- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Trypanosomatid, fluorescence-based in vitro U-insertion/U-deletion RNA-editing (FIDE)
Published: Vol 11, Iss 5, Mar 5, 2021 DOI: 10.21769/BioProtoc.3935 Views: 4671
Reviewed by: Manuel Miras MarinKarolina SubrtovaMarcelo S. da Silva
Original research article
The authors used this protocol in:

Sep 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Gene expression within the mitochondria of African trypanosomes and other protozoan organisms relies on a nucleotide-specific RNA-editing reaction. In the process exclusively uridine (U)-nucleotides are site-specifically inserted into and deleted from sequence-deficient primary transcripts to convert them into translatable mRNAs. The reaction is catalyzed by a 0.8 MDa multiprotein complex termed the editosome. Here we describe an improved in vitro test to quantitatively explore the catalytic activity of the editosome. The assay uses synthetic, fluorophore-derivatized oligoribonucleotides as editing substrates, which enable the automated electrophoretic separation of the reaction products by capillary electrophoresis (CE) coupled to laser-induced fluorescence (LIF) detection systems. The assay is robust, it requires only nanogram amounts of materials and by using multicapillary CE/LIF-instruments it can be executed in a highly parallel layout. Further improvements include the usage of phosphorothioate-modified and thus RNase-resistant substrate RNAs as well as multiplex-type fluorophore labeling strategies to monitor the U-insertion and U-deletion reaction simultaneously. The assay is useful for investigating the mechanism and enzymology of the editosome. However, it can also be executed in high-throughput to screen for RNA editing-specific inhibitors.
Graphic abstract:
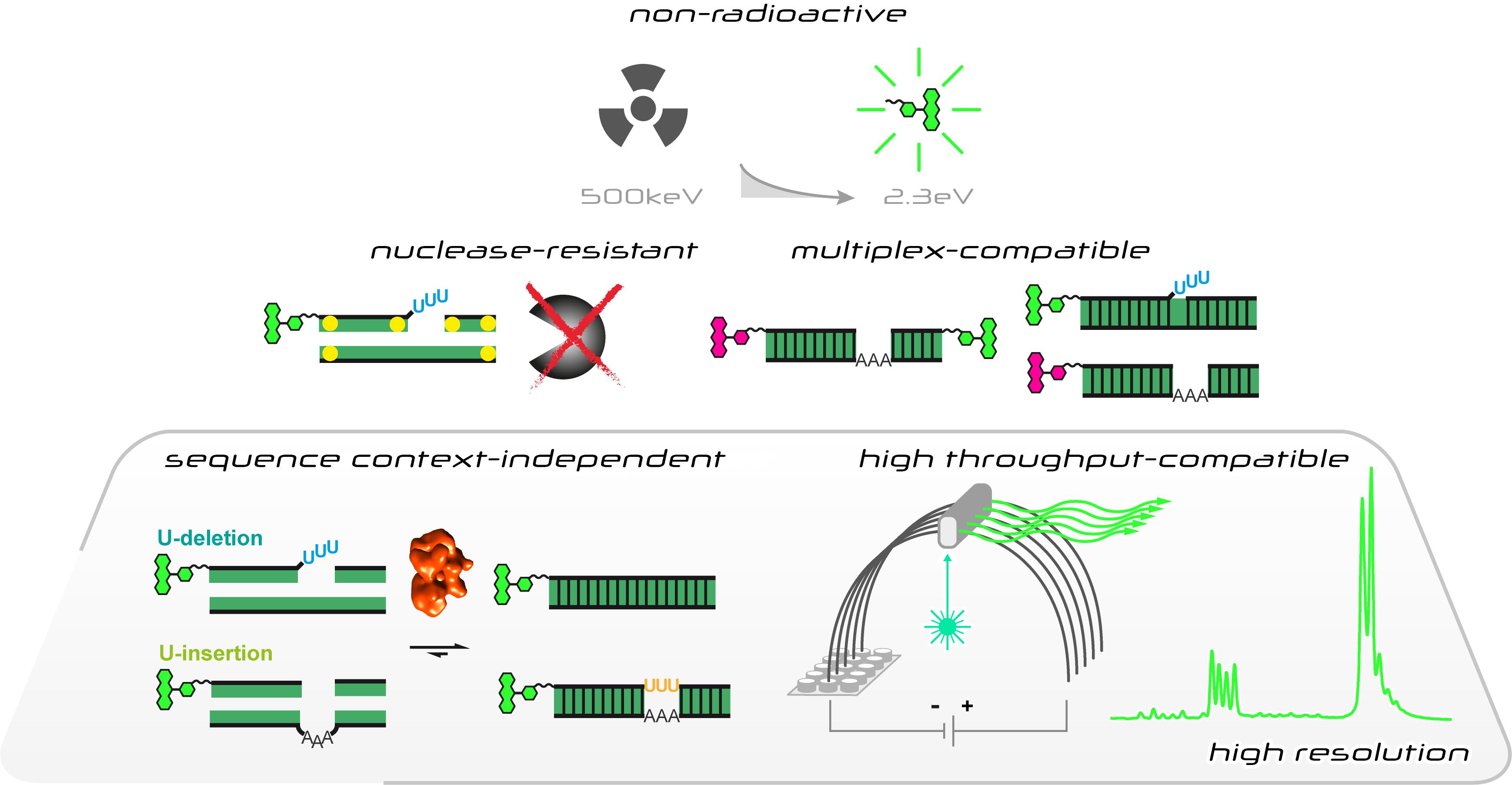
Characteristics of the fluorescence-based in vitro U-insertion/U-deletion RNA-editing (FIDE) assay
Background
The RNA editing reaction in kinetoplastid protozoa such as African trypanosomes and Leishmania represents one the most striking posttranscriptional modifications of messenger (m)RNAs (reviewed in Göringer, 2012a). The reaction takes place within the single mitochondria of the organisms, which is similar to other eukaryotes and holds the genetic information for components of the mitochondrial translation apparatus and subunits of the electron transport and chemiosmosis systems. However, unlike in other organisms, more than 50% of these genes are “incompletely” encoded: substantial sequence information is missing – in some cases more than 50% – in addition to the absence of start and stop codons and the presence of frame-shifts. As a consequence, precursor (pre)-mRNAs from these genes must be corrected, i.e., "edited" to generate functional mRNAs. Biochemically, the editing reaction is characterized by the insertion and/or deletion of U-nucleotides. In African trypanosomes, more than 3000 U-residues are inserted and about 300 U’s deleted at hundreds of editing sites in 12 different pre-mRNAs. The process relies on small, non-coding RNAs, known as guide (g)RNAs (Blum et al., 1990; Hermann et al., 1995). Guide RNAs are trans-acting genetic elements. They initiate the reaction cycle by antiparallel base pairing to the pre-edited mRNAs, which results in the formation of a gRNA/pre-mRNA hybrid. The hybrid-RNA has a 3-helix junction-topology including a so-called “anchor” – helix that borders the sequence to be edited. Unpaired gRNA nucleotides next to the anchor-helix specify U-insertion events, non-base paired uridylates in the pre-mRNA become deleted. The mitochondrial machinery that executes the processing reaction is a high molecular mass protein complex. In analogy to the ribosome and spliceosome it has been named the editosome (reviewed in Göringer, 2012b). Editosomes function as a catalytic platform for all steps of the reaction cycle and execute the processing reaction in a cascade of enzyme-driven steps. This includes endo- and exonucleases, a terminal uridylyltransferase (TUTase), RNA-ligases as well as RNA-chaperone activities.
The basal steps of the editing reaction cycle have been unraveled with the help of an in vitro RNA-editing assay (Seiwert and Stuart, 1994; Kable et al., 1996; Igo et al., 2000; Igo et al., 2002). The protocol represents a chemically simplified experimental system in which both, the substrate pre-edited mRNAs as well as the trans-acting gRNAs are mimicked by short RNA-oligonucleotides. Using purified editosome preparations, the pre-mRNA oligonucleotides are edited in a gRNA-dependent manner either by inserting 3 U-nt or deleting 4 U's. Importantly, in order to sidestep the rate-limiting endonucleolytic cleavage of the pre-mRNAs, the corresponding oligoribonucleotides are "pre-cleaved", i.e., they are provided as 5'- and 3'-pre-mRNA cleavage (Cl)-fragments. The 5'-Cl oligoribonucleotides are typically 5'-(32P)-labeled to enable a radioactive readout of the in vitro reaction.
Here we describe a new RNA-editing assay (Del Campo et al. 2020). The test system is a revised version of the established in vitro RNA-editing assays (Stuart et al., 1998), however, instead of using radioactively labeled substrate-RNAs it utilizes fluorophore-derivatized RNA-oligonucleotides. This allows the usage of automated, highly parallelized capillary electrophoresis (CE)-instruments coupled to laser-induced fluorescence (LIF) detection systems. It also enables multiplex-type fluorophore-labeling experiments to monitor multiple RNAs in one experiment. Furthermore, we designed new sets of pre-mRNA and gRNA-mimicking oligonucleotides. The different RNAs represent synthetic, non-natural sequences not found in the mitochondrial genomes of kinetoplastid organisms and they are, aside from the editing site, identical for both the U-insertion and the U-deletion reaction. This avoids potential sequence context issues and enables the side-by-side monitoring of both editing reactions in one reaction vial. Furthermore, we designed phosphorothioate (PS)-modified versions of the substrate RNAs. Phosphorothioate internucleotide linkages increase the RNase-stability of the pre-mRNA and gRNA-mimicking oligonucleotides, which permits the usage of editosome-enriched mitochondrial protein extracts instead of highly purified editosome preparations. Lastly, we optimized the work flow of the assay by adjusting the material quantities and reaction volume to multiwell-plate formats. The assay satisfies all signal-to-noise criteria for high-throughput screening methods (Del Campo et al., 2020) and is able to derive quantitative data for every step of the catalytic reaction cycle. As such the protocol should be helpful in the search for RNA-editing specific inhibitory compounds. The general workflow of the Fluorescence-based Insertion/Deletion Editing (FIDE)-assay is summarized in Figure 1.
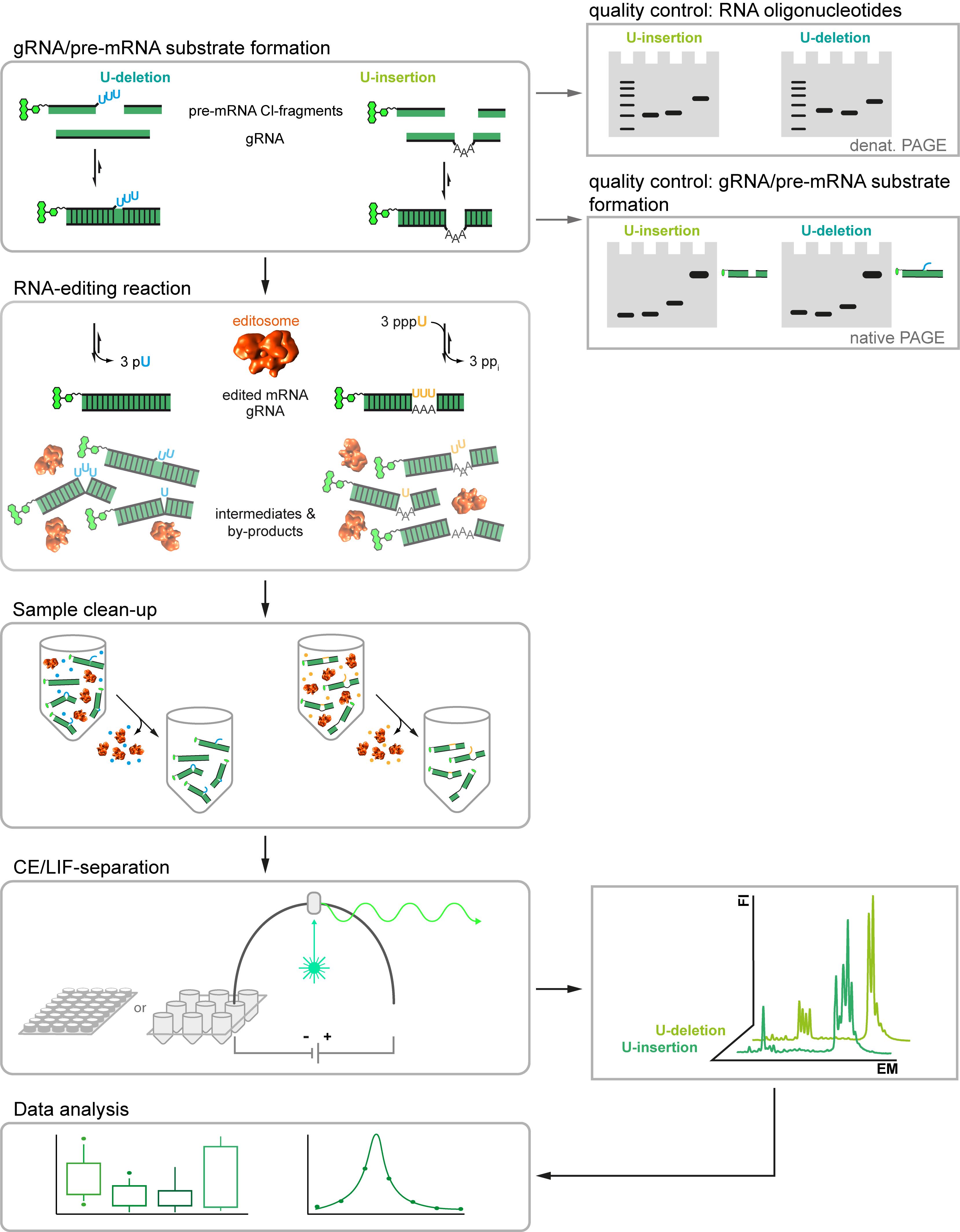
Figure 1. Workflow of the Fluorescence-based Insertion/Deletion Editing = FIDE-assay. Central steps of the protocol are on the left and boxed. Quality assessment steps and machine read-outs are illustrated on the right.
Materials and Reagents
All reagents, consumables and equipment are to be viewed as possible options. Unless stated otherwise, chemicals and consumables are stored at room temperature.
Solvents and Reagents
MeOH ≥99% p.a. (CARL ROTH, catalog number: 8388 )
EtOH ROTIPURAN® ≥99.8%, p.a. (CARL ROTH, catalog number: 9065 )
CHCl3 ≥99%, p.a (CARL ROTH, catalog number: 3313 )
Roti®-Phenol (TE-buffer equilibrated, pH 7.5-8.0) (CARL ROTH, catalog number: 0038 , store at 4 °C)
Isoamyl alcohol (3-Methyl-1-butanol), p.a. (Merck, catalog number: 100979 )
Glycerol ≥99.7%, p.a. (CARL ROTH, catalog number: 6962 , store at 4 °C)
Bradford assay reagent – detergent compatible (ThermoFisher Scientific, catalog number: 23246S , store at 4°C)
Phenol/Chloroform/Isoamylalcohol (PCI) (see Recipes, store at 4 °C)
Acids and Bases
Buffer Compounds and Salts
Tris-(hydroxymethyl)-amino methane (TRIS) PUFFERAN® (CARL ROTH, catalog number: 4855 )
N-2-Hydroxyethylpiperazine-N'-2-ethane sulphonic acid (HEPES) PUFFERAN® ≥99.5%, p.a. (CARL ROTH, catalog number: 9105 )
Imidazole PUFFERAN® ≥99%, p.a. (CARL ROTH, catalog number: X998 )
KCl (CARL ROTH, catalog number: 6781 )
MgCl2·6H2O (CARL ROTH, catalog number: 2189 )
NaCl (CARL ROTH, catalog number: 3957 )
Reducing and Chelating reagents
1,4-Dithiothreitol (DTT) ≥99%, p.a. (CARL ROTH, catalog number: 6908 , store at 4 °C)
Tris (2-carboxyethyl)phosphine hydrochloride (TCEP) ≥98% (Sigma-Aldrich, catalog number: C4706 , store at 4 °C)
Na2-ethylenediaminetetraacetate (Na2EDTA)·2H2O (CARL ROTH, catalog number: 8043 )
Ethylene glycol bis (β-aminoethylether) tetraacetic acid (EGTA) ≥99%, p.a. (CARL ROTH, catalog number: 3054 )
Ribonucleotides
Gel Electrophoresis Consumables
Acrylamide/Bisacrylamide (19:1) Rotiphorese®Gel 40 (CARL ROTH, catalog number: 3030 , store at 4 °C)
Urea (CARL ROTH, catalog number: 2317 )
Ammonium peroxydisulphate (CARL ROTH, catalog number: 9592 )
N,N,N',N'-Tetramethylethylendiamine (CARL ROTH, catalog number: 2367 )
Bromophenol Blue, Na+-salt (Merck, catalog number: 108122 )
Xylene cyanol FF, Na+-salt (Sigma-Aldrich, catalog number: X4126 )
Toluidine Blue O (Sigma-Aldrich, catalog number: T3260 )
6× Native PAGE gel-loading buffer (see Recipes)
2× Denaturing PAGE gel-loading buffer (see Recipes)
TBE buffer, pH 8.3 (see Recipes)
Gel staining solution (see Recipes)
Gel destaining solution (see Recipes)
Capillary Electrophoresis Consumables (see Note 1)
Glass capillary (Polymicro Technologies, catalog number: 1068150017 )
Performance optimized Polymer 6 (POP-6TM) (ThermoFisher Scientific, catalog number: 402837 , store at 4 °C)
10× CE/LIF-buffer (Sigma-Aldrich, catalog number: B4930 , store at 4 °C)
H2O LiChrosolv® (Merck Millipore, catalog number: 115333 )
Highly deionized formamide (Hi-DiTM Formamide) (ThermoFisher Scientific, catalog number: 4311320, store at -20 °C )
Editosomes and RNA-editing reaction mix
Affinity-purified editosomes in 10 mM HEPES/KOH, pH 7.5, 1 mM Mg(OAc)2, 75 mM NaCl, 0.1% (w/v) NP-40, 0.1 mM TCEP, 1 mM imidazole and 10 mM EGTA, 20% (v/v) glycerol (see Note 2, store at -20 °C)
Alternatively for HTS-applications: Editosome-enriched Trypanosoma brucei mitochondrial protein extracts in 20 mM HEPES/KOH, pH 7.5, 10 mM Mg(OAc)2, 30 mM KCl, 0.5 mM DTT, 20% (v/v) glycerol (see Note 2, store at -20 °C)
3x RNA-editing reaction mix (see Recipes, store at -20 °C)
Oligoribonucleotides (see Notes 3 and 4)
Standard-FIDE assay:
U-insertion
5’-FAM-synCl14ins: FAM-(CH2)6-AAAGGAAAUAUAGU
3'-synCl16: pAGGUGAUUCCAUUGAG-(CH2)6-NH2
syn-gRNAins: CUCAAUGGAAUCACCUAAAACUAUAUUUCCUUU
U-deletion
5'-FAM-synCl17del: FAM-(CH2)6-AAAGGAAAUAUAGUUUU
3'-synCl16: pAGGUGAUUCCAUUGAG-(CH2)6-NH2
syn-gRNAdel: CUCAAUGGAAUCACCUACUAUAUUUCCUUU
Standard-FIDE assay – phosphorothiate (PS)-modified (*) RNAs:
U-insertion
5’-FAM_modCl14ins: FAM-(CH2)6-AAAGGAAAU*A*U*A*GU
3’-modCl16: pAG*G*U*GAUUCCAUUGAG-(CH2)6-NH2
modgRNAins: C*U*C*AAUGGAAUCACCUAAAACUAUAUUUCC*U*U*U
U-deletion
5’-FAM_modCl17del: FAM-(CH2)6-AAAGGAAAU*A*U*A*GUUUU
3’-modCl16: pAG*G*U*GAUUCCAUUGAG-(CH2)6-NH2
modgRNAdel: C*U*C*AAUGGAAUCACCUACUAUAUUUCC*U*U*U
Multiplex ("one-pot") U-insertion/U-deletion assay:
U-insertion
5'-TAMRA_Cl18: TAMRA-(CH2)6-GGAAGUAUGAGACGUAGG
3'-Cl13: pAUUGGAGUUAUAG-(CH2)6-NH2
gRNAins: CUAUAACUCCGAUAAACCUACGUCUCAUACUUCC
U-deletion
5’-FAM_Cl22: FAM-(CH2)6-GGAAAGGGAAAGUUGUGAUUUU
3'-Cl15: pGCGAGUUAUAGAAUA-(CH2)6-NH2
gRNAdel: GGUUCUAUAACUCGCUCACAACUUUCCCUUUCC
Dual fluorophore assay:
U-insertion
5'-TAMRA_Cl18: TAMRA-(CH2)6-GGAAGUAUGAGACGUAGG
3'-Cl13_FAM: pAUUGGAGUUAUAG-(CH2)6-FAM
gRNAins: CUAUAACUCCGAUAAACCUACGUCUCAUACUUCC
U-deletion
5’-FAM_Cl22: FAM-(CH2)6-GGAAAGGGAAAGUUGUGAUUUU
3’-Cl15_TAMRA: pGCGAGUUAUAGAAUA-(CH2)6-TAMRA
gRNAdel: GGUUCUAUAACUCGCUCACAACUUUCCCUUUCC
RNA-size standards:
3'-FAM_13: AUUGGAGUUAUAG-(CH2)6-FAM
5'-FAM_22: FAM-(CH2)6-ggaaagggaaaguugugauuuu
5'-FAM_37: FAM-(CH2)6-ggaaagggaaaguugugauuuugcgaguuauagaaua
5’-FAM_44: FAM-(CH2)6-CUAGUACUCUCAUCAACAUAAGUCUCAUACUUCCGACAUGCA-CG
Reaction vials:
Equipment
Spectrophotometer (Thermo Scientific, model: NanoDrop 2000C )
Thermocycler (Biometra, T3 )
CE/LIF-based DNA-sequencer (Applied Biosystems, model: ABI PRISM 310 Genetic analyzer )
Benchtop Centrifuge (Eppendorf, model: 5417 R )
SpeedVac Vacuum Concentrator (Savant Instruments, model: SVC-100 )
Dry-Block Heating System (Grant, model: QBD2 )
Vortex Mixer (Scientific Instruments, model: Vortex Genie 2 )
DC-High Voltage Power Supply (Biometra, model: P30 )
Gel-Electrophoresis Chamber (BIO-RAD, model: Mini-PROTEAN® Tetra Cell)
Micropipettes (Gilson, models: Pipetman P10, P20, P200, P1000 )
Gel Documentation System (INTAS, model: Gel iX20 Imager )
Software
R (The R Project for Statistical Computing, https://www.r-project.org/)
Programmer-type text editor (such as Vim, https://www.vim.org or Atom, https://atom.io)
Procedure
gRNA/pre-mRNA substrate formation
Two thin-walled 0.5 ml polypropylene reaction tubes are used to separately assemble the trimeric U-insertion and U-deletion gRNA/pre-mRNA substrate RNAs. The final gRNA/pre-mRNA concentration is 10 nM.
Combine the 3'-pre-mRNA Cl-fragment, the fluorescently labeled 5'-pre-mRNA Cl-fragment and the corresponding gRNA oligoribonucleotide at a final concentration of 30 nM each in TE buffer (pH 7.5). You will need 10 µl of gRNA/pre-mRNA substrate for a 30 µl editing reaction.
Mix by vortexing (5 s) and collect the content of the tubes by brief centrifugation (10 s quick-spin).
Thermally unfold the oligoribonucleotides for 2 min at 75 °C and anneal the 3 RNAs by cooling to 27 °C with a rate of 0.08 °C/s.
Note: The formation of the gRNA/pre-mRNA hybrid must be verified in a separate analysis (see Note 5). The yield of the trimeric RNA-hybrid should be ≥90%.
RNA-editing reaction and sample clean-up
Add an equal volume of 3× editing reaction-mix to the annealed gRNA/pre-mRNA substrate from Step 1c.
Aliquot 20 µl of this pre-mix into thin-walled 0.2 ml polypropylene reaction tubes.
Adjust the volume with ddH2O, so that the final volume, including the volume of editosomes (to be added later), will be 30 µl.
Mix by vortexing (5 s) and collect the content of the tubes by brief centrifugation (10 s quick-spin).
Add editosomes and mix by gently pipetting up and down (see Note 6).
Incubate at 27 °C for 30 min to 3 h (see Note 6).
Transfer samples to 1.5 ml reaction tubes and terminate the reaction by adding 17 µl ddH2O, 3 µl 125 mM Na2EDTA and 50 µl PCI. Mix vigorously.
Spin for 5 min at >15,000 × g at 4 °C.
Transfer the H2O-phase to a new 1.5 ml reaction tube. Add 5 µl 3 M NaOAc/HOAc (pH 4.8) and 121 µl 100% (v/v) cold (-20 °C) EtOH. Mix and immediately spin at 4 °C for 30 min at >15,000 × g to pellet the RNA.
Aspirate the supernatant and add 0.75 ml 70% (v/v) cold (-20 °C) EtOH.
Wash RNA-pellets by vortexing and spin for 5 min at 4 °C at >15,000 × g.
Repeat Steps 2j-2k.
Remove any liquid by aspiration and dry RNA-pellets for 2 min in vacuo.
Dissolve RNA-pellets in 15 µl Hi-DiTM formamide. Collect the contents of the tube by brief centrifugation (10 s quick-spin).
Denature the RNA-samples by incubation at 95 °C for 2 min. Snap-cool on ice.
CE/LIF-separation
Transfer 3 µl of the samples to 0.5 ml reaction tubes (see Note 13).
Add 4 fmol of each of the 4 size-standard RNA oligonucleotides.
Add Hi-DiTM formamide to a final volume of 20 µl. Mix and spin down.
Cut off the lids and place the tubes into the autosampler of the CE/LIF-instrument.
Run capillary electrophoresis
On an CE/LIF-ABI PRISM® 310 DNA-Sequencer with a 50 cm glass capillary use the following settings: ModuleSeq POP6 (1 ml); laser power: 7 mW; injection voltage: 2.5 kV; run voltage: 12.2 kV; sample injection time: 10 s (see Note 7); run time: 25 min.
After completion of the CE/LIF-run, inspect the raw electropherograms. Since only non-saturated CE/LIF-traces can be quantitatively analyzed it may be necessary to re-run the samples at a higher dilution.
Data analysis
Output of the CE/LIF-separation is an unprocessed electropherogram, which is retrieved as a binary data file. For extracting the electropherogram data points and for downstream processing, we use the programming language R. R is a free software for statistical computing and graphics and it compiles and runs on UNIX, macOS and Windows operating systems.
Open R.
Commands can directly be typed into the R-console. By pressing ENTER/RETURN, the command will be executed. However, for more elaborate procedures, it is more convenient to type all commands into a text editor or the in-built editor of R (see Note 8) followed by copying the code to the R-console.
OS X: File → New document
Windows: File → New script
Install Bioconductor and the R packages “pracma” and “sangerseqR”.
Bioconductor provides the package “sangerseqR”, which is used to extract the fluorescence signal intensities (FI) in relative fluorescence units (RFU) from the binary sequencing files. The package “pracma” provides the functions “peakfind”. “pracma” is not included in the basic R-package collection.
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(version = "3.11")
install.packages("pracma")
install.packages("sangerseqR")
Create a folder for your FIDE-experiment as a working directory.
Move the CE/LIF-binary data files to this folder.
Set your experiment folder as working directory for the current R-session. All files generated during the active session will then be saved there.
setwd("/folder1/folder2/…/myExperimentFolder")
Upload all packages required for the downstream analysis.
library("pracma")
library("sangerseqR")
The binary CE/LIF-files are uploaded to the R-workspace using the function “read.abif” from the “sangerseqR” package provided within Bioconductor. They are converted to an abif class object, from which the CE/LIF-raw data (i.e., relative fluorescence units (RFU)) are extracted.
datafile <- read.abif("myAbiFile")ceData <- data.frame(datafile@data$DATA.1, datafile@data$DATA.2,
datafile@data$DATA.3, datafile@data$DATA.4)
Analyze and process the CE/LIF-raw data.
Visualize the electropherogram.
plot(ceData[,1], type = "l", col = "blue", xlab = "EM", ylab = "FI/RFU", main = "raw data")lines(ceData[,2], col = "green")
lines(ceData[,3], col = "orange")
lines(ceData[,4], col = "darkred")
legend("topleft", c("1", "2", "3", "4"), fill=c("blue", "green", "orange", "darkred"))
Select the appropriate trace and pass the channel number to the R-environment.
trace <- traceNumberForAnalysis (Note: The argument is a true integer)Perform baseline correction (see Note 9).
dataBg <- ceData[,trace] - median(ceData [500:2000,trace])
Identify peaks (see Note 10).
peaks <- data.frame(findpeaks(dataBg, threshold = 50, minpeakheight = 100, zero = "-"))colnames(peaks) <-c("height", "max", "start", "end")
Integrate peak areas.
area <-c()for(i in 1 : nrow(peaks)) {
start <- peaks [i,3]
stop <- peaks [i,4]-1
sum <- sum(dataBg [start:stop])
area <-round(c(area,sum),0)
}
results <-data.frame(peaks, area)
View the results in the R-console.
resultsWrite results to file.
myOutName <- "myNameForResults"write.table(data.frame(num=rownames(results), results), file = paste (myOutName, ".txt", sep = ""), append = F, sep = "\t", dec = ".", row.names = F, col.names = T, quote = F)
Visualize the peaks in the CE-profile (Figure 2).
plot(peaks[,2],peaks[,1],type="h",col="red", xlim = c(1000, 1700),ylim = c(0,max(dataBg)+200), xlab = "EM", ylab="FI/RFU")
text(peaks[,2],peaks[,1]+150, rownames(peaks), cex = 1)
lines(dataBg,type="l",col="black")
Save plot to file.
pdf(file = paste(myOutName, ".pdf", sep=""))plot(peaks[,2],peaks[,1],type="h",col="red", xlim = c(1000, 1700),
ylim = c(0,max(dataBg)+200), xlab = "EM", ylab="FI/RFU")
text(peaks[,2],peaks[,1]+150, rownames(peaks), cex = 1)
lines(dataBg,type="l",col="black")
dev.off()
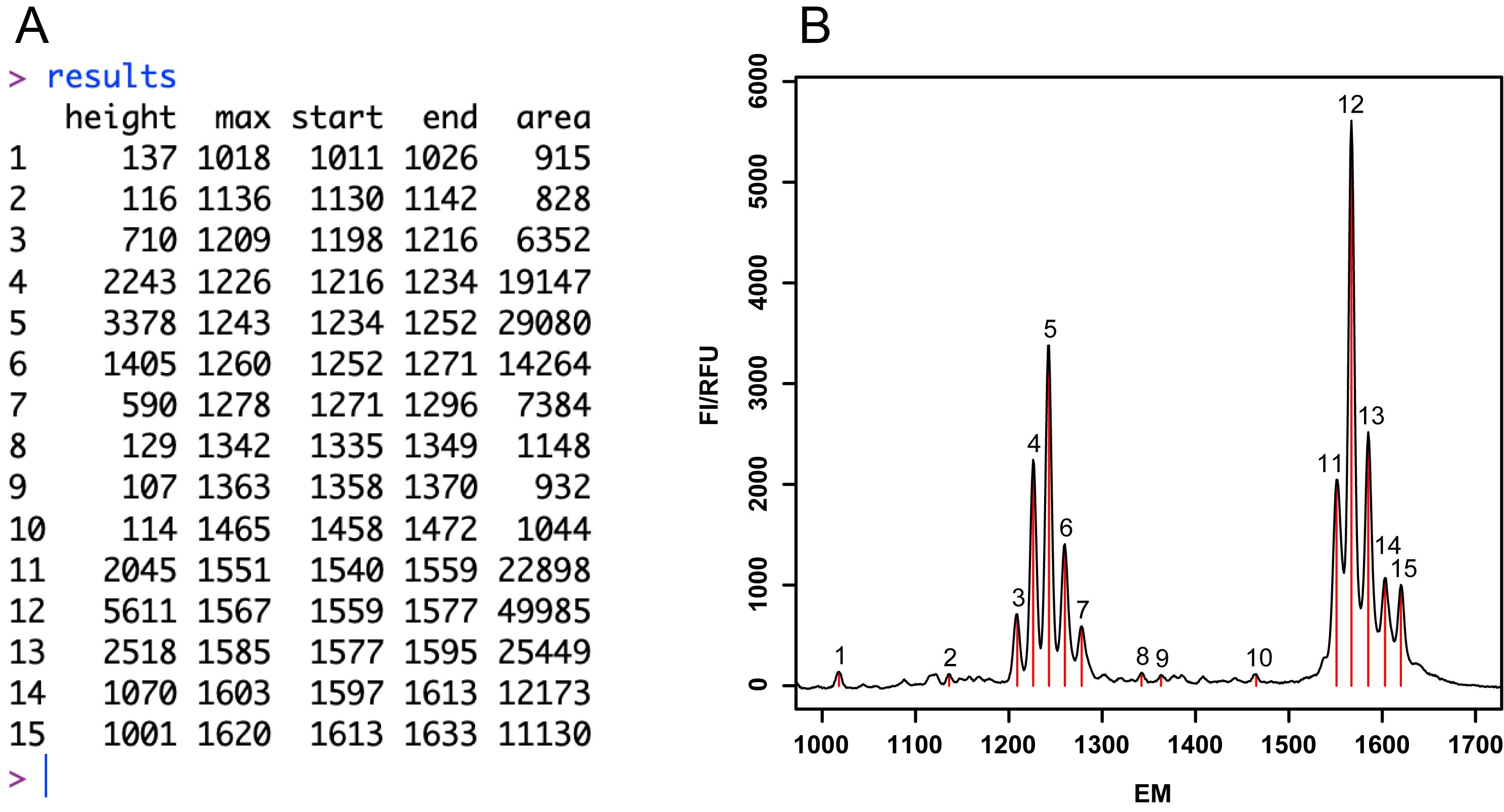
Figure 2. R-based data analysis output. A. Table representation of all peaks of the processed data specifying height, maximum (max), start, end and peak areas. B. Plot of the electropherogram (FI=f(EM)). FI=Fluorescence Intensity; RFU=Relative Fluorescence Unit; EM=Electrophoretic Migration (see Note 11).Peak assignment.
The assignment of the different peaks of the electropherogram is performed with the help of fluorophore-modified size-standard oligoribonucleotides (see Materials and Reagents, subitem I. Oligoribonucleotides). We recommend to use four FAM-derivatized RNA-oligonucleotides with a nucleotide-length of 13 nt, 22 nt, 37 nt and 44 nt. Small amounts (4 fmol each) of the four RNAs can be spiked into the samples and are used to extrapolate the nt-length of every RNA-species. As shown in Figure 3A, the electrophoretic migration of marker oligoribonucleotides is linear over a wide separation range. Figure 3B shows a typical example of a "spiked-in" FIDE U-deletion assay and Figure 3C shows representative FIDE-results for both, the U-insertion and the U-deletion assay in which the identity and nt-length of every RNA-species is specified.
Calculate editing activity.
Several possibilities exist to define and calculate the RNA-editing activity in order to compare different samples (see Note 12):
Ratio of the peak area of the fully edited RNA-product over the sum of all assigned peaks.
Peak area ratio of all ligated RNA-products over all assigned peaks.
Peak area ratio of all processed RNA-species compared to all assigned peaks.
Peak area ratio of all processed RNA-species compared to the area of the RNA-input peak.
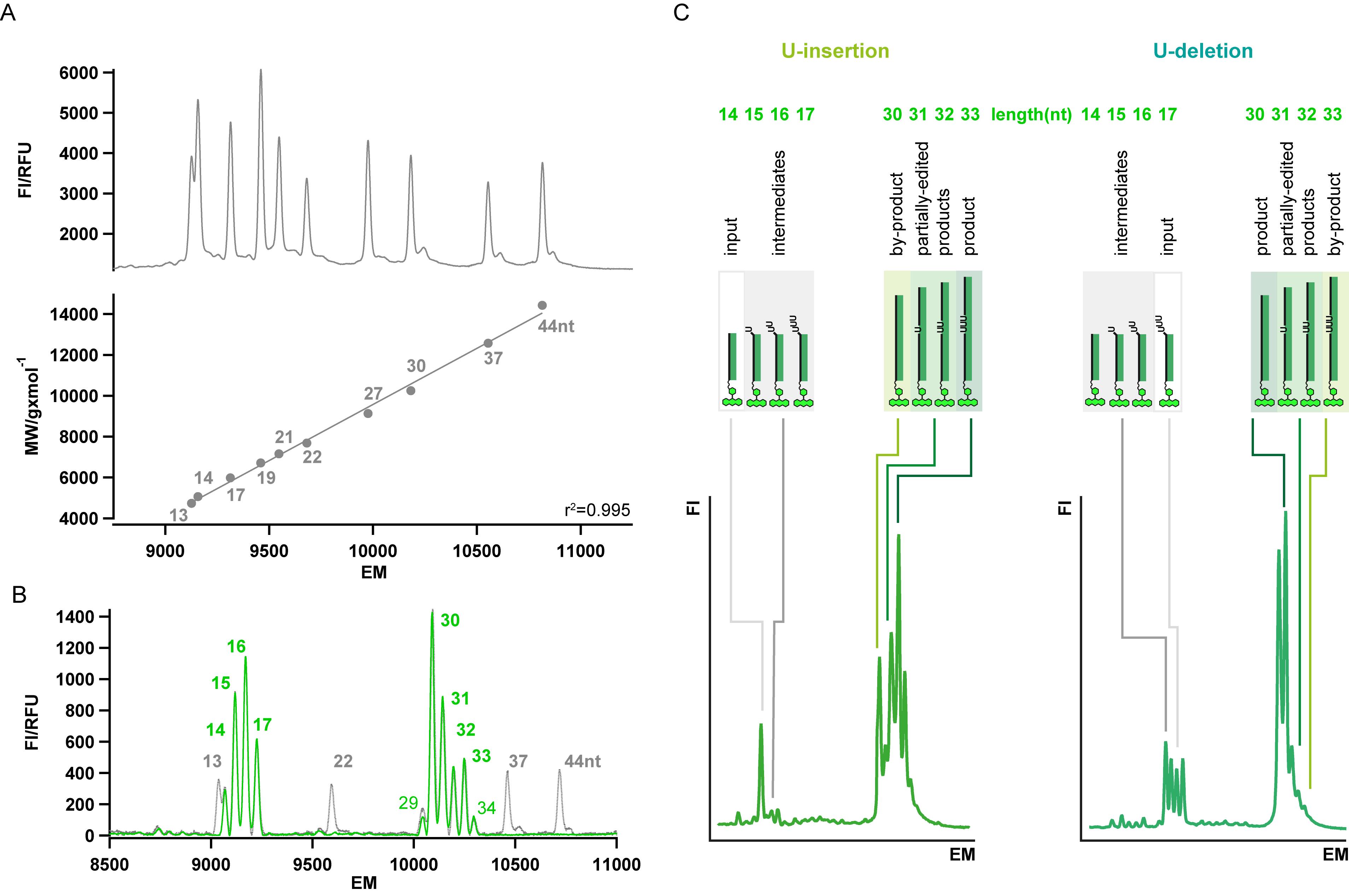
Figure 3. Representative FIDE-results. A. Top: Electropherogram of a CE/LIF-separation of 10 size-standard oligoribonucleotides ranging from 13 nt to 44 nt. Bottom: Plot of the molecular weight (MW) of the marker RNA-oligonucleotides in relation to their electrophoretic mobility (EM). A linear fit of the data points results in an R-squared (r2) of 0.995. B. Representative electropherogram of a FIDE U-deletion assay (green trace) with "spiked-in" marker RNA-oligonucleotides 13 nt, 22 nt, 37 nt and 44 nt in length (black trace). C. Typical electropherograms of FIDE U-insertion (left) and U-deletion (right) assays. The individual peaks are assigned as: RNA-input, reaction intermediates, partially edited RNAs, fully edited RNA-product and by-products. Numbers specify the nt-length of the different RNA-species. FI=Fluorescence Intensity. RFU = Relative Fluorescence Unit.
Modifications to the standard FIDE-protocol
High-throughput FIDE
For high-throughput screening (HTS)-applications FIDE can be downscaled to adapt the assay to multiwell plates. Lower limit for both editing reactions are roughly 5-10 fmol of each, editosomes and fluorophore-modified pre-mRNA/gRNA-hybrid in a reaction volume ≤5 µl. Because of the small amounts of protein and RNA in the samples, reactions can be stopped by directly adding Hi-DiTM formamide. This conveniently eliminates the phenol extraction and EtOH-precipitation steps prior to the CE/LIF-separation. Furthermore, FIDE can be performed with only partially purified mitochondrial extracts as editosome source. Due to the presence of RNases this requires the usage of phosphorothioate (PS)-modified and thus RNase-resistant substrate oligoribonucleotides as specified in the Materials and Reagents section.
"One-pot" U-insertion and U-deletion editing – "matrixing"
A notable advantage of FIDE is the possibility to monitor the U-insertion and U-deletion reaction simultaneously. This can simply be done by using different fluorophores for the two substrate RNAs (see Materials and Reagents, I. Oligoribonucleotides). However, since the emission spectra of fluorescent dyes usually spread into multiple channels of the CE/LIF-detector an additional data processing-step is required. This procedure is known as "matrixing" (Giddings et al., 1993; Giddings et al., 1998). For that the two fluorescently labeled oligoribonucleotides must be analyzed in separate, i.e., singleplex CE/LIF-runs (see Note 13). After baseline correction of the raw data, the fluorescence intensity (FI) of both fluorophores in every detector channel (i-n) is calculated as: FIchannel (i)/ΣFIchannel (i-n). The matrix can conveniently be created from the binary CE/LIF-files using the R-script “matrix.fun” provided in the Supplementary Material section. The function is loaded into the workspace using the command:
source("pathToScript/matrix.fun.R")
The mandatory argument is a character vector with the names of the singleplex CE/LIF-files:
myMatrix <- matrix.fun(c("myFileDye1", "myFileDye2"))The matrix is stored as a tab-delimited text and can be used for all multiplex FIDE-assays run on the same CE/LIF-device.
Data analysis for multiplex FIDE-assays
Matrixing
Read the binary CE/LIF-files to R and extract the raw data as described in: Data analysis, 1-7.
Upload the matrix file to the workspace, if the matrix has been created in another R-environment.
myMatrix <- data.matrix(read.delim("pathAndNameOfMatrixFile", header=FALSE))Perform baseline correction.
for (i in 1:ncol(ceData)) {ceData [,i] <- ceData[,i] - median(ceData[500:2000,i])
}
Matrixing and correction for negative values.
for (i in 1:nrow(ceData)) {
ceData [i,] = data.matrix(ceData)[i,]%*%t(solve(myMatrix))
}
ceData <- replace(ceData, ceData < 0, 0)
Peak detection and quantification
U-deletion and U-insertion assays are analyzed separately from the corresponding traces as described for the singleplex FIDE/CE-assay (see Data analysis 8, d-i) after passing the trace number to R.
trace <- traceNumberForAnalysis Dye channel number for U-deletion or U-insertion assay, respectively. (Note: The argument is a true integer).
dataBg <- ceData[,trace]
Continue as described in: Data analysis, 8, d-i.
Dual-fluorophore FIDE
By using a 5'-fluorophore-derivatized 5'-mRNA oligonucleotide and at the same time a 3'-fluorophore labelled 3'-mRNA oligonucleotide, it is possible to monitor the reaction pathway of both pre-mRNA-fragments in one experiment. This can be done for the U-insertion and the U-deletion reaction alike and the corresponding oligoribonucleotides are listed in the Materials and Reagents section. In this case the sequences of the different RNA-oligonucleotides are derived from the pre-mRNA encoding subunit 6 of the mitochondrial ATP synthase (A6) from Trypanosoma brucei. Similarly, the reaction pathway of the gRNA can be traced by using a 5'-fluorophore-modified gRNA-oligoribonucleotide.
Notes
For the CE/LIF-instrument we exclusively use reagents of the highest chemical grade. For instance, we use LiChrosolv®-H2O for cleaning purposes and for diluting the CE/LIF-buffer. Highly deionized formamide (Hi-DiTM formamide) is used for sample preparations.
Editosomes cannot be purchased commercially. They need to be purified from insect-stage African trypanosomes (Trypanosoma brucei) grown in axenic cell culture (Brun and Schönenberger, 1979). The complexes are either purified by tandem-affinity purification (TAP) using transgenic T. brucei cell lines that conditionally express TAP-tagged variants of editosomal proteins (Golas et al., 2009; Gerace and Moazed, 2015) or from mitochondrial detergent extracts of wildtype trypanosomes (Böhm et al., 2012). Typically, 5 L of mid-log phase T. brucei cell culture (5 × 1010-8 × 1010 cells) yield about 50 µg of TAP-purified editosomes. The same number of cells yield about 0.5 mg of editing-active mitochondrial protein extract. Protein concentrations are determined by Coomassie Blue G-250 binding (Bradford assay [Bradford, 1976]).
RNA-oligonucleotides are synthesized by solid-phase chemical synthesis on controlled pore glass (CPG)-beads using 2'-O-(tert-butyl)dimethylsilyl (TBDMS)-protected phosphoramidites. A 200 nmol synthesis scale typically yields ≥200 µg of pure oligoribonucleotide. RNA-oligonucleotides should be high-performance liquid chromatography (HPLC)-purified either by reversed phase or anion-exchange HPLC and verified by mass-spectrometry. We further recommend to scrutinize the purity of the synthesized RNAs in 8 M urea-containing polyacrylamide gels. Fluorophore modifications are introduced at the 5′- or 3’-termini of the different oligoribonucleotides. This can be done co- or post-synthetically. Internal fluorophore labelling strategies should be avoided to uphold the base pairing capacity of the RNA-molecules. We also recommend the introduction of linker sequences such as hexamethylene linkers between the fluorophore and the RNA to minimize sterical clashes with the nucleobases. Numerous fluorophores can be used as substituents. The specific choice depends on the wavelength specification of the laser-induced fluorescence (LIF)-detection system. Examples include 5-Carboxytetramethyl-rhodamine (TAMRA), 6-Hexachloro-fluorescein (HEX), 6-Carboxy-fluorescein (FAM), the Sulfo-cyanine dyes Cy5 and Cy5.5 as well as the Infrared fluorescent dye IRDye800 and WellRedD1. Additional modifications can be introduced such as phosphorothioate backbone modifications to increase the RNase-stability of the different RNAs. Exonucleolytic degradations from the 3'-end can be counteracted by introducing a 3'-terminal hexamethylene-amino-linker. Oligoribonucleotides representing 3'-pre-mRNA fragments require the introduction of a 5'-phosphate group using T4-polynucleotide kinase (T4-PNK) and ATP.
RNA-concentration are calculated from UV-absorbency measurements at a wavelength of 260 nm (A260). Molar extinction coefficients (ϵ) for the different RNAs can be derived from nearest neighbor calculations using the OligoAnalyzer® tool at: www.idtna.com/scitools. Measurements can be performed with the help of NanoDrop-type UV-spectrophotometer instruments. Oligoribonucleotide concentrations ≥0.2 µg/µl and ≤2.5 µg/µl are well within the dynamic linear range of the instruments generating reliable measurements without the need for more sensitive, reporter fluorophore-based methods. Oligoribonucleotides are stored in TE buffer (pH 7.5) (see Recipes) at -20 °C at a concentration of 100 µM.
In a separate set-up, it is necessary to confirm and quantify the formation/yield of the trimeric gRNA/pre-mRNA hybrid RNAs. For that combine 0.4 nmol of the fluorescently labeled 5‘-pre-mRNA Cl-oligoribonucleotides, the 3’-pre-mRNA Cl-oligoribonucleotides and the corresponding gRNAs. Thermally unfold and anneal the samples as described above (Procedure 1c). Add 6× Native PAGE gel-loading buffer containing 2 mM MgCl2 and separate the samples in 18% (w/v) polyacrylamide gels (gel dimensions: 7.2 cm × 8.3 cm × 0.1 cm). Run gels in TBE (pH 8.3), 2 mM MgCl2 at 4 °C and 8 W (constant) until the bromophenol blue marker has reached the bottom of the gel. As controls load all 3 oligoribonucleotides individually and optionally all bimolecular combinations of the RNA-oligonucleotides. Stain/destain the gels (recipes 2 and 3) and determine the degree of gRNA/pre-mRNA hybrid formation by densitometry. The majority of stained material (≥90%) should appear in the band with the lowest electrophoretic mobility, which represents the trimeric gRNA/pre-mRNA hybrid. Monomeric oligoribonucleotides should be negligible. A typical result is shown in Figure 4.
The required amount of editosomes and the incubation time will depend on the specific editing activity (EA) (EA/protein mass) of the editosome preparation. In our hands, 50 ng of TAP-purified editosomes typically yield ≥20% of fully edited product within 60 min in the FIDE U-deletion assay.
Prolonged injection times can result in peak anomalies/artifacts (Johansson et al., 2003).
Use a code/programmer-type text editor and be aware that R is case-sensitive.
The data region used for background correction is adapted to the CE/LIF-ABI PRISM® 310 DNA-Sequencer with a 50 cm glass capillary and may be adjusted for other CE/LIF-instruments.
The settings for "threshold" and "minpeakheight" will influence the sensitivity of peak detection.
The commands can be combined to enable the processing of multiple files in batch. A script is provided upon request from the authors.
For methods I-III, the ratios correlate linearly with the editing activity, if less than 50% of substrate is processed. Method IV displays a higher dynamic range, but there is no linear correlation to the editing activity. Methods III and IV do not account for ligation activity, i.e., editosome preparation lacking ligase activity will be considered as active.
It is of the essence that the peaks of the CE/LIF-electropherogram are below saturation. This can necessitate a rerun of the samples at higher dilution.
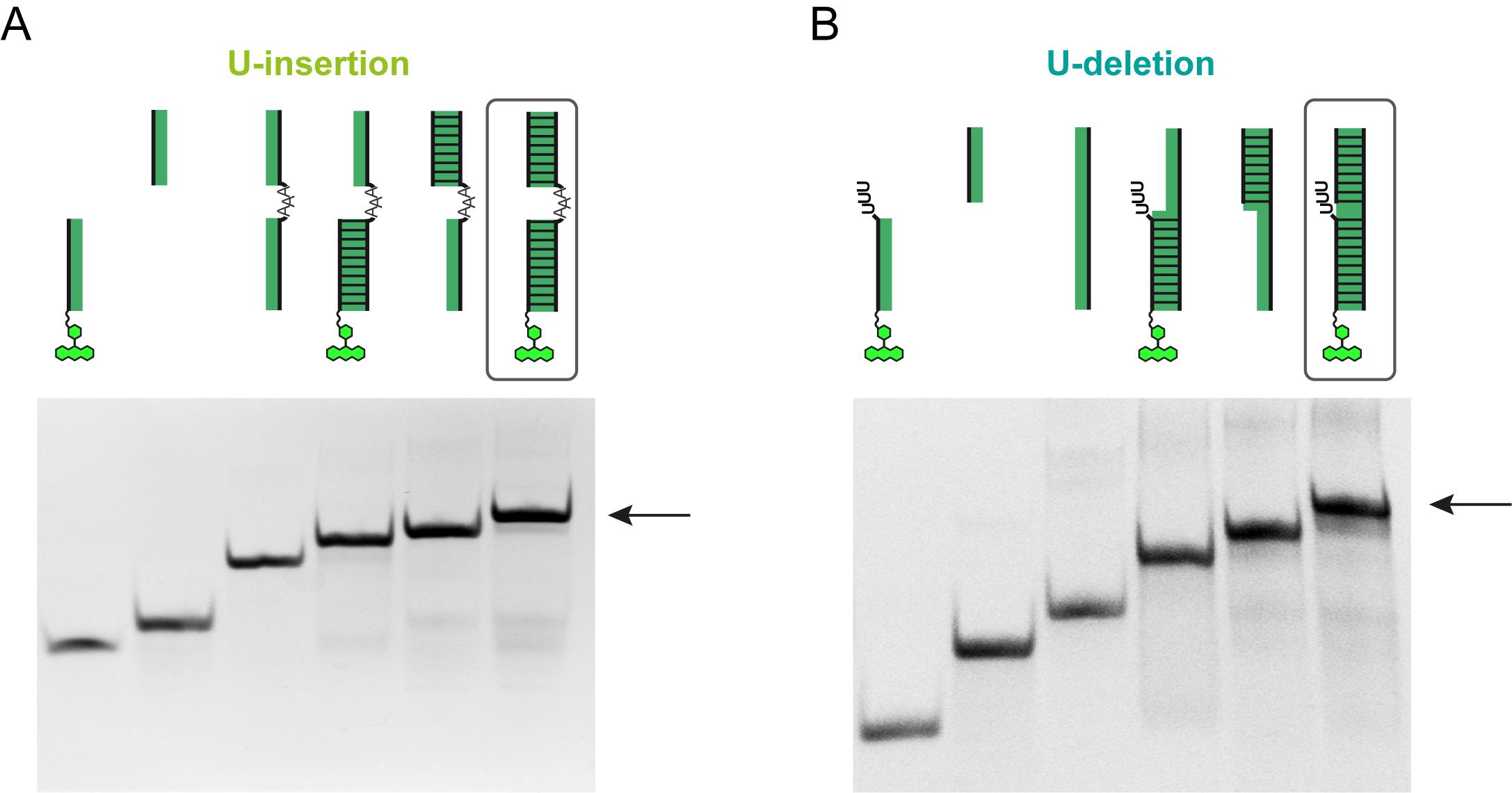
Figure 4. Guide RNA/pre-mRNA substrate formation. Gel electrophoretic verification of the formation of gRNA/pre-mRNA hybrid RNAs for the U-insertion reaction (A) and the U-deletion reaction (B). The two PAA-gels show from left to right the fluorophore-labeled 5’-pre-mRNA cleavage fragment (5'-Cl), the 3’-pre-mRNA cleavage fragment (3'-Cl), the corresponding gRNA-oligoribonucleotide next to the two bimolecular complexes (5'-Cl/gRNA; 3'-Cl/gRNA) and the final trimolecular (5'-Cl/3'-Cl/gRNA) editing substrate (boxed, arrow). The yield of the trimeric RNA should exceed 90%. Fluorophore-subtituents are shown as chemical ring systems.
Recipes
TBE buffer, pH 8.3
89 mM Tris-(hydroxymethyl)-amino methane
89 mM B(OH)3
2 mM Na2EDTA
Phenol/Chloroform/Isoamylacohol (PCI)
Phenol/CHCl3/Isoamylacohol (25/24/1) [v/v/v]
Gel staining solution
7.5% (v/v) HOAc in H2O
0.1% (w/v) Toluidine Blue O (C15H16ClN3S)
Gel destaining solution
10% (v/v) MeOH
1% (v/v) HOAc
TE buffer, pH 7.5
10 mM Tris/HCl, pH 7.5
1 mM Na2-EDTA
3× RNA-editing reaction mix
60 mM HEPES/KOH, pH 7.5
30 mM MgCl2
90 mM KCl
1.5 mM DTT
1.5 mM ATP
0.3 mM UTP
2× Denaturing PAGE gel-loading buffer
1× TBE buffer, pH 8.3
8 M Urea
0.01% (w/v) Bromophenol Blue (C19H10Br4O5S)
0.01% (w/v) Xylene cyanol FF, Na+-salt (C25H27N2NaO6S2)
6× Native PAGE gel-loading buffer
10 mM Tris/HCl, pH 7.6
0.03% (w/v) Bromophenol Blue, Na+-salt (C19H10Br4O5S)
0.03% (w/v) Xylene cyanol FF, Na+-salt (C25H27N2NaO6S2)
60 mM Na2EDTA
60% (v/v) Glycerol
Acknowledgments
This work was funded by the German Research Foundation (DFG-GO 516/8-1) and the Dr. Illing-Foundation for Molecular Chemistry. We thank Cristian Del Campo, Paul Reißig, Robert Knieß and Andreas Völker for experimental input. Del Campo et al. (2020) is the original paper from which this protocol was derived.
Competing interests
The authors have no competing interests to declare.
References
- Blum, B., Bakalara, N. and Simpson, L. (1990). A model for RNA editing in kinetoplastid mitochondria: "guide" RNA molecules transcribed from maxicircle DNA provide the edited information. Cell 60(2): 189-198.
- Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248-254.
- Brun, R. and Schönenberger, M. (1979). Cultivation and in vitro cloning or procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Acta Trop 36(3): 289-292.
- Böhm, C., Katari, V.S., Brecht, M. and Göringer, H.U. (2012). Trypanosoma brucei 20 S editosomes have one RNA substrate-binding site and execute RNA unwinding activity. J Biol Chem 287(31): 26268-26277.
- Del Campo, C., Leeder, W.M., Reißig, P. and Göringer, H.U. (2020). Analyzing editosome function in high- throughput. Nucleic Acids Res 48(17): e99.
- Gerace, E. and Moazed, D. (2015). Affinity purification of protein complexes using TAP tags. Methods Enzymol 559: 37-52.
- Giddings, M. C., Brumley Jr., R. L., Haker, M. and Smith, L. M. (1993). An adaptive, object oriented strategy for base calling in DNA sequence analysis. Nucleic Acids Res 21(19): 4530-4540.
- Giddings, M. C., Severin. J., Westphall, M., Wu, J. and Smith, L.M. (1998). A software system for data analysis in automated DNA sequencing. Genome Res 8(6): 644-665.
- Göringer, H. U. (2012a). RNA editing in African trypanosomes: A U-ser's G-U-ide. In: A. Bindereif (Ed.). RNA Metabolism in Trypanosomes. Nucleic Acids Mol Biol 28: 149-165.
- Göringer, H. U. (2012b). ‘Gestalt,’ composition and function of the Trypanosoma brucei editosome. Annu Rev Microbiol 66: 65-82.
- Golas, M. M., Böhm, C., Sander, B., Effenberger, K., Brecht, M., Stark, H. and Göringer, H. U. (2009). Snapshots of the RNA editing machine in trypanosomes captured at different assembly stages in vivo. EMBO J 28(6): 766-778.
- Hermann, T., Schmid, B., Heumann, H. and Göringer, H. U. (1997). A three-dimensional working model for a guide RNA from Trypanosoma brucei. Nucl Acids Res 25(12): 2311-2318.
- Igo, Jr. R. P., Palazzo, S. S., Burgess, M. L., Panigrahi, A. K. and Stuart, K. (2000). Uridylate addition and RNA ligation contribute to the specificity of kinetoplastid insertion RNA editing. Mol Cell Biol 20(22): 8447-8457.
- Igo. Jr. R. P., Lawson, S. D. and Stuart, K. (2002). RNA sequence and base pairing effects on insertion editing in Trypanosoma brucei. Mol Cell Biol 22(5): 1567-1576.
- Johansson, G., Isaksson, R. and Harang V. (2003). Migration time and peak area artifacts caused by systemic effects in voltage controlled capillary electrophoresis.J Chromatogr A 1004(1-2): 91-98.
- Kable, M. L., Seiwert, S. D., Heidmann, S. and Stuart, K. (1996). RNA editing: a mechanism for gRNA-specified uridylate insertion into precursor mRNA. Science 273(5279): 1189-1195.
- Seiwert, S. D. and Stuart, K. (1994). RNA editing: Transfer of genetic information from gRNA to precursor mRNA in vitro. Science 266(5182): 114-117.
- Stuart, K., Kable, M. L., Allen, T. E. and Lawson, S. (1998). Investigating the mechanism and machinery of RNA editing.Methods 15(1): 3-14.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Leeder, M., Kruse, E. and Göringer, H. U. (2021). Trypanosomatid, fluorescence-based in vitro U-insertion/U-deletion RNA-editing (FIDE). Bio-protocol 11(5): e3935. DOI: 10.21769/BioProtoc.3935.
Category
Molecular Biology > RNA
Systems Biology > Transcriptomics
Biochemistry > RNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.