlluminiationCalculate (Illumination function calculation: Regular): From the “Sensor” image, this module calculates a regular illumination correction function and creates an image with enhanced contrast, named “IllumSensor”.
CorrectIlluminiationCalculate (Illumination function calculation: Background): From the “Sensor” image, this module calculates a background-based illumination function called “IllumSensorbackground”.
RescaleIntensity: Converts the output image of the regular illumination function (“IllumSensor”) to an image with rescaled intensity (“RescaleSensor”).
CorrectIlluminationApply: This module applies the “IllumSensorbackground” correction function to the “RescaleSensor” image and generates an enhanced image named “CorrSensor”.
CorrectIlluminationApply: This second module of correction works on the original “Sensor” image, applies the “IllumSensorbackground” correction function but keeping the original intensity values and generates an output image called “mSensor”.
IdentifyPrimaryObjects: This module works with the enhanced image “CorrSensor” to identify the objects Cells. You may modify the Typical diameter to obtain the correct cell segmentation with other cell lines or parameters.
MeasureObjectIntensity: Measures the intensities of the identified objects Cells but on the image with the original intensity values only corrected by illumination (“mSensor”).
TrackObjects: This module tracks cells over the time-lapse microscopy and generates an output image called “TrackedCells”.
GraytoColor, OverlayOutlines, SaveImages: These modules create and save color images with the outlines of the primary objects identification for documentation and validation of the cell segmentation by the researcher’s eye and criteria.
ExportToSpreadsheet: Saves the selected measurements to an excel spreadsheet (e.g., Integrated intensity).
Note: Together with this protocol we supply the Zip file “SingleCellsTracking pipeline and dataset” which contains the applied pipeline (“SingleCellsTracking.cpproj”) and an example dataset composed of a single input image to run the pipeline (“Input_image_GFP”) and two output files to corroborate the expected results (“Output_image_GFP” and “Infected_Cells”).
Kinetic plaque assay in reporter cells
The identification and tracking of viral plaques were performed using two different pipelines programmed in CellProfiler. For single-plaque recognition use our “PlaqueIdentification.cpproj” pipeline (Figure 5) to generate images such as those depicted in the upper panel of Figure 3B. To track the plaques over a time series and determine the cell counts for every recognized plaque, first apply the “PlaqueIdentification.cpproj” pipeline to generate an image with the identified plaques and then use that image and the “PlaqueTracking.cpproj” pipeline (Figure 6) to obtain tracking images such as those showed in the lower panel of Figure 3B. Depending on the cell type and/or density, some modifications to several parameters may be done for an optimal plaque identification using the option Start Test Mode and running each module step by step. The modules of the above mentioned pipelines are the following:
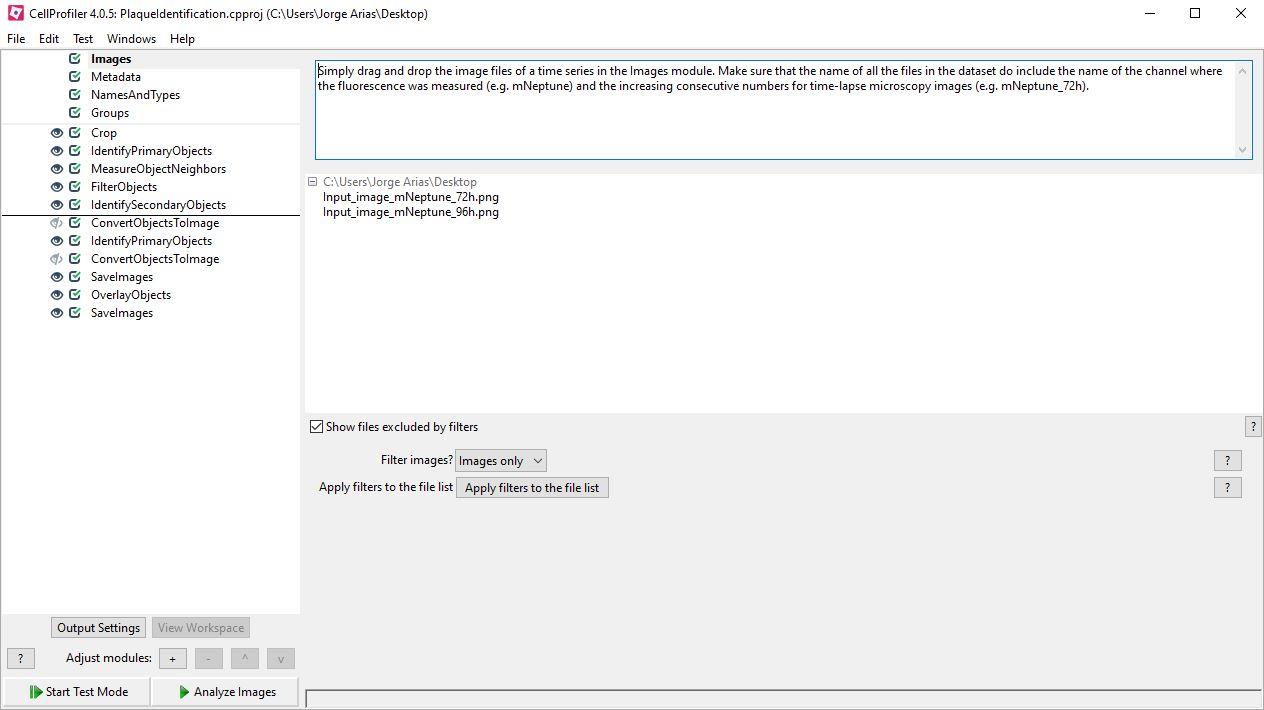
Figure 5. CellProfiler image analysis pipeline for flaviviral plaques identification in BHK-21/mNeptune reporter cells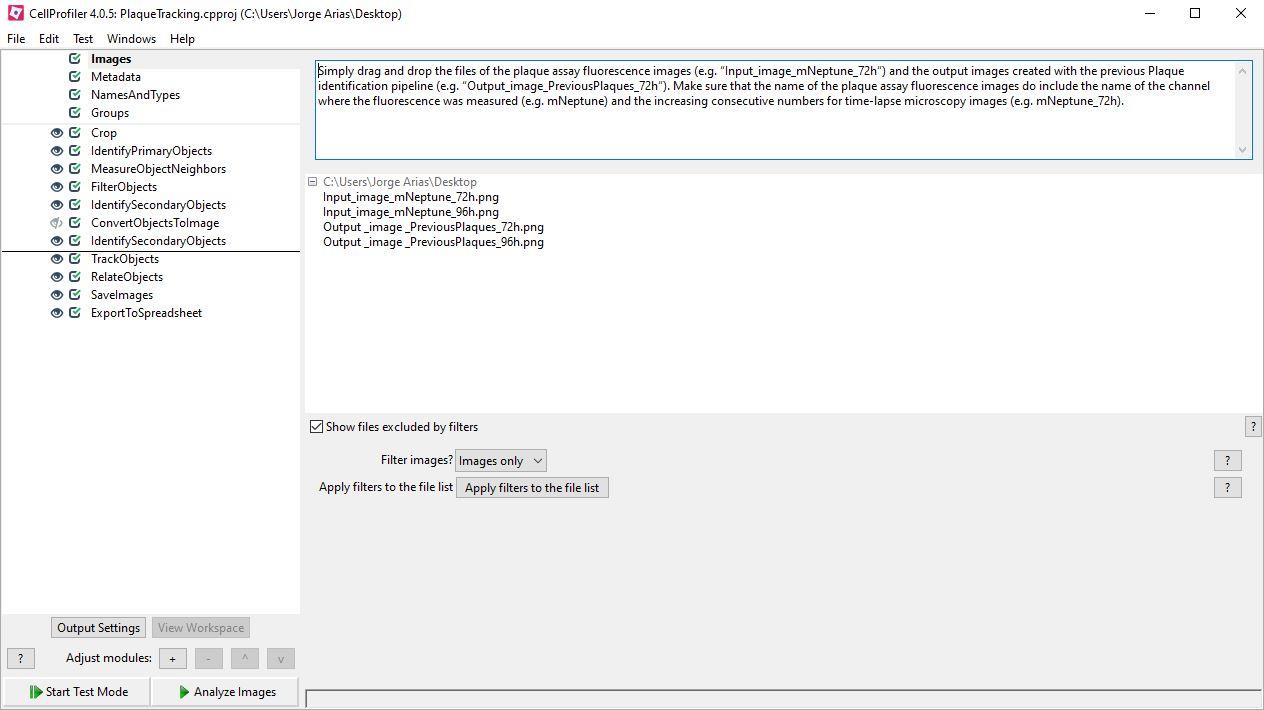
Figure 6. CellProfiler image analysis pipeline for flaviviral plaques tracking in BHK-21/mNeptune reporter cells
Plaque identification pipelineImages: Simply drag and drop the image files of a time series in the Images module. Make sure that the name of all the files in the dataset do include the name of the channel where the fluorescence was measured (e.g., mNeptune) and the increasing consecutive numbers for time-lapse microscopy images (e.g., mNeptune_72h).
NamesAndTypes: Select the rule criteria with an expression contained in the name of all the files in the dataset, in this case, indicating the channel of the fluorescence (e.g., mNeptune). For each file of the dataset it creates an image called “Sensor”.
Crop: This is an optional module in case you need to remove part of the image (e.g., distance bars). It generates an output image named “CropSensor”.
IdentifyPrimaryObjects: This module identifies the individual objects Nuclei from the “CropSensor” image. You may need to change the Typical diameter of objects or the Threshold strategy and its parameters to obtain an optimal identification of your cells.
MeasureObjectNeighbors: Measures the number of neighbors for each Nuclei object within a specified Neighbor distance. You may need to modify this parameter to obtain a good optimal range of neighbor numbers to differentiate the plaque-belonging cells from the surrounding cells. This module creates an image called “ObjectNeighborCount”.
FilterObjects: This module sets a threshold to identify plaque-belonging Nuclei objects using the range of number of neighbors calculated in the previous module. You may need to set this Minimum value according to the range of neighbors for an optimal identification of plaque belonging Nuclei objects. It generates the output objects FilteredNuclei.
IdentifySecondaryObjects: Extends the above FilteredNuclei by a determined number of pixels to fill the gaps between the plaque-belonging cells in the “CropSensor” image. You may need to modify the Number of pixels by which to expand the primary objects in order to fill most of the gaps. This module identifies the output objects Cells.
ConvertObjectstoImage: The expanded Cells objects constitute the basis to create a new image named “CellImage”.
IdentifyPrimaryObjects: Uses the previous “CellImage” to identify the new Plaque objects. You may need to modify the range of Typical diameter of objects to obtain the correct plaque identification with images from different post-infection times, as shown in the colored frame Plaque, within the visual output generated by this module. In such an instance images must be analyzed one by one, like in the case of our example dataset.
ConvertObjectstoImage: Converts the Plaque objects to an image called “PlaqueImage”.
SaveImages: Uses the “PlaqueImage” to create and save an image called “Output_image_PreviousPlaques”, that constitutes one of the input images for the plaque tracking with the pipeline “PlaqueTracking.cpproj”.
OverlayObjects: This module fuses the “CropSensor” image with the Plaque objects to make a composite image named “OverlayImage”.
SaveImages: Creates an image called “Output_image_OverlayPlaques” based on the “OverlayImage”.
Note: Together with this protocol we supply the Zip file “PlaqueIdentification tracking pipeline and dataset” which contains the applied pipeline (“PlaqueIdentification.cpproj”) and an example dataset composed of two input images to run the pipeline and practice the required adjustments (“Input_image_mNeptune_72h” and “Input_image_mNeptune_96h”) and four output files to corroborate the expected results (“Output_image_PreviousPlaques_72h”, “Output_image_PreviousPlaques_96h”, “Output_image_OverlayPlaques_72h”, and “Output_image_OverlayPlaques_96h”).
Plaque tracking pipeline
Images: Simply drag and drop the files of the plaque assay fluorescence images (e.g., “Input_image_mNeptune_72h”) and the output images created with the previous Plaque identification pipeline (e.g., “Output_image_PreviousPlaques_72h”). Make sure that the name of the plaque assay fluorescence images do include the name of the channel where the fluorescence was measured (e.g., mNeptune) and the increasing consecutive numbers for time-lapse microscopy images (e.g., mNeptune_72h).
NamesAndTypes: Select the rule criteria with an expression contained in the name of all the plaque assay fluorescence images, in this case, indicating the channel of the fluorescence (e.g., mNeptune). For each fluorescence image it creates an image called “Sensor”.
The modules 3-8 are the same as those described in the Plaque identification pipeline.
IdentifySecondaryObjects: This module identifies the new Plaque objects in the “CellImage” as secondary objects around the previously identified PreviousPlaques primary objects.
TrackObjects: This module tracks the plaques over time using an overlap criteria across the time-resolved images and creates an output image named “TrackedPlaques”. You may adjust the Maximum distance to consider matches for an optimal plaque tracking.
SaveImages: Saves the “Tracked_Plaques” image for your validation and final results.
RelateObjects: This module correlates the new Plaque objects with the previously identified Nuclei objects.
ExporttoSpreadSheet: This module exports to an excel spreadsheet the data of the number of cells (Nuclei objects) that compose every recognized plaque in all the analyzed images.
Note: Together with this protocol we supply the Zip file “PlaqueIdentification tracking pipeline and dataset” which contains the applied pipeline (“PlaqueTracking.cpproj”) and an example dataset composed of four input images to run the pipeline (“Input_image_mNeptune_72h”, “Input_image_mNeptune_96h”, “Output_image_Previous Plaques_72h”, and “Output_image_PreviousPlaques_96h”) and three output files to corroborate the expected results (“Tracked_Plaques_72h”, “Tracked_Plaques_96h” and “Viral_Plaque”).
Recipes
Complete DMEM
500 ml DMEM (Gibco)
5 ml Antibiotic-antimycotic 100×
5 ml (for 2%) or 50 ml (for 10%) of FBS
Homogenize by hand rotation (20 times, gently to avoid formation of foam)
Store at 4 °C. Stable for 4 months
PEI solution (1 mg/ml)
1 mg of PEI powder
1 ml of HCl 0.2 M in distilled water
Heat and shake at 60 °C, 600 rpm in a ThermoMixer® block
Sterilize by filtration (0.2 µm pore size)
Prepare 45 µl aliquots into 2 ml reaction tubes and store at -80° C. Stable for 4 months
Polybrene solution (10 mg/ml)
10.6 mg of hexadimethrine bromide powder
Dissolve in 1 ml of destilled water
Sterilize by filtration (0.2 µm pore size)
Prepare 50 µl single use aliquots into 0.5 ml reaction tubes and store at -20 °C. Stable for 1 year
Complete MEM
500 ml MEM (Gibco)
5 ml Sodium pyruvate 100 mM
5 ml Antibiotic-antimycotic 100×
5 ml (for 2%) or 50 ml (for 10%) of FBS
Homogenize by hand rotation (20 times, gently to avoid formation of foam)
Store at 4 °C. Stable for 4 months
PBS 1% FBS
45 ml PBS, pH 7.4
5 ml complete MEM 10% FBS
Store at 4 °C. Stable for 4 months
Puromycin solution (10 mg/ml)
10 mg of puromycin
Dissolve in 1 ml of destilled water
Sterilize by filtration (0.2 µm pore size)
Prepare 20 µl single use aliquots into 1.5 ml light protection reaction tubes and store at -20 °C. Stable for 2 years
FluoroBriteTM DMEM 2% FBS
500 ml FluoroBriteTM DMEM (Gibco)
5 ml GlutaMAXTM supplement
5 ml Sodium pyruvate 100 mM
5 ml Antibiotic-antimycotic 100×
5 ml of FBS
Homogenize by hand rotation (20 times, gently to avoid formation of foam)
Store at 4 °C. Stable for 4 months
Plaque media 2% FBS
9.4 g MEM (Sigma)
900 ml Milli-Q water
Adjust pH to 4.0
10 g Carboxymethylcellulose sodium salt
Autoclave (121 °C at 100 kPa for 15 min)
30 ml 7.5% sodium bicarbonate solution in Milli-Q water (sterilized by filtration – 0.2 µm pore size)
10 ml GlutaMAXTM supplement
10 ml Sodium pyruvate 100mM
10 ml Antibiotic-antimycotic 100×
20 ml of FBS
Homogenize by hand rotation (20 times, gently to avoid formation of foam)
Store at 4 °C. Stable for 4 months
Acknowledgments
We want to thank Dr. Jeanne A. Hardy from the Department of Chemistry, University of Massachusetts, for her kindness and scientific advice during our outstanding collaboration. We also want to acknowledge our funding sources at Universidad de Costa Rica (project VI-803-B9-505) and International Centre for Genetic Engineering and Biotechnology (Grant CRP/CRI18-02).
Competing interests
The authors declare that they do not have any conflicts of interests.
References
- Arias-Arias, J. L., MacPherson, D. J., Hill, M. E., Hardy, J. A. and Mora-Rodriguez, R. (2020). A fluorescence-activatable reporter of flavivirus NS2B-NS3 protease activity enables live imaging of infection in single cells and viral plaques.J Biol Chem 295(8): 2212-2226.
- Balsitis, S. J., Coloma, J., Castro, G., Alava, A., Flores, D., McKerrow, J. H., Beatty, P. R. and Harris, E. (2009). Tropism of dengue virus in mice and humans defined by viral nonstructural protein 3-specific immunostaining. Am J Trop Med Hyg 80(3): 416-424.
- Freppel, W., Mazeaud, C. and Chatel-Chaix, L. (2018). Production, titration and imaging of Zika virus in mammalian cells. Bio-protocol 8(24): e3115.
- Gould, E. A. and Solomon, T. (2008). Pathogenic flaviviruses.Lancet 371(9611): 500-509.
- Hawley, T. S., Herbert, D. J., Eaker, S. S. and Hawley, R. G. (2004). Multiparameter flow cytometry of fluorescent protein reporters. Methods Mol Biol 263: 219-237.
- Hsieh, M. S., Chen, M. Y., Hsieh, C. H., Pan, C. H., Yu, G. Y. and Chen, H. W. (2017). Detection and quantification of dengue virus using a novel biosensor system based on dengue NS3 protease activity. PLoS One 12(11): e0188170.
- Kümmerer, B. M. (2018). Establishment and application of flavivirus replicons. Adv Exp Med Biol 1062: 165-173.
- Li, S. H., Li, X. F., Zhao, H., Deng, Y. Q., Yu, X. D., Zhu, S. Y., Jiang, T., Ye, Q., Qin, E. D. and Qin, C. F. (2013). Development and characterization of the replicon system of Japanese encephalitis live vaccine virus SA14-14-2. Virol J 10: 64.
- McFadden, M. J., Mitchell-Dick, A., Vazquez, C., Roder, A. E., Labagnara, K. F., McMahon, J. J., Silver, D. L. and Horner, S. M. (2018). A Fluorescent Cell-Based System for Imaging Zika Virus Infection in Real-Time.Viruses 10(2).
- Medin, C. L., Valois, S., Patkar, C. G. and Rothman, A. L. (2015). A plasmid-based reporter system for live cell imaging of dengue virus infected cells. J Virol Methods 211: 55-62.
- Medina, F., Medina, J. F., Colon, C., Vergne, E., Santiago, G. A. and Munoz-Jordan, J. L. (2012). Dengue virus: isolation, propagation, quantification, and storage. Curr Protoc Microbiol Chapter 15: Unit 15D 12.
- Schmid, B., Rinas, M., Ruggieri, A., Acosta, E. G., Bartenschlager, M., Reuter, A., Fischl, W., Harder, N., Bergeest, J. P., Flossdorf, M., Rohr, K., Höfer, T. and Bartenschlager, R. (2015). Live cell analysis and mathematical modeling identify determinants of attenuation of dengue virus 2'-o-methylation mutant. PLoS Pathog 11(12): e1005345.
- Soto-Garita, C., Somogyi, T., Vicente-Santos, A. and Corrales-Aguilar, E. (2016). Molecular Characterization of Two Major Dengue Outbreaks in Costa Rica. Am J Trop Med Hyg 95(1): 201-205.
- Tamura, T., Fukuhara, T., Uchida, T., Ono, C., Mori, H., Sato, A., Fauzyah, Y., Okamoto, T., Kurosu, T., Setoh, Y. X., Imamura, M., Tautz, N., Sakoda, Y., Khromykh, A. A., Chayama, K. and Matsuura, Y. (2018). Characterization of recombinant flaviviridae viruses possessing a small reporter tag. J Virol 92(2).
- Tiscornia, G., Singer, O. and Verma, I. M. (2006). Production and purification of lentiviral vectors. Nat Protoc 1(1): 241-245.
- Xie, X., Zou, J., Shan, C., Yang, Y., Kum, D. B., Dallmeier, K., Neyts, J. and Shi, P. Y. (2016). Zika virus replicons for drug discovery. EBioMedicine 12: 156-160.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Arias-Arias, J. L. and Mora-Rodríguez, R. (2021). Generation and Implementation of Reporter BHK-21 Cells for Live Imaging of Flavivirus Infection. Bio-protocol 11(5): e3942. DOI: 10.21769/BioProtoc.3942.
- Arias-Arias, J. L., MacPherson, D. J., Hill, M. E., Hardy, J. A. and Mora-Rodriguez, R. (2020). A fluorescence-activatable reporter of flavivirus NS2B-NS3 protease activity enables live imaging of infection in single cells and viral plaques.J Biol Chem 295(8): 2212-2226.
Category
Microbiology > Microbial cell biology > Cell-based analysis > Reporter assay
Cell Biology > Cell imaging > Live-cell imaging
Cell Biology > Cell-based analysis > Viral infection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.