- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Retention Using Selective Hooks (RUSH) Cargo Sorting Assay for Protein Vesicle Tracking in HeLa Cells
(*contributed equally to this work) Published: Vol 11, Iss 5, Mar 5, 2021 DOI: 10.21769/BioProtoc.3936 Views: 9312
Reviewed by: Vikash VermaAndrew L. EagleYoshihiro Adachi
Original research article
The authors used this protocol in:

Aug 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Monitoring vesicle trafficking is an excellent tool for the evaluation of protein dynamics in living cells. Such study is key for the understanding of protein sorting and secretion. Recent developments in microscopy, as well as new methodologies developed to study synchronized trafficking of proteins, allowed a better understanding of signaling, regulation and trafficking dynamics at the secretory pathway. One of the most helpful tools so far developed is the Retention Using Selective Hooks (RUSH) system, a methodology that facilitates the evaluation of synchronized cargo trafficking by monitoring fluorescent vesicles in cells upon biotin addition. Here we present a protocol that allows the quantitative evaluation of protein cargo trafficking at different fixed time points and an analytic approach that enables a better examination of specific cargo trafficking dynamics at the secretory pathway.
Graphic abstract:
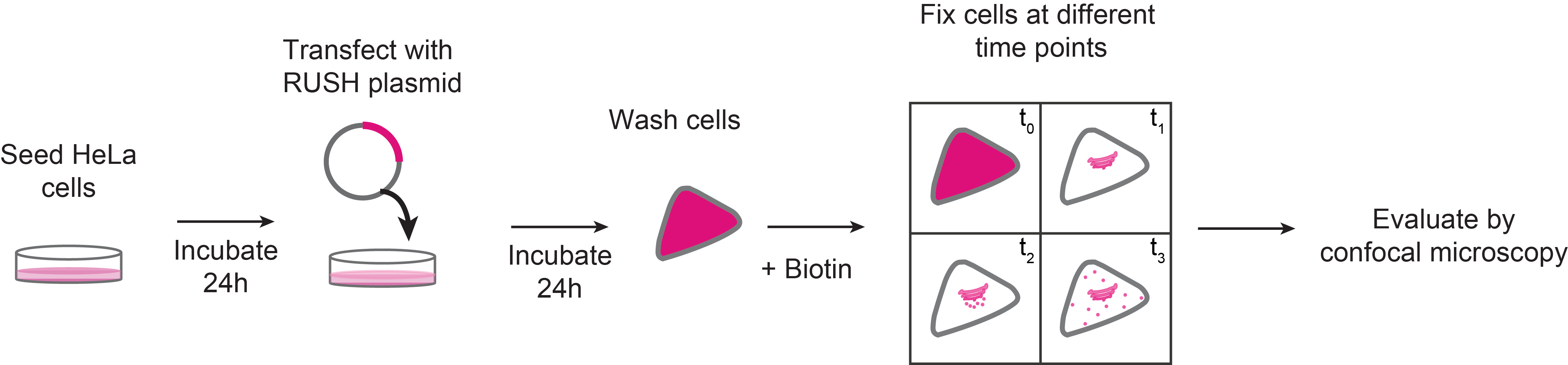
Schematic representation of RUSH sorting assay in mammalian cells
Background
Monitoring vesicle trafficking is an excellent tool for the evaluation of secretory protein dynamics in living cells. Given that around 30% of the total amount of newly synthesized proteins in mammals follow the secretory pathway (Pfeffer, 2010; Boncompain and Weigel, 2018), the study of its trafficking dynamics is key for the understanding of protein sorting and secretion. Proteins that follow the secretory pathway go through several maturation steps, starting at the ER, they are transported along the different Golgi stacks until they reach the trans-Golgi network (TGN), a sorting station. In the TGN they are finally packed into vesicles and initiate their route to their final destination (Glick and Luini, 2011; Pantazopoulou and Glick, 2019).
Recent developments in microscopy, as well as new methodologies developed to study the synchronized trafficking of proteins allowed to better understand signaling, regulation and trafficking dynamics at the secretory pathway (Stephens and Perez, 2013). One of the most helpful tools so far developed is the Retention Using Selective Hooks (RUSH) system, a methodology that facilitates the evaluation of synchronized cargo trafficking by monitoring fluorescent vesicles in cells upon biotin addition (Boncompain et al., 2012).
The RUSH system has two main elements: a protein of interest (POI) tagged with a fluorophore and bound to a streptavidin binding peptide (SBP); and a streptavidin molecule bound to a retention signal (known as the “hook”, e.g., a KDEL sequence for retention in the ER; Figure 1). This complex, in the absence of biotin will be retained in the donor compartment, but once biotin is added to the culture medium, the POI-fluorophore-SBP complex will be released to the next compartment. This set-up allows to follow up the POI at different time points by confocal microscopy (Boncompain et al., 2012).
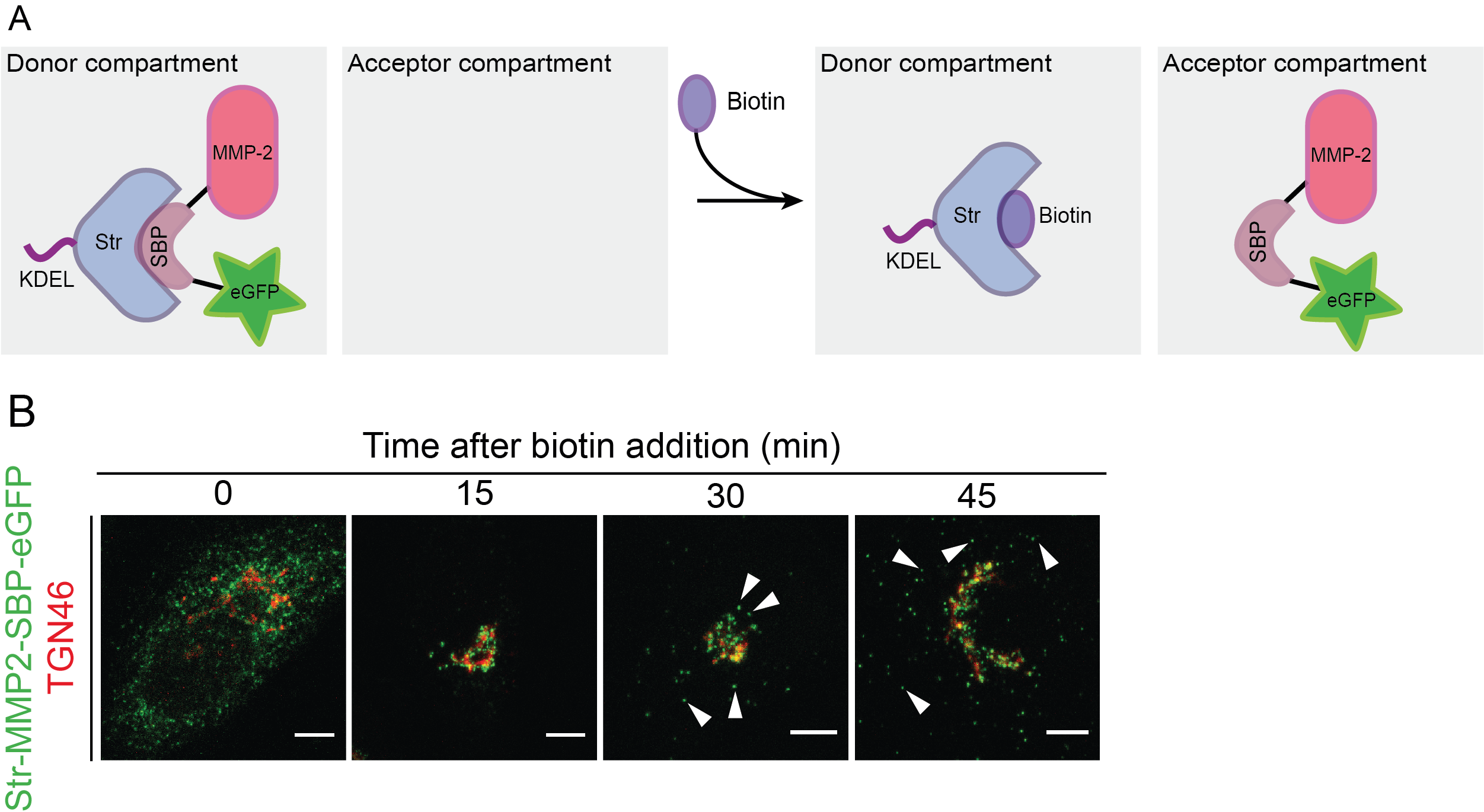
Figure 1. Scheme representing the RUSH system. A. Protein complex containing the protein of interest (here matrix metalloprotease 2, MMP2), a streptavidin binding peptide (SBP) and a fluorescent protein (eGFP). The complex is bound via SBP to streptavidin (Str), which is linked to a KDEL sequence for its retention in the endoplasmic reticulum (donor compartment). Without biotin addition, the complex remains in the donor compartment; however, once biotin is added to the media, it binds to streptavidin and enables the trafficking of the MMP2-SBP-eGFP protein complex to the acceptor compartment. Figure adapted from Boncompain et al. (2012). B. Immunofluorescence images showing the trafficking of MMP2 across the secretory pathway, with MMP2 localizing at the ER when biotin is absent, and later localizing at the Golgi (15 and 30 min) and in post-Golgi vesicles (45 min) upon biotin addition. TGN46: trans-Golgi marker. Figure taken from Pacheco-Fernandez et al. (2020).
Among other techniques, the RUSH system has the advantage of enabling the monitoring of cells under physiological conditions. Given that biotin is a non-toxic small molecule, the synchronization of cargo release does not lead to high stress for the cells (Boncompain and Perez, 2013). Also, compared to other techniques such as the light-triggered protein secretion system (Chen et al., 2013), it does not require a previous aggregation of the protein, which may lead to trafficking through different routes, higher protein degradation rates, and consequently, higher cell toxicity (Boncompain and Perez, 2013).
Building on the original RUSH methodology, here we present a protocol that allows to quantitatively evaluate protein cargo trafficking at different fixed time points. Importantly, in this protocol we provide a quantitative analysis to better examine intra-Golgi trafficking dynamics by evaluating specific cargo trafficking dynamics along the secretory pathway using confocal microscopy. This allows to evaluate the impact of knocking-out or silencing secretory pathway proteins on cargo trafficking (Crevenna et al., 2016; Deng et al., 2018; Pacheco-Fernandez et al., 2020).
Materials and Reagents
Circle coverslips, thickness 1 (ThermoScientificTM, catalog number: CB00120RA120MNT0 )
6-well cell culture plates (Corning®, Costar®, catalog number: CLS3516-1EA )
SuperfrostTM Microscopy Slides (ThermoScientificTM, catalog number: AA00008332E00MNT10 )
Polyethylenimine, linear, MW 25,000, transfection grade (PEI 25KTM; Polysciences, catalog number: 23966-1).
60-70% confluent HeLa cells
1× Dulbecco’s Phosphate Buffered Saline (DPBS), no calcium, no magnesium (Life Technologies, Gibco, catalog number: 14190144 ). Storage temperature: 4 °C.
pIRESneo3-Str-KDEL-MMP2-SBP-EGFP vector (Figure 2). Storage temperature: -20 °C
Important note: This plasmid was generated by replacing the ST (ST6GAL1) sequence by MMP2 (our protein of interest) in the Addgene plasmid number 65264 (Str-KDEL_ST-SBP-EGFP).
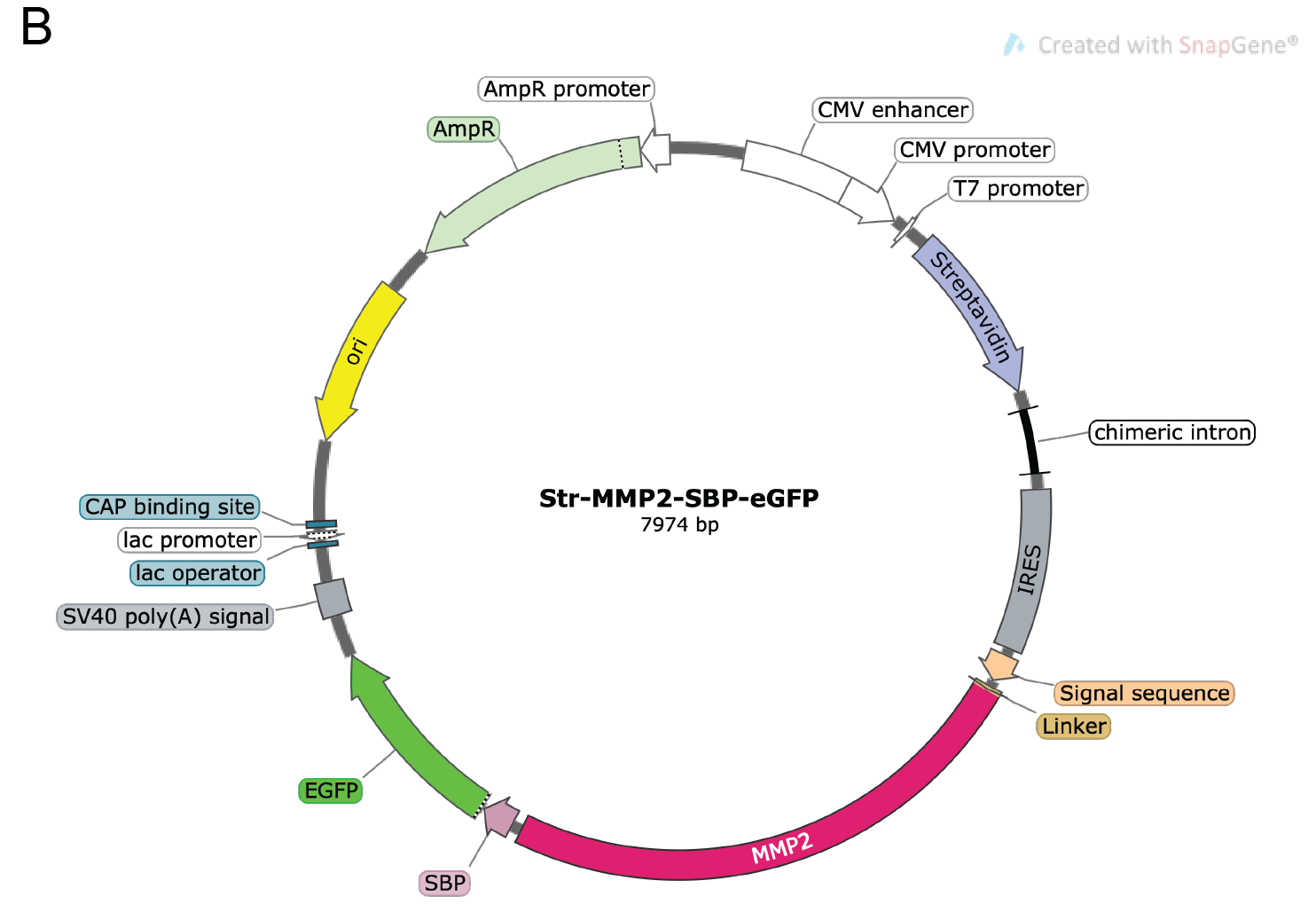
Figure 2. Vector map for the pIRESneo2-Str-KDEL-MMP2-SBP-eGFP construct used as an example in this protocol. The plasmid depicts the KDEL-bound streptavidin and the reporter MMP2-SBP-eGFP. An IRES element in the plasmid enables the simultaneous expression of both Str-KDEL and MMP2-SBP-eGFP in one single plasmid. For more details about the construct, please see Boncompain et al. (2012). The map was generated with SnapGene®.Opti-MEM® reduced serum media (Life Technologies, Gibco, catalog number: 31985070 ). Storage temperature: 4 °C
500 mM d-biotin (Merck, SUPELCO, catalog number: 47868 ). Storage temperature: 4 °C
4% paraformaldehyde (PFA) in 1× PBS. Prepared using the formaldehyde solution, ROTIPURAN® 37%, p.a., ACS (Roth, catalog number: 4979.1 )
Primary antibodies
For the protein of interest
Depending on the analysis, specific organelle markers can be used. For example, calnexin can be used as an ER marker, whereas, depending on the analytical evaluation, different Golgi markers could be used (e.g., GM-130 for cis-Golgi and TGN-46 for trans-Golgi network)
Secondary antibodies
Our studies were performed with Alexa-Fluor antibodies 488, 594 and 633 targeting different species (Life Technologies, ThermoScientific, catalog number varies according to species and wavelength). Storage temperature: 4 °C.
ProLong Gold antifade reagent (Life Technologies, Invitrogen, catalog number: P36934 )
PEI solution at 1 mg/ml (see Recipes)
Dulbecco’s Modified Eagles Medium (DMEM) complete medium (see Recipes)
Permeabilizing solution (see Recipes)
Triton X-100 (Carl Roth, catalog number: 3051.2 )
Sodium dodecyl sulphate (SDS) 10% solution
Blocking solution (see Recipes)
Bovine Serum Albumin (BSA) lyophilized powder (PAN Biotech, catalog number: P06-1391050 )
Phosphate buffered saline (PBS) powder (Sigma, catalog number: P3813-1PAK ): Prepare a 1× solution according to manufacturer’s instructions
Equipment
Tweezers
Laminar flow hood
Cell incubator set at 37 °C, 5% CO2
Water bath set at 37 °C
Zeiss laser scanning LSM780 confocal microscope (Carl Zeiss)
Zeiss 100× (NA 1.46, oil) objective (Carl Zeiss)
488-nm laser line
Software
Zeiss Zen software 2010 (Zeiss, https://www.zeiss.com/microscopy/int/products/microscope-software/zen.html)
ImageJ v.1.37 (National Institutes of Health [NIH], https://imagej.nih.gov/ij/)
ImageJ macro for “RUSH-Vesicle-Analysis” (https://github.com/MehrshadPakdel/RUSH-Vesicle-Analysis)
Procedure
Cell culture and plasmid transfection
All steps involving cell culture must be performed in a laminar flow hood.
Seed 3 × 104 HeLa cells in a 6-well plate containing 2-3 small coverslips using 2 ml DMEM complete medium per well. Prepare 1 well per time point (see Note 1).
Incubate the cells at 37 °C, 5% CO2 for 24 h.
After 24 h incubation, set up PEI transfection reaction. Per well:
200 µl OptiMEM
2 µg RUSH construct, e.g., pIRESneo3-SS-Str-KDEL-MMP2-SBP-EGFP
15 µl 50mM PEI solution
Vortex the transfection reaction and incubate it at room temperature for 20 min.
Add dropwise to the cells and incubate at 37 °C, 5% CO2 for at least 16 h. Avoid incubation periods longer than 24 h. If they are necessary, change cell medium after 24 h.
RUSH experiment
After incubation, prepare 2 ml of DMEM complete with d-biotin to a final concentration of 40 µM per well. Prepare enough volume according to the number of wells to analyze (for example, for 4 wells (2 ml medium per well), prepare 9 ml of DMEM complete + 7.2 µl d-biotin 500 mM). Keep it warm at 37 °C.
Remove medium from cells either by aspiration with a micropipette or with a vacuum pump and wash once with 1× DPBS.
Add 2 ml DMEM complete without biotin to all the time points except the latest one (see Note 2). For example, for MMP2 four time points are analyzed (0, 15, 30 and 45 min). Add medium only to the wells labeled as 0, 15 and 30 min.
Add 2 ml DMEM complete + biotin (from Step B1) to the latest time point (for MMP2, to the well labeled as 45 min. Figure 3, “start of the experiment”).
Incubate cells at 37 °C, 5% CO2.
The incubation time is determined by the trafficking kinetics of each construct and in order to facilitate the experimental handling we recommend to start adding biotin first to the well that needs to be incubated the longest time (45 min for MMP2) and then continue with the earlier time points (30 and 15 min, see scheme in Figure 3).
Important:Here is recommended to take the cells out of the incubator at least 2 min before triggering cargo trafficking in the next time point (for MMP2, the plate is taken out of the incubator 13 min after adding biotin to the previous well, namely, 2 min before starting the next time point. See note 2 for a more detailed explanation).
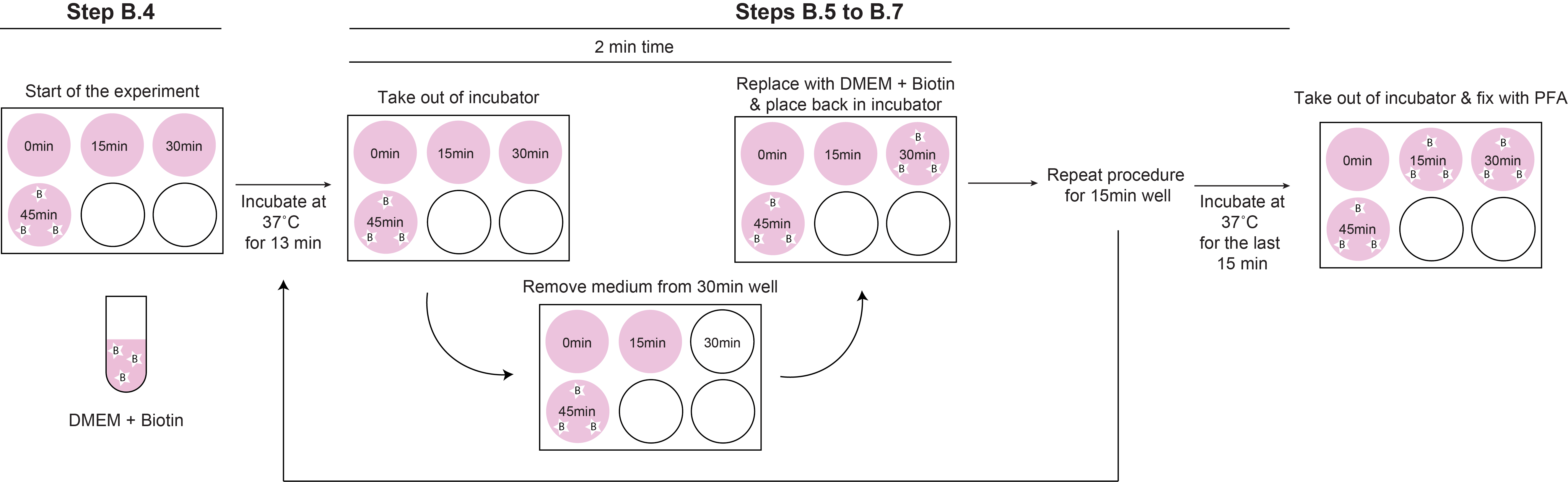
Figure 3. Scheme depicting the handling times during time intervals for the RUSH experiment. The scheme depicts the handling time for an experienced person using the MMP2 construct with the described time points of 0, 15, 30 and 45 min.Remove media from the next time point to be analyzed and add 2 ml DMEM complete + biotin.
Repeat Steps B2, B4 and B5 until completing the full-time cycle (for MMP2, 45 min with a last biotin addition at 15 min before fixation, Figure 3).
Coverslip montage
Right before incubation ends, wash the cells (all the wells) 2 times with 1× PBS.
Incubate cells for 10 min in 4% PFA in PBS under low light conditions. Make sure that the coverslips are completely covered with the PFA solution.
Wash 3 to 5 times with 1× PBS.
If no co-staining is performed: add a small amount of PBS to the well to facilitate the handling of coverslips and mount them using the ProLong antifade Gold reagent. For this purpose:
Take a microscopy glass slide.
Put one drop of ProLong antifade Gold reagent on the slide.
Take carefully one of the coverslips with tweezers.
Place the coverslip facing down on top of the ProLong antifade Gold reagent (i.e., the side of the coverslip with cells must be in contact with the antifade reagent).
Carefully store them in a dark place until imaging. It is recommended to allow the montaged slides to dry for at least 24 h, ideally for 48 h.
If co-staining is performed:
Permeabilize the cells with permeabilization solution for 5 min under low light conditions.
Wash 3 to 5 times with 1× PBS.
Incubate with blocking solution for 1 h at room temperature or overnight at 4 °C under low light conditions.
Freshly prepare the primary antibody solutions by diluting the antibody in blocking solution. Depending on the antibody, this information may be recommended by the manufacturer or require previous characterization in the lab to determine the ideal time and temperature.
Wash 3 to 5 times with 1× PBS.
Incubate with primary antibody for 1 h at room temperature or overnight at 4 °C under low light conditions.
Freshly prepare the secondary antibody solutions by diluting the amounts of antibody in blocking solutions recommended by the manufacturer.
Wash 3 to 5 times with 1× PBS.
Incubate with secondary antibody for 1 h at room temperature under low light conditions.
Wash 3 to 5 times with 1× PBS.
Mount the glass slides using the ProLong antifade Gold reagent (see Step C4).
Imaging
Start the microscope and warm up the required lasers. This process can take up to around 30 min depending on the lasers and the microscope.
Set up a z-stack acquisition experiment. We used a 0.35 µm distance per slice with typically 10 to 15 planes to cover most of the cell volume.
Focus a single cell (or several clearly defined and healthy single cells per field of view) and set up the number of stacks to be imaged using the signal from the RUSH channel.
Adjust gain and intensity from the other channels, if necessary.
Start imaging.
Repeat Steps D3 and D4 until completing at least 15 cells per time point.
Save files as *.lsm.
Data analysis
Open Fiji.
Open in Fiji our custom-made ImageJ macro for “RUSH-Vesicle-Analysis” available at https://github.com/MehrshadPakdel/RUSH-Vesicle-Analysis. For more details about the ImageJ functions used in this macro, see Note 7.
Open the microscopy (*.lsm) file in Fiji. Other microscopy files such as *.lif can also be used.
Click on run for the macro to initiate the analysis (Figure 4).
Figure 4. Screenshot of the RUSH-Vesicle analysis macro window in FijiSelect the number of cells to analyze (Figure 5).
Figure 5. Screenshot of step 1 from RUSH-analysis macroSelection of regions of interest (ROIs) to analyze: Identify each cell by drawing a polygon around it (Figure 6). The selected area will be saved automatically as an ROI. Click ok to select the next cell.
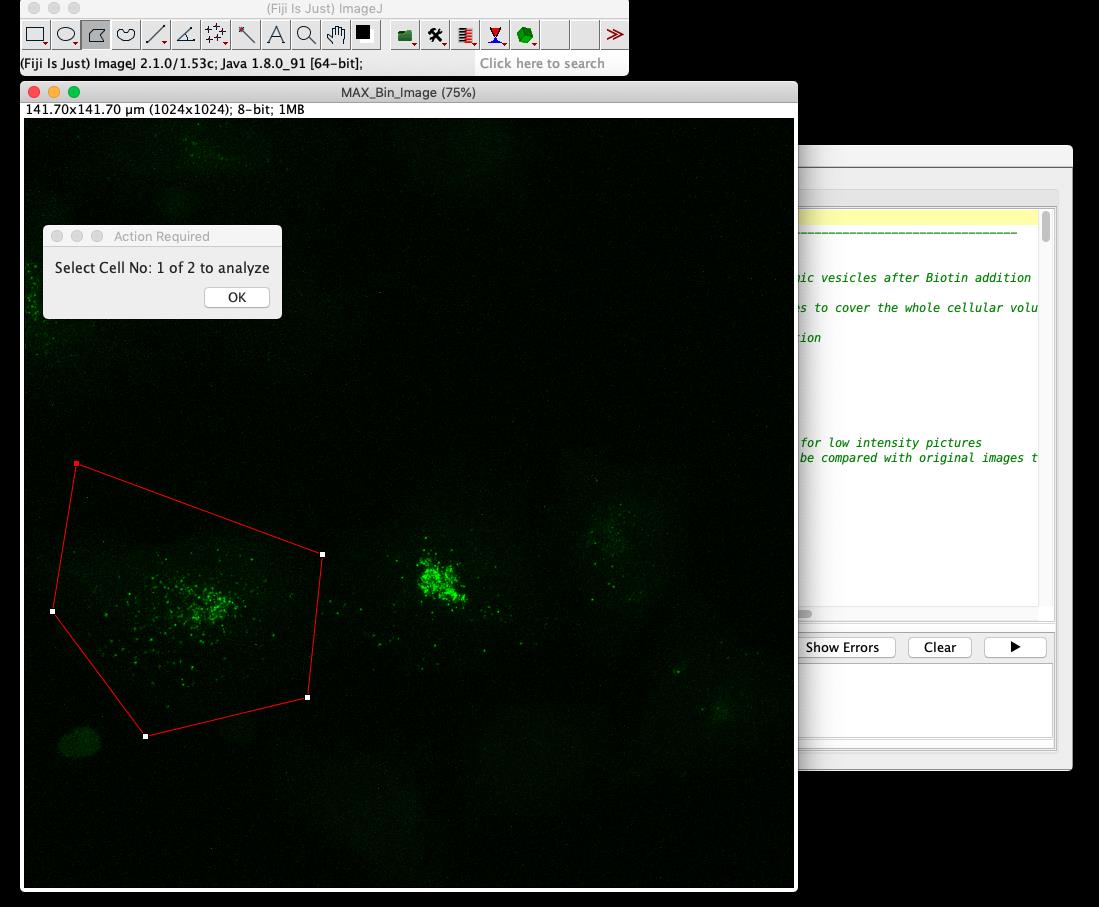
Figure 6. Screenshot of step 2 from RUSH-analysis macroGeneration of binary image and threshold adjustment: After selecting the last cell and clicking ok, you will see 2 windows appearing automatically in your screen (Figure 7, before): one showing the binary generated image (left window, title: MAX_Bin_Image) and another one showing the original maximal projection (right window, title: MAX_Image). In addition, two control windows will appear: one named “Threshold” that enables manual adjustment of black and white balance for the MAX_Bin_Image, and another one named “Action Required”. In order to avoid losing low-intensity vesicles in the counting, you need to visually adjust the threshold of the generated binary image in comparison to the original one (Figure 7). To do so, slide the bars in the “Threshold” window until MAX_Bin_Image resembles MAX_Image (Figure 7, after). Once both images are identical, click “Apply” in the threshold window. Importantly, once you click apply it is not possible to undo the threshold modification. After you click apply, click OK on the “Action Required” window to confirm the changes and continue with the macro (Figure 7, after).
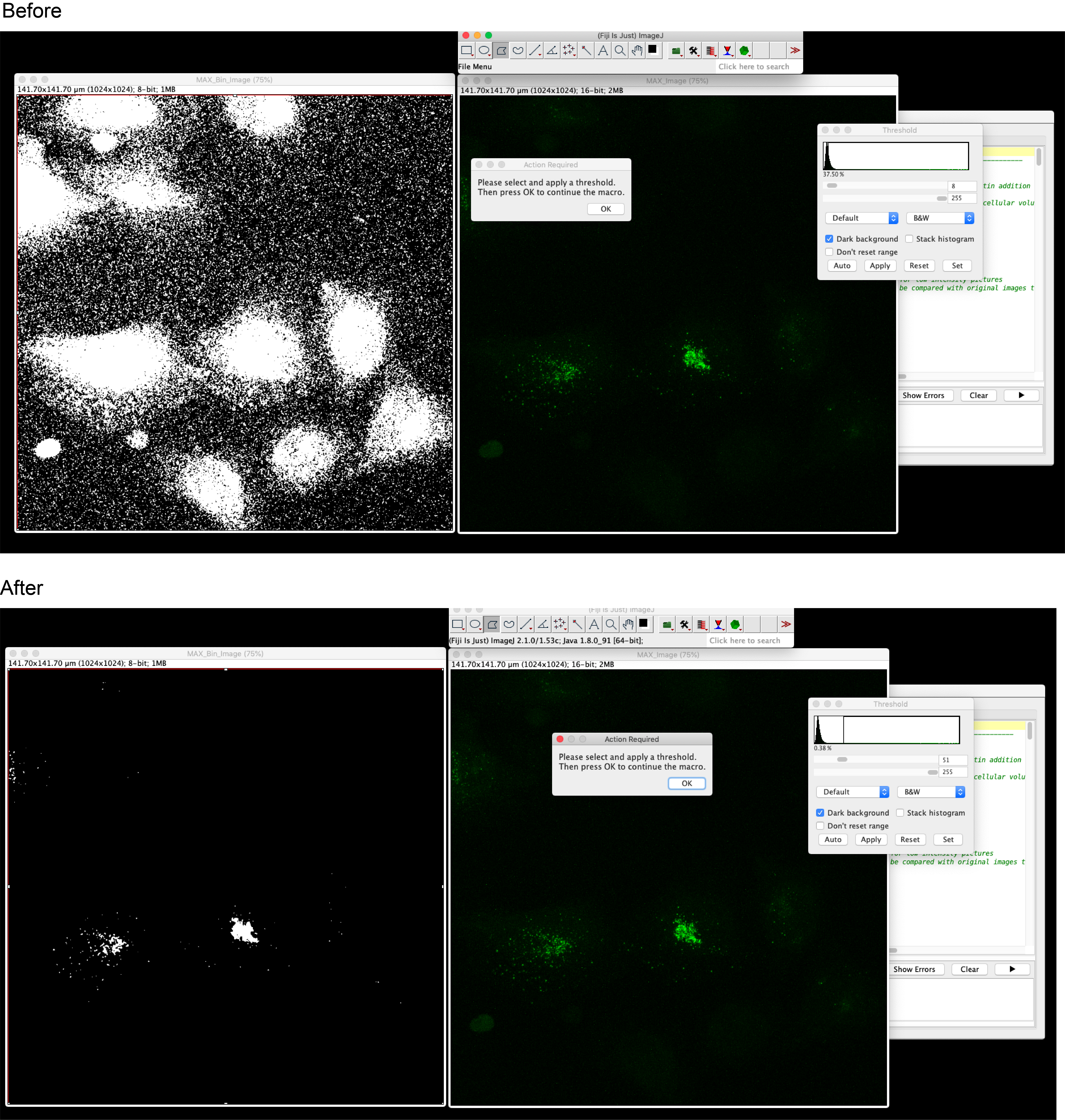
Figure 7. Screenshot of step 3 from RUSH-analysis macro. The figure “Before” shows the binary image generated (MAX_Bin_Image, left window), the original maximal projection (MAX_Image), the threshold control window and the “Action Required” window. To avoid losing low-intensity vesicles in the count you have to adjust manually the black and white balance of the MAX_Bin_Image having the fluorescent maximal projection as a reference (MAX_Image). Proper adjustment is illustrated in the figure named as “After”.Analyze particles: Once you click ok on the “Action Required” window, the macro will start automatically counting the number of vesicles on each one of the selected ROIs. Therefore, 3 new windows will appear: a window with the drawing (blue square, Figure 8 here named as Cell1_Example_eGFP_45min_Quanti.tif), a log window indicating the number of vesicles counted per cell (magenta square, Figure 8), and the ROI manager window showing the selected ROIs for the cells (green square, Figure 8). These numbers can be copied to an Excel file or similar spreadsheet file for further analysis.
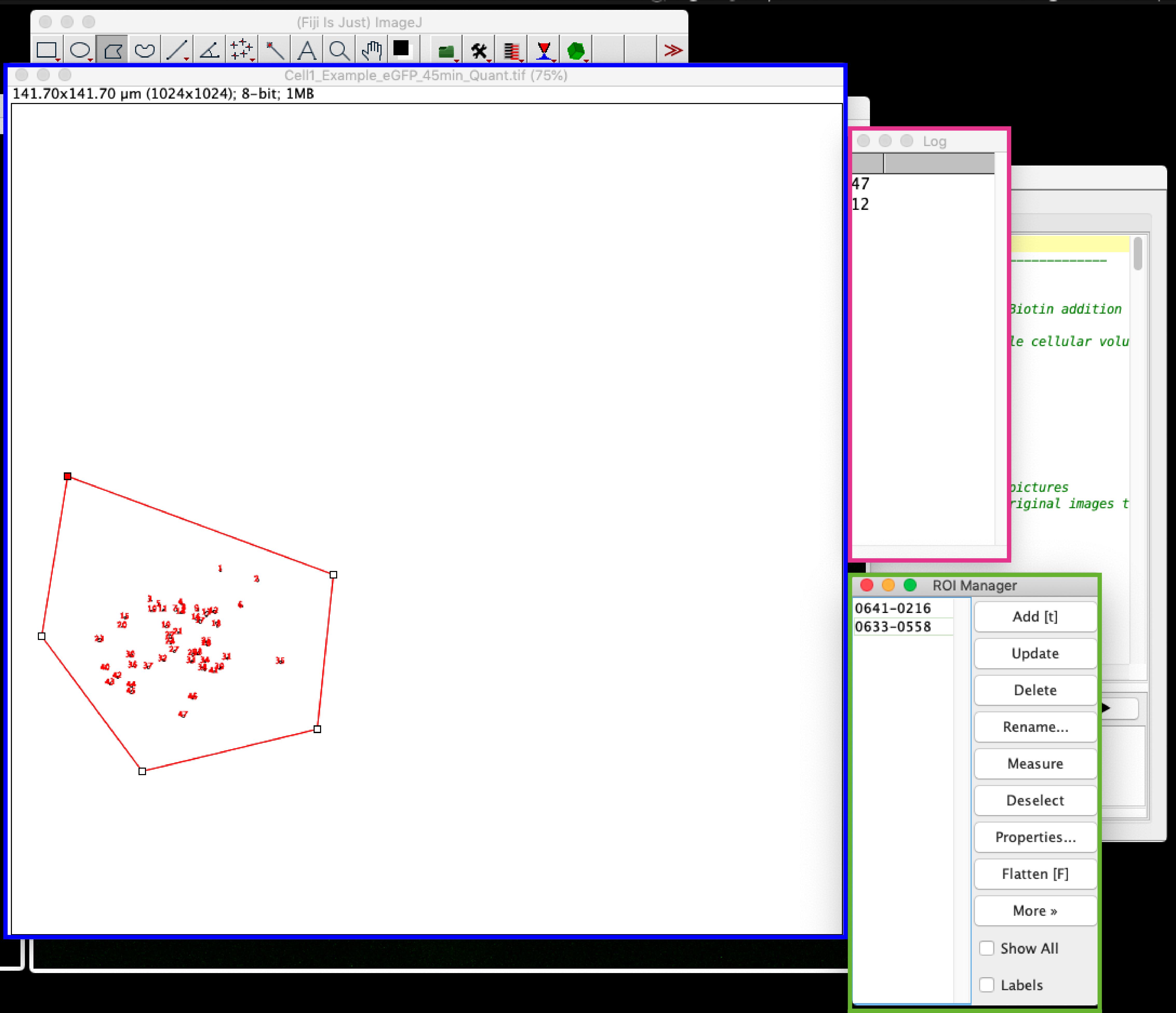
Figure 8. Screenshot of the final step from quantitative analysis. Three windows will appear automatically after clicking ok in step no. 7: one with a draw of the counted vesicles per cell (generated for each analyzed cell, blue square), a log window that depicts the number of vesicles counted per cell (magenta square) and a ROI window containing the information of the selected ROIs (selected cells, green square). The log window values can be copied to an excel file or statistical analysis software for further evaluation.Once the macro finishes running, all the results are saved automatically in a new results folder in the original file directory (Figure 9). The files saved are the original file (Orig_filename), the maximal projection of the original image (MAX_filename), the generated binary image (Binary_filename), one drawing file showing the number of counted vesicles for each cell selected (CellNumber_filename) and the log file containing the total number of counted vesicles per cell (log_filename).
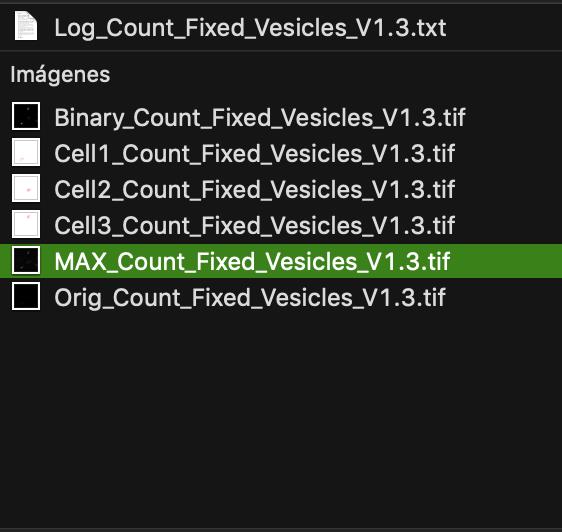
Figure 9. Screenshot of the files generated after running the macroThe number of vesicles can be further compared performing statistical tests such as t-test, ANOVA or their corresponding non-parametric test, to evaluate differences between time points and treatments. Figure 10 shows an example of vesicle quantification for MMP2eGFP positive vesicles in HeLa cells.
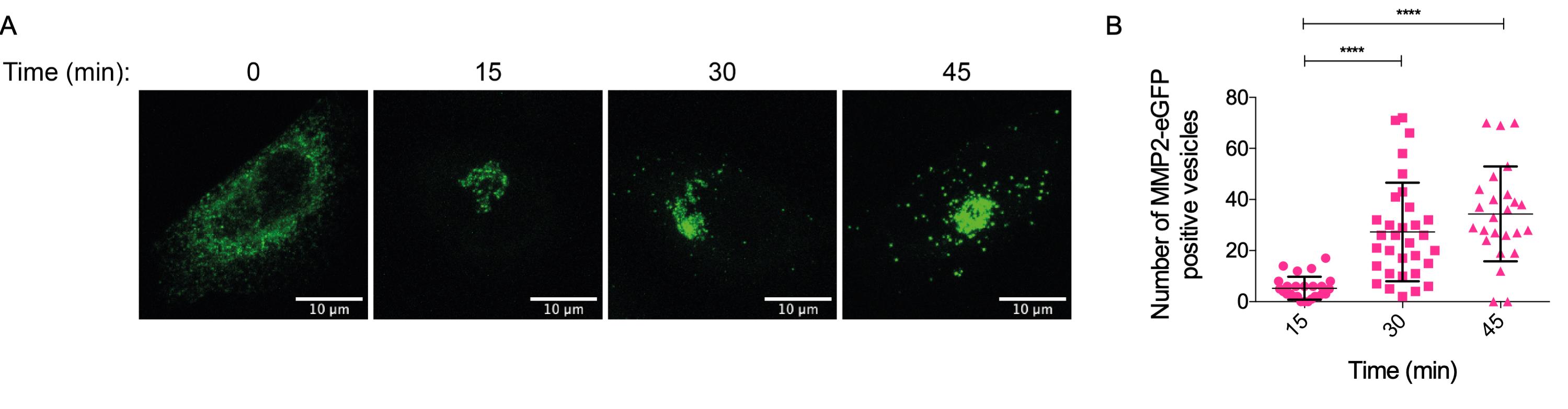
Figure 10. Final result from MMP2 RUSH vesicle analysis. Panel (A) depicts immunofluorescent images of HeLa cells expressing the RUSH MMP2eGFP construct at different time points. Scale bars: 10 µm. Panel (B) shows the accumulated number of MMP2-positive vesicles from at least 25 analyzed cells. Differences between time points were evaluated with a Kruskall-Wallis test. **** P-value < 0.0001.When using secretory pathway markers, other statistical tests could be used to evaluate co-localization, such as Pearson’s or Manders coefficients. For a more detailed description of such analysis, please refer to Pacheco-Fernandez et al. (2020). Figure 11 shows an example of MMP2 colocalization with the Golgi marker TGN 46, visualized by confocal microscopy.
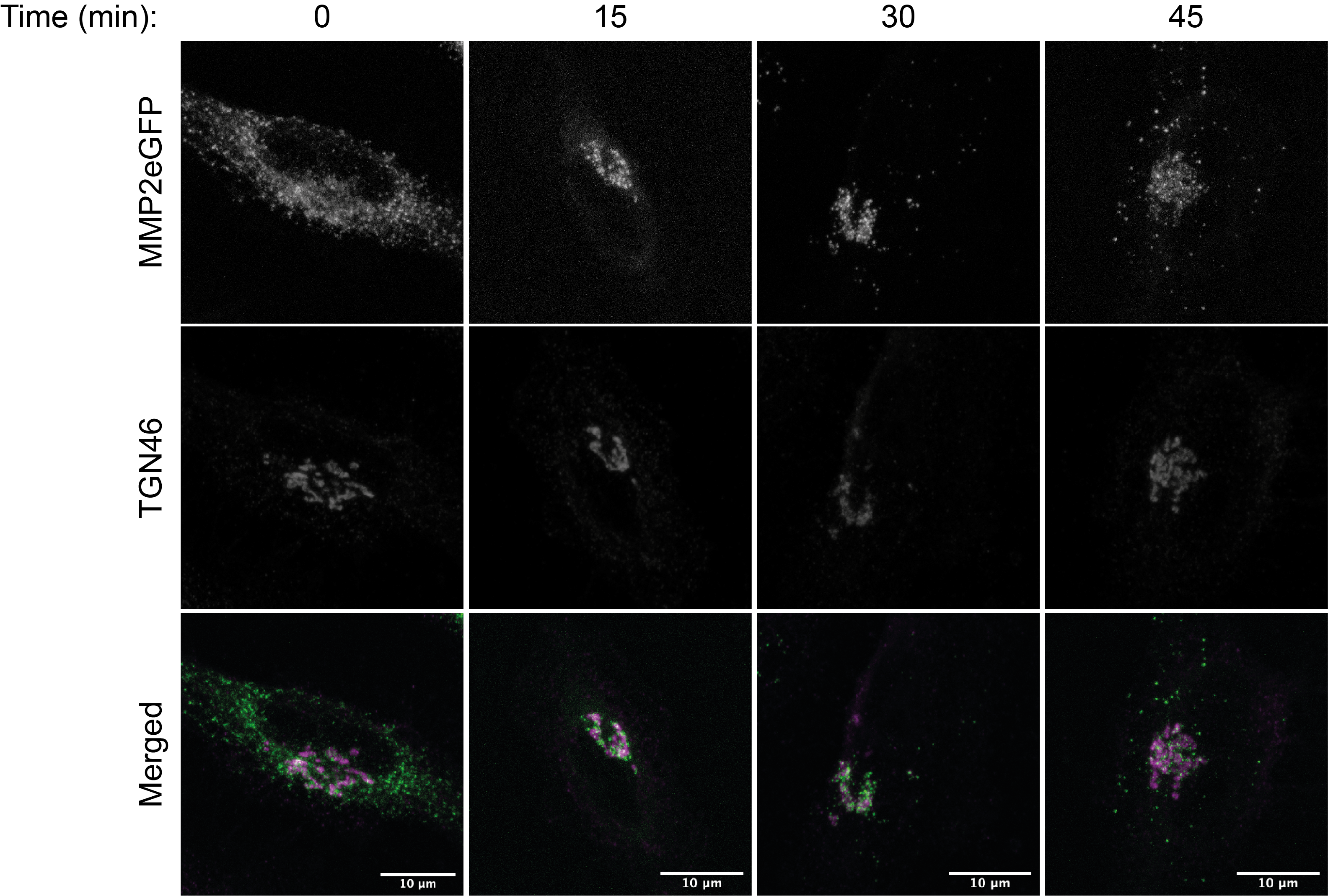
Figure 11. Immunofluorescent images of HeLa cells transfected with MMP2-RUSH and co-stained with TGN46. Cells were fixed at the indicated time points, permeabilized and incubated with GFP and anti-TGN46 antibodies (TGN-46 is a trans-Golgi marker). Green: MMP2-SBP-eGFP, magenta: trans-Golgi network. Scale bars: 10 µm.
Notes
Time points are defined according to the cargo evaluated. If you are using an uncharacterized cargo, a previous experiment with a known cargo and its described time points should be performed and used as a positive control. In our experience, the soluble secretory cargo we have evaluated in the lab (such as Lysozyme C and Cathepsin D) traffics through the secretory pathway in a time span of 60 to 75 min. The unknown cargo must be firstly evaluated in different intervals to establish the kinetics of its trafficking. For example, when we assessed the trafficking dynamics of MMP2 for the first time, we evaluated 5 min intervals to determine the critical points of transport (from ER to Golgi and from Golgi to extracellular space). However, the time points to evaluate will always depend on the desired kind of analysis.
This protocol refers to the latest incubation time as the last time point evaluated in the experiment (i.e., in the present protocol 45 min) and earlier incubation times as any time below such time point (i.e., 0, 15 and 30 min).
Keep in mind that the process of taking the plate out of the incubator, removing medium and adding medium with biotin takes at least 2 min for experienced people. For the first time, a working time window of 5 min between time points is recommended. Therefore, take the plate out of the incubator either 2 or 5 min (according to experience in handling) before the start of the next time point. For example, with the MMP2 construct here described, once DMEM + biotin has been added to the 45 min time point, take the plate out of the incubator after 13 min incubation so you have enough time to place the plate in the hood, remove medium from the 30 min well, replace with new DMEM + biotin and place the plate back in the incubator at 37 °C exactly 15 min after the 45 min incubation has started (Figure 3).
Protein trafficking is a temperature sensitive process; therefore, it is recommended to adjust your experimental conditions to avoid periods shorter than 5 min of cell incubation at 37 °C.
The set up for confocal microscopy can also be done in 35-mm dishes. We recommend counting at least 15 cells for a valid statistical analysis.
A control is necessary for the validation of this assay. In this case, a RUSH construct without a protein cargo (i.e., containing Str, the retention hook, SBP and the fluorophore to be analyzed) can be used as a control for the validation of any construct. Importantly, this control is suitable to evaluate the efficacy of the assay but for further kinetic dynamics comparisons, it is recommended to use a construct encoding a protein cargo with well-described trafficking dynamics as a control.
The macro here used for the analysis uses 7 functions (in the following order):
Background subtraction: Uses ImageJ subtract background function that is based in a rolling ball algorithm (https://imagej.net/Rolling_Ball_Background_Subtraction). This algorithm adjusts the pixel size to the radius of the largest object in the image that is not part of the background. For the present analysis, this radius was adjusted to the Golgi size.
Enhance contrast: This function facilitates the visibility of small vesicle particles. It enhances the contrast by stretching the image histogram without altering the pixel values (https://imagej.net/docs/guide/146-29.html#sub:Enhance-Contrast).
Z Project: Generates a 2D image with the flattened maximum intensities of the z-stack series (https://imagej.nih.gov/ij/docs/guide/146-28.html#sub:Z-Project ...).
Image processing filter “median”: The filter reduces the noise in the selection. In this macro is used to smoothen the vesicles and to generate less noisy objects after thresholding (https://imagej.nih.gov/ij/docs/guide/146-29.html#sub:Median ...).
Threshold: Segments grayscale images (binary image in this analysis) into feature of interest and background. Selecting threshold generates a binary image that defines which objects should not be count as background, and is required for subsequent particle quantification (https://imagej.nih.gov/ij/docs/guide/146-28.html#sub:Threshold...[T]).
Analyze particles: Detects and quantifies particles within a given pixel size (https://imagej.nih.gov/ij/docs/guide/146-30.html#toc-Subsection-30.2).
Recipes
PEI solution
Dissolve 50 mg of Polyethylenimine in 40 ml of distilled water
Mix well the solution with a magnetic stirrer on a slightly warmed plate
Adjust pH to 7.4
Complete volume up to 50 ml. Aliquot in 500 µl and store at -20 °C
DMEM complete medium
Prepare 500 ml of DMEM medium by adding FCS and P/S to a final concentration of 10% and 1%, respectively
Permeabilizing solution
Mix in 1× PBS: 0.2% Triton X-100 and 0.5% sodium dodecyl sulphate (SDS; dilute from a 20% or 10% SDS solution).
Make 5 ml aliquots and store long term at -20 °C. Once thawed, store at 4 °C and warm up at room temperature for working. If some precipitate forms, briefly warm up at 37 °C until the solution becomes clear.
Blocking solution
Dissolve 20 g of bovine serum albumin (BSA, molecular biology grade) in 500 ml of 1× PBS. Aliquot in 50 ml and store long term at -20 °C. Once thawed, store at 4 °C and keep on ice while working at room temperature.
Acknowledgments
This protocol was previously documented in the publications (Deng et al., 2018) and (Pacheco-Fernandez et al., 2020). This work was supported by grants from the Deutscher Akademischer Austauschdienst (DAAD), Deutsche Forschungsgemeinschaft (DFG), National Institutes of Health (NIH), the Perspective Program (Boehringer Ingelheim Fonds) and the Max Planck Institute of Biochemistry.
Competing interests
The authors declare no competing financial interests.
References
- Boncompain, G., Divoux, S., Gareil, N., de Forges, H., Lescure, A., Latreche, L., Mercanti, V., Jollivet, F., Raposo, G. and Perez, F. (2012). Synchronization of secretory protein traffic in populations of cells. Nat Methods 9(5): 493-498.
- Boncompain, G. and Perez, F. (2013). Fluorescence-based analysis of trafficking in mammalian cells. Methods Cell Biol 118: 179-194.
- Boncompain, G. and Weigel, A. V. (2018). Transport and sorting in the Golgi complex: multiple mechanisms sort diverse cargo. Curr Opin Cell Biol 50: 94-101.
- Chen, D., Gibson, E. S. and Kennedy, M. J. (2013). A light-triggered protein secretion system. J Cell Biol 201(4): 631-640.
- Crevenna, A. H., Blank, B., Maiser, A., Emin, D., Prescher, J., Beck, G., Kienzle, C., Bartnik, K., Habermann, B., Pakdel, M., Leonhardt, H., Lamb, D. C. and von Blume, J. (2016). Secretory cargo sorting by Ca2+-dependent Cab45 oligomerization at the trans-Golgi network. J Cell Biol 213(3): 305-314.
- Deng, Y., Pakdel, M., Blank, B., Sundberg, E. L., Burd, C. G. and von Blume, J. (2018). Activity of the SPCA1 Calcium Pump Couples Sphingomyelin Synthesis to Sorting of Secretory Proteins in the Trans-Golgi Network. Dev Cell 47(4): 464-478 e468.
- Glick, B. S. and Luini, A. (2011). Models for Golgi traffic: a critical assessment. Cold Spring Harb Perspect Biol 3(11): a005215.
- Pacheco-Fernandez, N., Pakdel, M., Blank, B., Sanchez-Gonzalez, I., Weber, K., Tran, M. L., Hecht, T. K., Gautsch, R., Beck, G., Perez, F., Hausser, A., Linder, S. and von Blume, J. (2020). Nucleobindin-1 regulates ECM degradation by promoting intra-Golgi trafficking of MMPs. J Cell Biol 219(8).
- Pantazopoulou, A. and Glick, B. S. (2019). A Kinetic View of Membrane Traffic Pathways Can Transcend the Classical View of Golgi Compartments.Front Cell Dev Biol 7: 153.
- Pfeffer, S. R. (2010). How the Golgi works: a cisternal progenitor model. Proc Natl Acad Sci U S A 107(46): 19614-19618.
- Stephens, D. and Perez, F. (2013). Preface. In: Perez, F. and Stephens, D. (Eds.). Methods for Analysis of Golgi Complex Function (Vol. 118, pp. xix–xx). Academic Press.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Pacheco-Fernandez, N., Pakdel, M. and von Blume, J. (2021). Retention Using Selective Hooks (RUSH) Cargo Sorting Assay for Protein Vesicle Tracking in HeLa Cells. Bio-protocol 11(5): e3936. DOI: 10.21769/BioProtoc.3936.
- Pacheco-Fernandez, N., Pakdel, M., Blank, B., Sanchez-Gonzalez, I., Weber, K., Tran, M. L., Hecht, T. K., Gautsch, R., Beck, G., Perez, F., Hausser, A., Linder, S. and von Blume, J. (2020). Nucleobindin-1 regulates ECM degradation by promoting intra-Golgi trafficking of MMPs. J Cell Biol 219(8).
Category
Cell Biology > Cell imaging > Confocal microscopy
Biochemistry > Protein > Self-assembly
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.