- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Expression and Purification of Yeast-derived GPCR, Gα and Gβγ Subunits for Structural and Dynamic Studies
(*contributed equally to this work) Published: Vol 11, Iss 4, Feb 20, 2021 DOI: 10.21769/BioProtoc.3919 Views: 7326
Reviewed by: Sneha RayAbhinit NagarAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2016

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
In the last several years, as evidence of a surged number of GPCR-G complex structures, the expressions of GPCRs and G proteins for structural biology have achieved tremendous successes, mostly in insect and mammalian cell systems, resulting in more than 370 structures of over 70 GPCRs have been resolved. However, the challenge remains, particularly in the conformational transition and dynamics study area where a much higher quantity of the receptors and G proteins is required even in comparison to X-ray and cryo-EM (5 mg/ml, 3 μl/sample) when NMR spectroscopy (5 mg/ml, 250 μl /sample) is applied. As a result, the expression levels of the insect and mammalian systems are also difficult to meet this demand, not to mention the prohibitive cost of producing GPCRs and G proteins using these systems for a vast majority of laboratories. Therefore, exploration of an effective, affordable, and practical approach with broad applicability is demanded. Pichia pastoris expression system has shown its promise in the GPCR preparation with many merits that other eukaryotic expression systems can’t compete with. GPCRs expressed in this system are inexpensive, easy-to-manipulate, and capable of isotopically labeling. Herein, we present related protocols recently developed and upgraded in our lab, including expressions and purifications of P. pastoris derived GPCR along with Gα and Gβγ proteins. We anticipate that these protocols will advance the conformational transition and dynamics studies of the GPCR and its complexes.
Keywords: GPCRBackground
G protein-coupled receptor (GPCR), the biggest membrane protein family, plays critical roles in many (patho) physiological activities. Dysfunctions of GPCRs or their effectors will result in various pathologies, including neuro-degenerative diseases, cancers, and chronic inflammations (Overington et al., 2006). Although more than 350 structures (Congreve et al., 2020) of over 70 GPCRs have been resolved in complex with ligands and/or transducins, our understanding of how the signaling is propagated through the receptor upon ligand binding to downstream signaling partners is still elusive, in part due to the resolved structures merely reflect static snapshots of the dynamics signaling processes for these complexes. In addition, limitations of the structures themselves include missing parts of flexible domains and side-chain information of many residues that were not able to be resolved in both X-ray and cryo-EM spectroscopies. The development of complementary approaches to acquire the missing information would provide invaluable insights that broaden our understanding of the sequential signaling process of the GPCRs and guide us to the discovery of drugs that target specific malfunctional signaling.
Obviously, NMR is a superb tool to interrogate the conformational transition and dynamics of the GPCRs and their signaling partners with minimal structure-function perturbation beyond static structures. In the NMR study, a wild-type or minimally modified construct is often used, which can maximally reflect the genuine behaviors of the receptor when conducted in a mimic physiological environment such as detergent MNG-3, micelle, or high-density lipoprotein particle (HDL) systems after reconstitution. The specific requirements for the NMR study also propose the challenges for a regular expression system such as E. coli heterogenous expression system. Although it is the simplest and most efficient system, the expressed proteins (especially human-derived ones) lack post-translational modifications, which often results in the loss of functionality. Insect cells such as sf9, sf21, or Hi5, and mammalian expression systems such as HEK293 and CHO cells are increasingly used for structural biology especially for X-ray and cryo-EM spectroscopic studies where a relatively small quantity of the receptors is needed. However, for NMR study, the need for a large protein quantity along with isotopic labeling makes insect and mammalian systems challenging to be employed in this type of study. Therefore, we developed a yeast-based expression system to produce receptors and their transducins meet this requirement. In this protocol, we are particularly focused on a broadly applicable strategy of preparing GPCRs, Gα and Gβγ, in which the protocol for GPCR preparation is further upgraded based on our previous publications (Ye et al., 2016, 2018a and 2018b) with a significantly improved productivity. The Gα and Gβγ preparations have been incorporated into this set of protocols as a complete system for GPCR-G protein complex study. The strategy described in the manuscript can also be applied to other signaling partners such as GRKs and β-arrestins. It is also easy to switch to isotopically labeling if needed. The advantages of expressing these receptors and effectors in the P. pastoris system are obvious in comparison with other systems aforementioned.
Materials and Reagents
1.5 ml microcentrifuge tubes (Fisher Scientific, catalog number: 05-408-130 )
2 mm electroporation cuvette (Fisher Scientific, catalog number: FB102 )
Nitrocellulose membrane (Bio-Rad, catalog number: 1620112 )
NalgeneTM Rapid-FlowTM Sterilize Disposable Filter Units with Nylon Membrane (Thermo Scientific, catalog number: 09-740-24A )
FisherbrandTM bulk-packed HDPE 7 ml scintillation vial (Fisher Scientific, catalog number: 03-337-20 )
WhatmanTM binder-free glass microfiber filters, grade GF/C (Cytiva, catalog number: 09-874-32A )
DWK Life Sciences KimbleTM solid borosilicate glass beads (Fisher Scientific, catalog number: 10-310-3 )
PuritanTM hospital standard cotton swab (Fisher Scientific, catalog number: 22-029-684 )
SartorisTM VivaspinTM 20 centrifugal concentrator (Sartorius, catalog number: 14-558-501 )
Thermo ScientificTM NalgeneTM Rapid-FlowTM sterilize disposable filter units with nylon membrane (Fisher Scientific, catalog number: 0974024A )
Slide-A-LyzerTM MINI Dialysis Devices, 3.5K MWCO, 0.5 ml (Fisher Scientific, catalog number: 88400 )
14 ml round-bottom tubes (Fisher Scientific, catalog number: 12-565-971 )
250 ml Nalgene® centrifuge bottles (Sigma-Aldrich, catalog number: Z353736 )
Pichia pastoris SMD 1163 strain (Invitrogen)
pPIC9K vector (Invitrogen)
pPIC9K_ADORA2A (Engineered from pPIC9K)
XL-10-GoldTM ultracompetent E. coli cells (Agilent, catalog number: 200314 )
BCA Protein Assay Kit (Pierce, catalog number: PI23227 )
Imidazole (Fisher Chemical edge, catalog number: 03196-500 )
EDTA (Sigma-Aldrich, catalog number: E5134 )
Glacial acetic acid (Fisher Scientific, catalog number: A385-500 )
Methanol (Fisher Scientific, catalog number: 412-500 or A412P4 )
Glycerol (Fisher Scientific, catalog number: G33-500 )
D-sorbitol (Fisher Scientific, catalog number: S459 )
G418 sulfate (Fisher Scientific, catalog number: BP6735 or Wisent Inc., catalog number: 400-130-XG )
Ampicillin sodium salt (Wisent Inc., catalog number: 400-110-EG or Fisher Scientific, catalog number: BP176025 )
LB miller broth (Wisent Inc, catalog number: 800-061-1K )
LB agar miller powder (Fisher Scientific, catalog number: BP14252 )
Yeast extract (Wisent Inc., catalog number: 800-150-IK )
Bacteriological peptone (Wisent Inc., catalog number: 800-157-IK )
Yeast nitrogen base w/o amino acid (Wisent Inc., catalog number: 800152018WG )
Dextrose (D-Glucose) (Fisher Scientific, catalog number: D16-500 )
Agar (Fisher Scientific, catalog number: BP1423-500 )
D-Biotin (Sigma-Aldrich, catalog number: 2031 )
L-histidine (Sigma-Aldrich, catalog number: H8000-5G or Fisher Scientific, catalog number: BP382100 )
DMSO (Fisher Scientific, catalog number: D1391 )
Theophylline (Sigma-Aldrich, catalog number: T1633 )
NaCl (Fisher Scientific, catalog number: S271-500 )
Bis-Tris (Fisher Scientific, catalog number: BP301-500 )
Lauryl maltose neopentyl glycol (MNG-3) (Anatrace, catalog number: MG310 )
Cholesterol hemisuccinate (CHS) (Sigma-Aldrich, catalog number: C6512 )
Talon Metal Affinity Resin (TaKaRa, catalog number: 635503 )
(Optional) Liquid Nitrogen
BamHI-HF (New England BioLabs, catalog number: R3136S )
NotI-HF (New England BioLabs, catalog number: R3189S )
PmeI (New England BioLabs, catalog number: R0560S )
T4 DNA ligase (New England BioLabs, catalog number: M0202S )
Quick CIP (New England BioLabs, catalog number: M0525S )
GenEluteTM Plasmid Miniprep Kit (Sigma-Aldrich, catalog number: PLD35-1KT )
100% ethanol (Fisher Scientific, catalog number: A4094 )
HRP-conjugated 6*His, His-Tag Monoclonal Antibody (Proteintech, catalog number: HRP-66005 )
DDDDK TAG Polyclonal Antibody (Equivalent to FLAG® Antibodies) (Proteintech, catalog number: HRP-20543-1-AP )
Zymolyase-20T (Amsbio, catalog number: 120493-1 )
Xanthine amine congener (Sigma-Aldrich, catalog number: X103 )
Affi-GelTM 10 (Bio-Rad, catalog number: 1536099 )
Isopropanol (Fisher Scientific, catalog number: A426P-4 )
Antifoam A Concentrate (Sigma-Aldrich, catalog number: A5633 )
Thermo ScientificTM Yeast DNA Extraction Kit (Fisher Scientific, catalog number: PI78870 )
Thermo ScientificTM Y-PERTM Yeast Protein Extraction Reagent (Fisher Scientific, catalog number: PI78991 )
Universal-ES liquid scintillation cocktail (MP BiomedicalsTM, catalog number: 88248001 )
3,3’,5,5’-tetramethylbenzidine solution (Alfa Aesar, catalog number: J61325 )
PageRuler Plus Prestained Protein Ladder (Fisher Scientific, catalog number: 26619 )
BioAcryl-P (30%, 29:1) liquid (Alfa Aesar, catalog number: J63279 )
QIAquick Gel Extraction Kit (Qiagen, catalog number: 28704 )
10× Phosphate buffer (see Recipes)
Immunoblotting Blocking buffer (see Recipes)
Immunoblotting Incubation buffer (see Recipes)
Immunoblotting Washing buffer (see Recipes)
LB plates (see Recipes)
YNBD plates (see Recipes)
YPD medium (see Recipes)
YPD agar plates (see Recipes)
BMGY medium (see Recipes)
BMMY medium (see Recipes)
Common Washing Buffer P1 (see Recipes)
Receptor Lysis Buffer P2 (see Recipes)
Receptor Preparation Buffer P3 (see Recipes)
Receptor Preparation Column Washing Buffer P4 (see Recipes)
Column Elution Buffer P5 (see Recipes)
XAC Column Elution Buffer P6 (see Recipes)
G Protein Washing Buffer P1 (see Recipes)
G Protein Lysis buffer P2 (see Recipes)
G Protein Elution Buffer P3 (see Recipes)
Q Buffer A-Gα (see Recipes)
Q Buffer B-Gα (see Recipes)
Q Buffer A-Gβγ (see Recipes)
Q Buffer B-Gβγ (see Recipes)
Immunoblotting Blocking Buffer (see Recipes)
Immunoblotting Washing Buffer (see Recipes)
Radioligand Binding/Washing Buffers (see Recipes)
Equipment
(Optional) -80 °C freezer
Thermo ScientificTM SorvallTM Centrifuge (Thermo Scientific, model: ST 40R )
EppendorfTM Microcentrifuge (Eppendorf, model: Legend Micro 21R )
Applied BiosystemsTM SimpliAmpTM Thermal Cycler (Applied Biosystems, model: A24812 )
GenesysTM UV-vis spectrometer (Genesys, model: 10S )
Thermo ScientificTM Heratherm Microbiological Incubator 117L (Thermo Scientific, model: IGS100 )
NanoDropTM spectrophotometer (NanoDrop, model: Lite )
ÄKTApurifier FPLC (GE, model: Purifier 100 )
Cell electroporation system electroporator (Bio-Rad, model: Gene Pulser X )
FisherbrandTM Heavy-Duty Vortex Mixer (Fisherbrand, model: STD )
5 L New Brunswick bioreactor (New Brunswick, model: BIOFLO III )
MicrofluidizerTM Processor (IDEX Material Processing, model: LM20 )
Procedure
This protocol was modified and finalized based on our prior (Ye et al., 2016 and 2018b) as well as current work. Different from our previous work, all target genes presented in this protocol were codon optimized using online codon optimization software from the IDT website and respective genes were synthesized prior to integration into the genome of P. pastoris via BamHI and NotHI restriction enzyme. In particular, both human derived adenosine A2AR (35.1KDa) and A1 receptor (36.5KDa) genes had FLAG and His10 tags in the N- and C-terminal ends, respectively. In addition, following the FLAG tag, a TEV protease cleavage site as well as α-Factor peptide were added in the front of genes (Figure 1). In contrast, there was no FLAG tag and α-Factor sequences in Gα (45.7KDa) as well as Gβ (38.7KDa) and Gγ (7.6KDa) constructs; in particular, there was no His-tag sequence in Gγ considering Gβ and Gγ were co-expressed together.
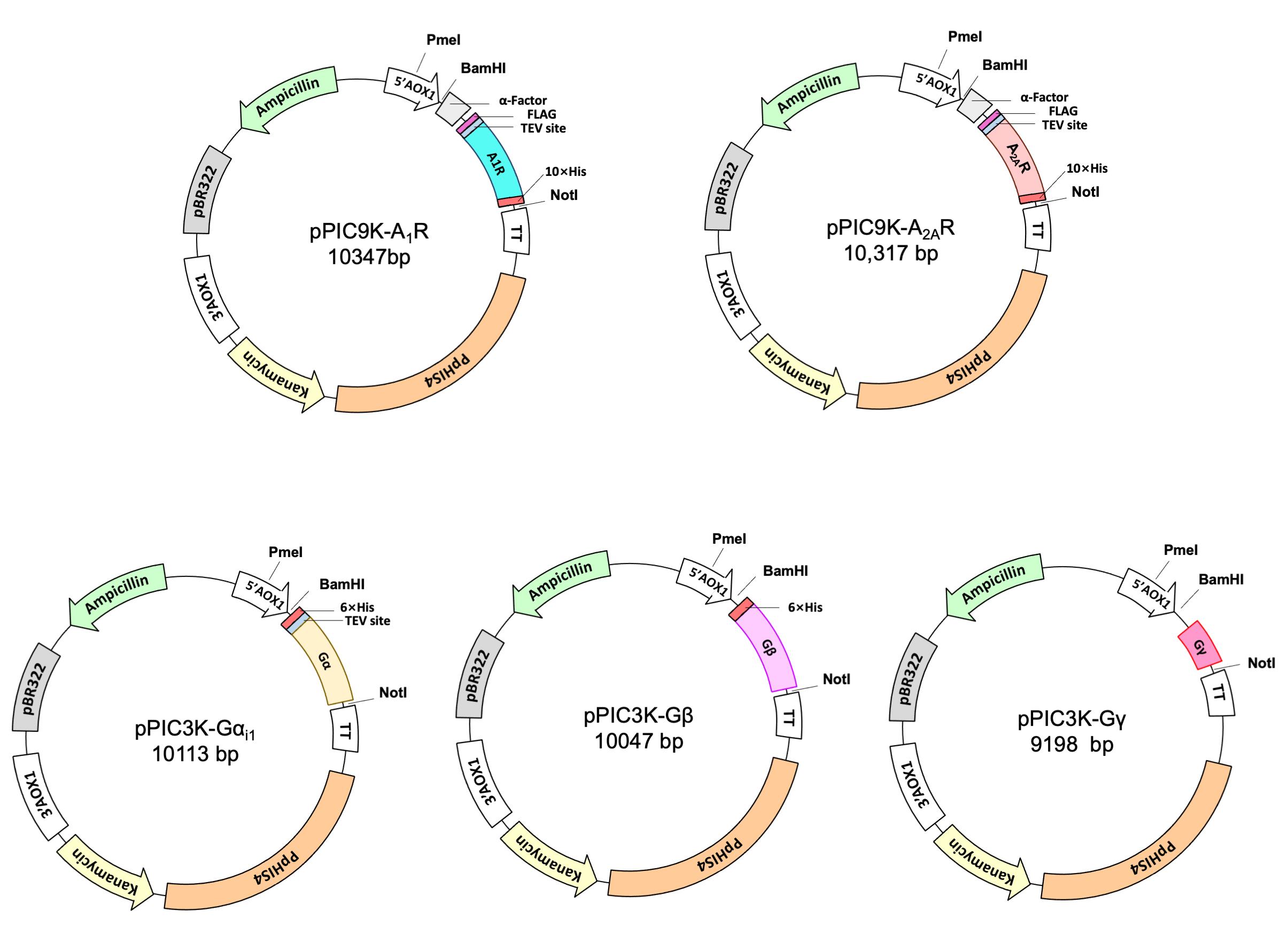
Figure 1. Constructs for GPCRs and G proteins described in this protocol. The pPIC9K-A1R/pPIC9K-A2AR had α-Factor peptide, the FLAG tag, a TEV protease cleavage. In contrast, there was no FLAG tag and α-Factor sequences in the G protein constructs; in particular, there was no His-tag motif in Gγ considering Gβ and Gγ were co-expressed together.
Plasmid Preparation for GPCRs and G proteins
1 μl of the construct pPIC9K_ADORA2A which was a recombinant vector of pPIC9K containing the ADORA2A receptor sequence, in a concentration of ~100 ng/μl, generously provided by T. Kobayashi (Japan) (Andre et al., 2006), was chemically transformed into 2 μl XL-10 Gold competent cells by heat shock for 45 s at 42 °C.
200 μl of LB medium was immediately added into the mixture and the transformed cells were spread on the 25 ml LB plates containing 50 μg/ml ampicillin. Incubated at 37 °C overnight.
One colony was picked from the plate and inoculated into a 4 ml LB medium containing 50 μg/ml ampicillin and cultured overnight at 37 °C.
The plasmid was extracted using GenEluteTM Plasmid Miniprep Kit following the instruction.
The extracted plasmids were digested with 1 μl BamHI-HF and1 μl NotI-HF with 1× NEB CutSmart® Buffer for 1.5 h in a 1.5 ml Eppendorf tube. 1 μl Quick CIP was added for additional 30 min digestion and phosphorylation to create pPIC9K backbone bearing BamHI and NotI restriction sites on each side. The total volumes were varied on the amounts of plasmids used. Usually, 1 μl of each restriction enzyme was used for the digestion of 20 μg plasmids.
All target gene fragments including A2AR, A1R, Gαs, Gβ, and Gγ (~200 ng/μl) were synthesized in accordance with P. pastoris codon optimized sequences. Of note, all gene fragments beared BamHI and NotI restriction enzyme sites at N- and C-termini.
The gene fragments were also digested with 1 μl of each BamHI-HF and NotI-HF restriction enzymes with 1× NEB CutSmart® Buffer for 2 h without Quick CIP treatment.
Both digested pPIC9K plasmid and gene fragments were run DNA electrophoresis (Krettler et al., 2013) and purified using QIAquick Gel Extraction Kit and the concentrations were measured using NanoDropTM Lite spectrophotometer.
The ligation process was processed with various ratios between backbone plasmid and gene fragments using 1 μl T4 DNA Ligase with 1× NEB T4 DNA Ligase Buffer in a 0.2 ml PCR tube. The molar ratios between gene fragments and empty plasmids were set to 1:3, 1:1, and 3:1 with concentrations of 10-100 ng/μl.
The ligated plasmids containing different gene fragments were transformed into XL-10 Gold competent cell (Smbrook et al., 1989; Dubnau, 1999) and spread on the LB plates containing 50 μg/ml ampicillin. Incubated overnight at 37 °C.
The plasmids for each constructs were extracted using GenEluteTM Plasmid Miniprep Kit. 20 μg plasmid in 100 μl for each construct was linearized with 1 μl PmeI restrict enzymes with 1× NEB CutSmart® Buffer for 2 h in a 0.2 ml PCR tube.
200 μl of 100% ethanol was added into the 100 μl linearized plasmids and incubated on the ice for 5 min.
The linearized plasmid was then centrifuged for 5 min at a speed of 16,200 × g at 4 °C.
The supernatant was discarded, and the linearized DNA pellet was dried under a Fume hood for 20 min before re-suspended in 20 μl of distilled water with DNA concentration around 1 μg/μl.
10 μl of linearized plasmids were mixed gently with 80 μl P. pastoris competent cells in 1.5 ml Eppendorf tube and kept on ice for 5 min prior to transfer into a 2 mm electroporation cuvette.
The transformations were performed using a Gene Pulser II electroporation with the condition of 1,500 V charging voltage, 25 μF capacitance, and 400 Ω resistance.
1 ml of ice-cold 1 M sorbitol was immediately added into the electroporation cuvette and transferred into 14 ml round-bottom tubes.
The samples were then incubated for 3 h at 30 °C without shaking prior to spreading them onto YNBD plates.
Of note, the linearized Gβ and Gγ plasmids (10 μl each) were transformed into the P. pastoris together while all other linearized plasmids were transformed separately.
Preparation of High-yield Constructs for GPCRs and G Proteins
As shown in Figure 2A, the colonies grown on the YNBD plates after 3-5 days incubation were transferred onto YPD plates containing 1 mg/ml G418 and incubated 3-5 days at 30 °C. YPD plates with gradient G418 concentration were prepared and stored at 4 °C for subsequent screening.
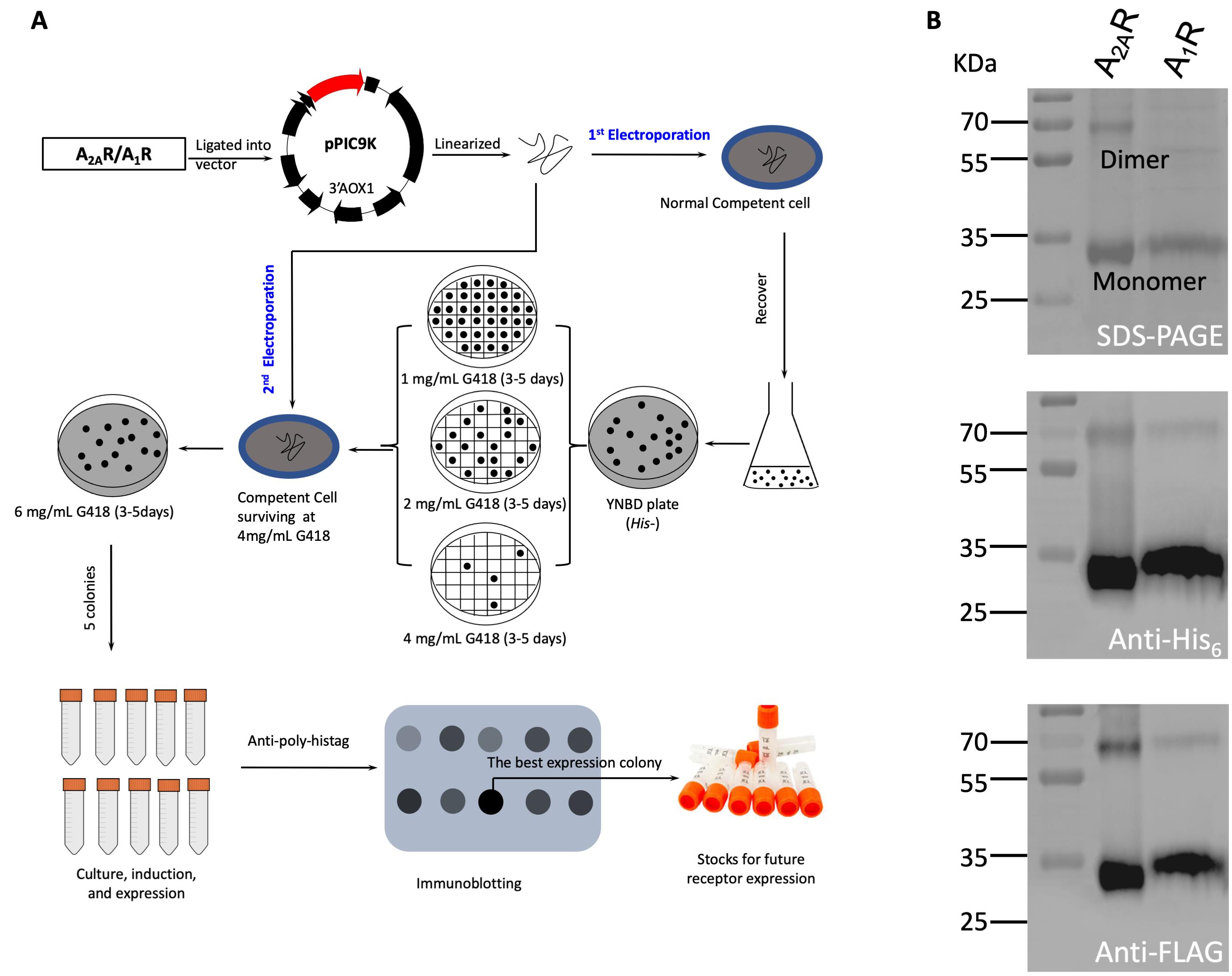
Figure 2. Schematic procedure of electroporation and high-yield construct screening for adenosine A2AR and A1R receptors (A) and their identifications using SDS-PAGE and western blotting assays (B)The colonies grown on YPD plates containing 1 mg/ml G418 were further transferred onto second YPD plates containing 2 mg/ml G418. Incubated 3-5 days at 30 °C.
Consecutively, the colonies grown on YPD plates containing 2 mg/ml G418 were finally transferred onto YPD plates containing 4 mg/ml G418. Incubated additional 3-5 days at 30 °C.
5-10 colonies on YPD plates containing 4 mg/ml G418 were picked for expression level evaluation using the immunoblotting assays against His10 and FLAG tags. Multiple integrated copies of pPIC9K can increase the Geneticin® resistance level from 0.5 mg/ml (1-2 copies) up to 4 mg/ml (7-12 copies).
The single colonies were inoculated into 4 ml BMGY medium in 14-ml Falcon tubes at 30 °C for at least 24 h with shaking (275 rpm) until OD595 = 2.0-6.0.
The medium was then transferred into 200 ml BMMY medium in 500 ml shake flasks covered by a cotton plug.
The cells were continued culturing 60 h at 22 °C with shaking at 275 rpm.
0.5% methanol/12 h was added into the medium in order to induce and maintain the receptor or G protein expression.
At the end of induction, the cell pellets were collected after centrifugation at 3,800 × g for 10 min at 4 °C in 250 ml centrifuge bottles.
The cell pellets were washed once with Common Washing Buffer P1 in a ratio of 1:2.
The cell pellets were then resuspended into Receptor Lysis Buffer P2 using the ratio of cell pellets to buffer equal to 1:4.
The cell pellets suspensions were then vortexed at 2,000 rpm for 2 h at 4 °C in the presence of a slurry of 5 mm glass beads.
The disrupted cell pellets were centrifuged at 9,720 × g for 30 min at 4 °C and the unbroken cells and cellular debris were discarded.
The supernatant containing cell membrane was collected and applied to immunoblotting assays (Gallagher and Chakavarti, 2008).
For accuracy, immunoblotting was performed for both anti-His and anti-FLAG in response to the FLAG-tag and His-tags, respectively.
1 μl of supernatant was blotted on nitrocellulose membrane and allowed to dry.
A second 1 μl of supernatant was applied onto the previous position and let it dry.
The membrane was placed in 20 ml Immunoblotting Blocking Buffer for 1 h at room temperature.
The membrane was then transferred to 20 ml Immunoblotting Incubation Buffer containing either His-tag antibody (1:2,000) or FLAG-tag antibody (1:2,000) for 2 h.
The membrane was then washed three times with Immunoblotting Washing Buffer, followed by distilled water.
The membrane was finally visualized by BM Blue POD substrate (3,3’,5,5’-tetramethylbenzidine solution).
The strongest intensity colonies were selected for further expressions. If the expression level was less than 0.5 mg/L cell culture, a second-cycle plasmid transformation was conducted following the instruction described in Procedure A and B. The colonies were directly screened on YPD plates containing 6 mg/ml G418 (Figure 2A).
The stocks of screened high-yield expression constructs were made with 20% autoclaved glycerol and frozen in the -80 °C freezer or Liquid Nitrogen.
Expression of GPCRs and G proteins
The expressions of GPCRs and G proteins were using the similar procedure with slight variances described in the following section.
Glycerol stocks of the transformants were inoculated onto YPD agar plates containing 0.1 mg/ml G418.
3-5 days later, a single colony was inoculated into 4 ml autoclaved YPD at 30 °C in 14 ml round bottom tubes with shaking at 275 rpm for 24 h.
4 ml medium was subsequently transferred into 200 ml autoclaved BMGY medium and cultured at 30 °C in 500 ml shake flask covered by aluminum foil with a shaking speed of 275 rpm till OD595 reached 2-6, which would take about 24 h.
To induce expression, 200 ml cell pellets were spun down and transferred into 1 liter of autoclaved BMMY medium in 2.8 L flasks and cultured at 20 °C with a shaking speed of 275 rpm. Filtered methanol was added every 12 h with 0.5% (5 ml/L medium) if the baffled flasks were used. If the fermenter was used, the rate of methanol addition was controlled at maximal 1 ml/h for each liter, which was dependent on the culture volume
The cells for receptors expression were collected after 80 h fermentation while the cells for G proteins expression were collected after 60 h.
Purifications of GPCRs and G proteins
Purifications of GPCRs, Gα, and Gβγ proteins were different in this protocol. Therefore, we described them separately in the following sections.
Purification of GPCRs
After 80 h expression, the cell pellets were collected at 3,800 × g for 10 min at 4 °C in 250 ml centrifuge bottles.
Cell pellets were washed once with Common Washing Buffer P1 in the ratio of 1 g cell pellets to 2 ml P1 and centrifuged at 3,800 × g for 10 min at 4 °C in 250 ml centrifuge bottles.
The washed cell pellets were resuspended in Receptor Lysis buffer P2 in a ratio of 4:1 to ensure the suspension was sufficient in 250 ml centrifuge bottles.
The cells were then disrupted using the Microfluidizer for four cycles on the ice at the working pressure of 15,000 psi. The Microfluidizer was balanced with buffer P2 prior to the lysis.
Intact cells and cell debris were separated from the membrane suspension at 9,720 × g for 30 min at 4 °C in 250 ml centrifuge bottles.
The supernatant was then collected and centrifuged at 100,000 × g for 75 min using Type 45 Ti rotor for the Beckman Ultracentrifuge with corresponding tubes.
The supernatant from ultracentrifugation was discarded and the membranes from different runs were collected together and suspended in Common Washing Buffer P1 to remove the EDTA.
The supernatant from ultracentrifugation was discarded and the membrane pellets were dissolved in a 50 ml conical centrifuge tube containing Receptor Preparation Buffer P3 and shaking at 4 °C until all membranes were dissolved sufficiently.
The solution was centrifuged at 1,980 × g for 5 min at 4 °C to remove the undissolved membrane.
The dissolved membranes were mixed with pre-balanced Talon Resin using Receptor Preparation Column Washing Buffer P4. Usually, 2 ml Talon Resin was used for 6 g membrane. Incubated for 2 h at 4 °C. During the incubation, the imidazole was added to the final concentration of 100 μM.
Two hours later, the Talon resin was packed onto a disposal column and washed with 5 column volumes of Receptor Preparation Column Washing Buffer P4.
Subsequently, the receptors were eluted from the column using the Column Elution Buffer P5 at a gravity rate.
The eluted receptors were concentrated to 1 ml by Ultra-15 Centrifugal filters 3K and buffer change with 10 ml Receptor Preparation Column Washing Buffer P4 one time with a dilution factor of 10 at 4 °C with the speed of 3,846 × g.
The receptor then went through the XAC ligand column, which was pre-balanced using Receptor Preparation Column Washing Buffer P4. Repeated three times.
Wash the receptor bound XAC column for 2 column volumes using Receptor Preparation Column Washing Buffer P4 to remove the non-functional receptors.
The receptors were then eluted out using XAC Column Elution Buffer P6 consisting of Receptor Preparation Column Washing Buffer P4 with 25 mM theophylline.
The eluted receptors were concentrated into 1 ml by Ultra-15 Centrifugal filters 3K and dialyzed against Receptor Preparation Column Washing Buffer P4 by Slide-A-LyzerTM MINI Dialysis Devices 3.5K with a dilution factor of 106 to remove all ligands to bring the receptor into the apo state.
The purified receptors usually with 0.5-2 mg/L productivity were validated by SDS-PAGE (Laemmli, 1970) as well as immunoblotting, as shown in Figure 2B.
Cell pellets were harvested by centrifugation at 3,800 × g for 10 min using the centrifuge at 4 °C in 250 ml centrifuge bottles.
Cell pellets were washed once with Common Washing Buffer P1 in the ratio of 1 g cell pellets to 2 ml P1 and centrifuged with the speed of 3,800 × g for 10 min at 4 °C in 250 ml centrifuge bottles.
Cell pellets were resuspended in G Protein Lysis buffer P2 in a ratio of 1:4.
Cells were broken by Microfluidizer using 4 cycles at 15,000 psi for completely disrupting the yeast cell wall. The Microfluidizer was balanced with buffer P2 prior to the lysis.
The lysate was centrifuged at 4 °C for 30 min for 9,720 × g in 250 ml centrifuge bottles.
The supernatant was applied to Talon resins for 2 h, in which imidazole was added with a final concentration of 100 μM in order to decrease non-specific binding.
The G protein bound Talon resins were applied to a disposal column.
The packed column was then washed with 5 column volumes of G Protein Washing buffer P1.
The target Gα was eluted with 10 ml G Protein Elution Buffer P3 at a gravity rate. Strategically recycling the elution buffer to reduce the elution volume was optional.
MgCl2 was added to the final concentration of 1 mM as well as GDP of 50 μM.
The Gα was concentrated to 2 ml by Ultra-15 Centrifugal filters, 3K at 4 °C with the speed of 3,846 × g. The FPLC system was balanced with Buffer A- Gα for Q Sepharose Column prior to applying the eluted Gα protein into the system.
Q Buffer A- Gα: 50 mM HEPES, pH 8.0, 50 μM GDP, 1 mM MgCl2.
Q Buffer B- Gα: 50 mM HEPES, pH 8.0, 50 μM GDP, 1 mM MgCl2, 1,000 mM NaCl.
The Gα protein should be eluted with 20% of Q buffer B.
The FPLC program for target Gα elution is shown in Figure 3 and the Gα was eluted out as shown in the graph, in which the flow rate, maximum column pressure, and fraction were set to 1.0 ml/min, 0.5 MPa, and 2.0 ml, respectively.
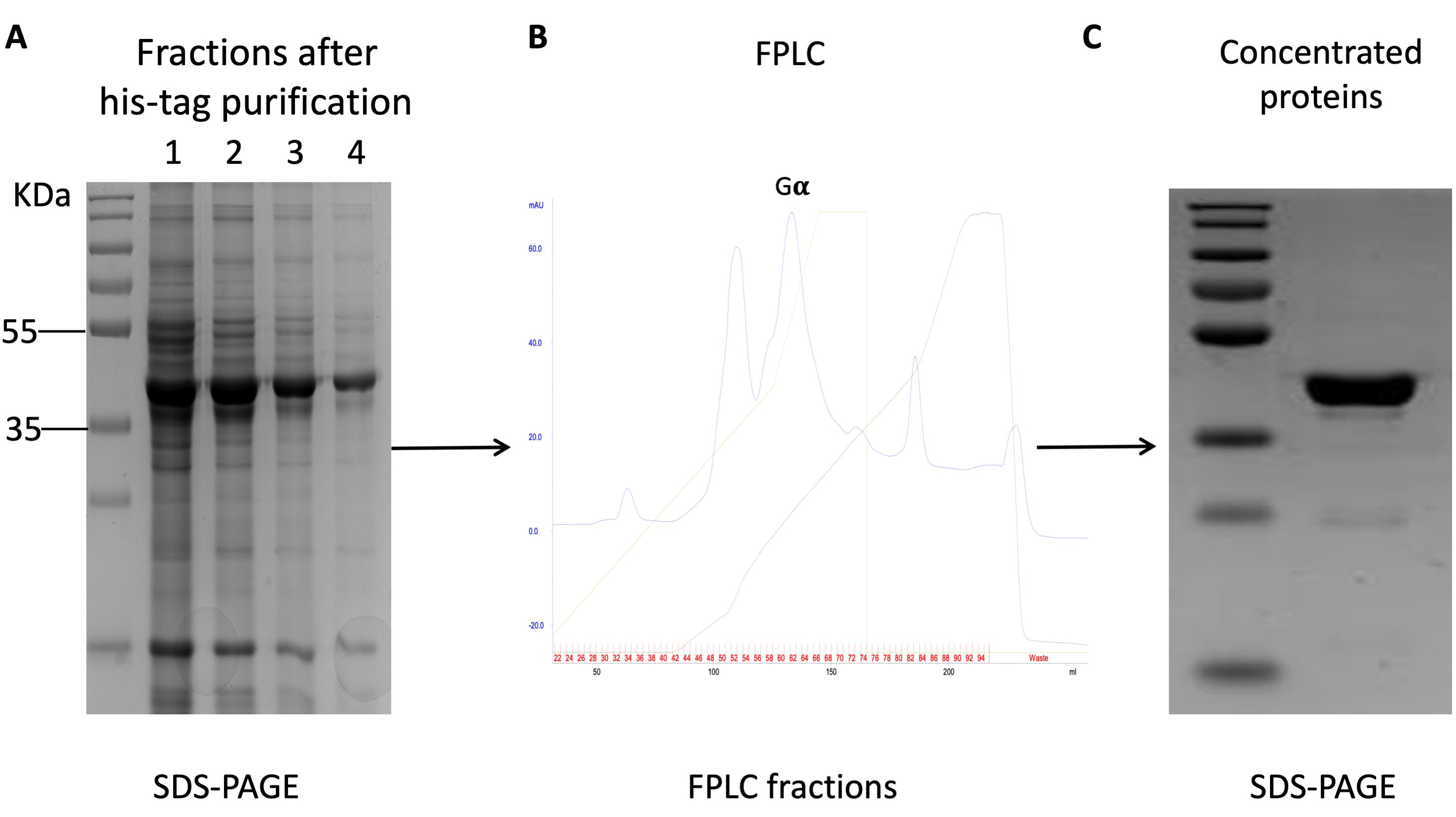
Figure 3. Purification of yeast-derived Gα proteins. A. SDS-PAGE for the fractions after his-tag purification. B. The elution program and resultant elution profile for Gα. C. SDS-PAGE for concentrated fraction from FPLC marked in (B).The corresponding fractions were collected and concentrated to 1 ml using Ultra-15 Centrifugal filters 3K at 4 °C with the speed of 3,846 × g.
The concentration of the Gα was measured using BCA kit with a productivity of 2-5 mg/L cell culture.
Cell pellets were harvested by centrifugation at 3,800 × g for 10 min using the centrifuge at 4 °C in 250 ml centrifuge bottles. The supernatant was discarded.
Cell pellets were washed once with Common Washing Buffer P1 in the ratio of 1 g cell pellets to 2 ml P1 and centrifuge at 3,800 × g for 10 min at 4 °C in 250 ml centrifuge bottles.
Cell pellets were washed one time with 1:2 ratio Common Washing Buffer P1 and centrifuged at 4 °C with the speed of 3,800 × g for 10 min.
The cell pellets were suspended in ice-cold G Protein Lysis Buffer P2.
The cell pellets were lysed by Microfluidizer for 4 cycles at a pressure of 15,000 psi. The Microfluidizer was balanced with Buffer P2 prior to lysis.
The lysed cell pellets were centrifuged at 9,720 × g for 30 min to remove all debris and intact cells in 250 ml centrifuge bottles.
The supernatant was applied to Talon resin for 2 h.
Note: Imidazole was added to the final concentration of 100 μM in order to decrease non-specific binding proteins.
The G protein bound resins were applied onto a disposal column.
The column was washed with 5 column volumes of Common Washing buffer P1.
The Gβγ was eluted with 10 ml G Protein Elution Buffer at a gravity rate. Strategically recycling the elution buffer to reduce the elution volume was optional.
The Gβγ was concentrated to 2 ml by Ultra-15 Centrifugal filters 3K at 4 °C with the speed of 3,846 × g. and changed the buffer into Q Sepharose High performance Buffer A-Gβγ.
The sample was applied onto the FPLC and eluted with gradient elution with Q Sepharose High performance Buffer B-Gβγ (Figure 4A).
Fractions were collected using the same parameters for the Gα; a typical elution profile was as follows (Figure 4):
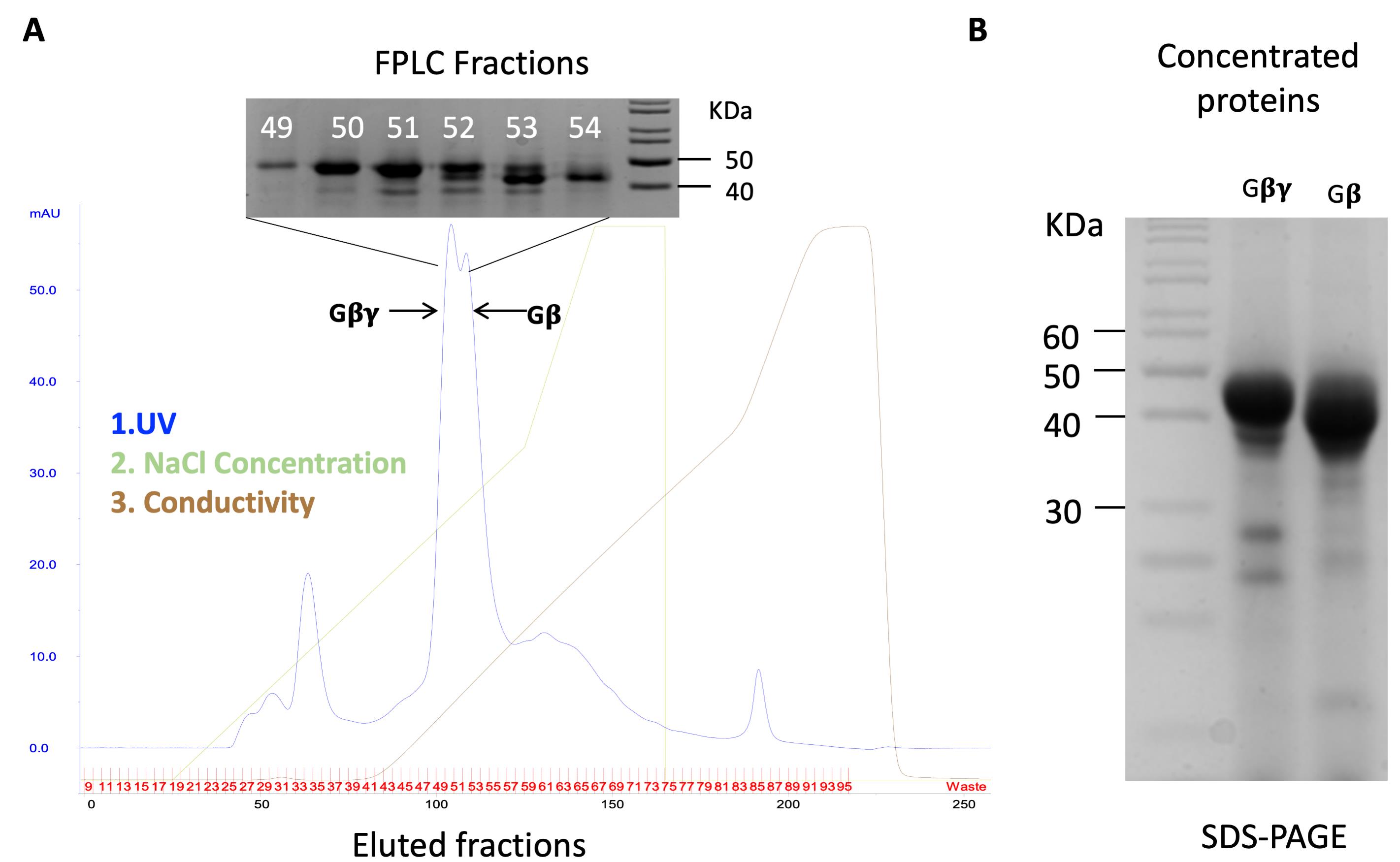
Figure 4. Purification of yeast-derived Gβγ proteins. A. The elution program and resultant elution profile for Gβγ. B. SDS-PAGE for the concentrated fractions marked in (A).The eluted fractions were collected and concentrated with a productivity of 2-5 mg/L cell culture similar to Gα. The purity and molecular weight of proteins were validated using SDS-PAGE as shown in Figure 4B.
Recipes
10× Phosphate buffer
49.7 g Na2HPO4
98.0 g NaH2PO4 in 1,000 ml dH2O, pH 6.5
Immunoblotting Blocking buffer
125 mM NaCl
25 mM Tris base, pH 7.5, 0.3% Tween-20 and 3% non-fat milk
Immunoblotting Incubation buffer
Anti-His/Anti-Flag antibody diluted to 1:2,000 with blocking buffer
Immunoblotting Washing buffer
125 mM NaCl
25 mM Tris base, pH 7.5
0.3% Tween-20
LB plates
2.5% LB broth
1.5% Agar
YNBD plates
1.34% Yeast Nitrogen based w/o amino acid
0.0004% D-Biotin
1% Dextrose
1.5% Agar
YPD medium
1% Yeast extract
2% Peptone
2% Dextrose
YPD agar plates
1% Yeast extract
2% Peptone
2% Glucose
2% Agar
BMGY medium
1% (w/v) Yeast extract
2% (w/v) Peptone
1.34% (w/v) YNB without amino acids
0.00004% (w/v) Biotin
1% (w/v) Glycerol
0.1 M Phosphate buffer at pH 6.5
BMMY medium
1% (w/v) Yeast extract
2% (w/v) Peptone
1.34% (w/v) Yeast nitrogen base without amino acids
0.00004% (w/v) Biotin
0.5% (w/v) Methanol
0.1 M Phosphate buffer at pH 6.5
0.04% (w/v) Histidine and 3% (v/v) DMSO
10 μM Theophylline
Common Washing Buffer P1
20 mM Bis-Tris, pH 6.5 (50 mM HEPES, pH 7.4)
Receptor Lysis Buffer P2
20 mM Bis-Tris, pH 6.5 (50 mM HEPES, pH 7.4)
2.5 mM EDTA
10% Glycerol
Receptor Preparation Buffer P3
20 mM Bis-Tris, pH 6.5 (50 mM HEPES, pH 7.4)
100 mM NaCl
1% MNG-3 and 0.02% CHS
Receptor Preparation Column Washing Buffer P4
20 mM Bis-Tris, pH 6.5 (50 mM HEPES, pH 7.4)
100 mM NaCl
0.1% MNG-3 and 0.002% CHS
Column Elution Buffer P5
20 mM Bis-tris, pH 6.5 (50 mM HEPES, pH 7.4)
100 mM NaCl
0.1% MNG-3
0.002% CHS
300 mM Imidazole
XAC Column Elution Buffer P6
20 mM theophylline in Receptor Receptor Preparation Column Washing Buffer P4
G Protein Washing Buffer P1
50 mM HEPES, pH 8.0
G Protein Lysis buffer P2
50 mM HEPES, pH 8.0
10% Glycerol
100 mM NaCl
G Protein Elution Buffer P3
50 mM HEPES, pH 8.0, 300 mM imidazole
Q Buffer A- Gα
50 mM HEPES, pH 8.0
50 μM GDP
1 mM MgCl2
Q Buffer B- Gα
50 mM HEPES, pH 8.0
50 μM GDP
1 mM MgCl2
1,000 mM NaCl
Q Buffer A-Gβγ
20 mM Bis-Tris, pH 6.5 (50 mM HEPES, pH 8.0)
Q Buffer B-Gβγ
20 mM Bis-Tris, pH 6.5 (50 mM HEPES, pH 8.0)
1,000 mM NaCl
Immunoblotting Blocking Buffer
125 mM NaCl
25 mM Tris base, pH 7.5
0.3% Tween-20
3% Non-fat milk
Immunoblotting Washing Buffer
125 mM NaCl
25 mM Tris base, pH 7.5
0.3% Tween-20
Radioligand Binding/Washing Buffers
25 mM HEPES, pH 7.4
100 mM NaCl
Acknowledgments
The article was supported by Nexus Initiative (L. Y.) from University of South Florida (USF) as well as startup funding (L.Y) from the department of Cell Biology, Microbiology and Molecular Biology at USF.
Competing interests
There are no conflicts of interest or competing interest.
References
- Andre, N., Cherouati, N., Prual, C., Steffan, T., Zeder-Lutz, G., Magnin, T., Pattus, F., Michel, H., Wagner, R. and Reinhart, C. (2006). Enhancing functional production of G protein-coupled receptors in Pichia pastoris to levels required for structural studies via a single expression screen. Protein Sci 15(5): 1115-1126.
- Congreve, M., de Graaf, C., Swain, N. A. and Tate, C. G. (2020). Impact of GPCR Structures on Drug Discovery. Cell 181(1): 81-91.
- Dubnau, D. (1999). DNA uptake in bacteria. Annu Rev Microbiol 53: 217-244.
- Gallagher, S. and Chakavarti, D. (2008). Immunoblot analysis. J Vis Exp (16). doi:10.3791/759.
- Krettler, C., Reinhart, C. and Bevans, C. G. (2013). Expression of GPCRs in Pichia pastoris for structural studies. Methods Enzymol 520: 1-29.
- Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T. Nature 227(5259): 680-685.
- Overington, J. P., Al-Lazikani, B. and Hopkins, A. L. (2006). How many drug targets are there. Nat Rev Drug Discov 5(12): 993-996.
- Smbrook, J., Fritsch, E. F. and Maniatis, T. (1989). Molecular cloning: a laboratory manual. Cold Spring harbor laboratory.
- Ye, L., Neale, C., Sljoka, A., Lyda, B., Pichugin, D., Tsuchimura, N., Larda, S. T., Pomes, R., Garcia, A. E., Ernst, O. P., Sunahara, R. K. and Prosser, R. S. (2018). Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat Commun 9(1): 1372.
- Ye, L., Orazietti, A. P., Pandey, A. and Prosser, R. S. (2018). High-Efficiency Expression of Yeast-Derived G-Protein Coupled Receptors and 19F Labeling for Dynamical Studies. Methods Mol Biol 1688: 407-421.
- Ye, L., Van Eps, N., Zimmer, M., Ernst, O. P. and Prosser, R. S. (2016). Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 533(7602): 265-268.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Zhao, W., Wang, X. and Ye, L. (2021). Expression and Purification of Yeast-derived GPCR, Gα and Gβγ Subunits for Structural and Dynamic Studies. Bio-protocol 11(4): e3919. DOI: 10.21769/BioProtoc.3919.
Category
Biochemistry > Protein > Isolation and purification
Biochemistry > Protein > Expression
Biophysics > NMR spectroscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.